一种水不溶性温度pH双敏感微凝胶及其制备方法
一种水不溶性温度ph双敏感微凝胶及其制备方法
技术领域
1.本发明属于日用化工技术领域,具体涉及到一种水不溶性温度ph双敏感微凝胶及其制备方法。
背景技术:
2.水凝胶是一种天然的亲水性高聚物,一般由物理或化学行为交联形成三维网状结构,在水中具有极强的溶胀性能。其主要分为两类,一类为环境响应性凝胶,可根据外界温度、ph、电磁等变化发生亲水疏水变化;一类为环境不响应性水凝胶,这类水凝胶对外界变化没有明显变化。由于环境响应性凝胶的特殊性能使其受到广泛关注,其中温度/ph双重刺激相应微凝胶由于其特殊性能可作为生物组织和外界环境交互的媒介,成为近些年研究的热点,其常利用于载药、医学、生物传感、催化等领域。目前温度/ph双重刺激智能微凝胶主要采用的制备方法有无皂乳液聚合、乳液聚合、沉淀聚合。大部分聚合过程中通常引入金属元素或卤化物,这使产物在生物应用领域的应用受到限制。
3.缓释型微胶囊通常以具有缓释特性的材料作为壁材,采用微胶囊化技术将芯材包裹,以达到减缓芯材释放的目的。其中乙基纤维素由于多孔且具有缓释骨架结构,常在微胶囊中用作壁材。使微胶囊具有较好的缓释性能,从而提高药物利用率。
4.现有微胶囊主要研究方向在于利用一种或多种温度、ph、离子敏感性凝胶进行共聚,或对单体进行改性,制得不同条件下两种甚至更多种的刺激响应性微凝胶。但制备出的微凝胶多为水溶性,目前没有报道涉及制备油溶性多重响应性微凝胶,其药物包封条件受到限制,释放速率较高,在生物应用和药品包封性能上仍有较大待提高空间。
技术实现要素:
5.本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本申请的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
6.鉴于上述和/或现有技术中存在的问题,提出了本发明。
7.因此,本发明克服现有技术中存在的不足,提供一种水不溶性温度ph双敏感微凝胶及其制备方法,使得制备出的微凝胶具有水不溶性,大幅提升微凝胶的缓释效果和其在油性环境下的应用。
8.为解决上述技术问题,本发明提供了如下技术方案:一种水不溶性温度ph双敏感微凝胶的制备方法,包括,
9.将温敏性水凝胶单体、ph敏感性水凝胶单体、交联剂置于水中溶解,得到水相溶液;
10.向所述水相溶液中加入引发剂溶液,得到混合溶液;
11.将高分子聚合物置于油相中溶解,向溶液中通入氮气排除氧气,得到油相溶液;
12.将所述混合溶液加入所述油相溶液中,剪切混合,得到乳液;
13.将所述乳液经紫外光照交联,洗涤后冷冻干燥,得到本发明的水不溶性温度ph双敏感微凝胶。
14.作为本发明水不溶性温度ph双敏感微凝胶的制备方法的一种优选方案,其中:所述水相溶液中,所述温敏性水凝胶单体的质量分数为2~40mas%,所述ph敏感性水凝胶单体的质量分数为5~50mas%,所述交联剂的质量分数为0.1~5mas%。
15.作为本发明水不溶性温度ph双敏感微凝胶的制备方法的一种优选方案,其中:所述油相溶液中,所述高分子聚合物的质量分数为0.5~10mas%。
16.作为本发明水不溶性温度ph双敏感微凝胶的制备方法的一种优选方案,其中:所述引发剂溶液,由引发剂单体于水中溶解得到,其中,所述引发剂单体的质量分数为0.1~5mas%。
17.作为本发明水不溶性温度ph双敏感微凝胶的制备方法的一种优选方案,其中:所述将水相溶液加入油相溶液,所述油相溶液与所述水相溶液的体积比为0.5~10:1。
18.作为本发明水不溶性温度ph双敏感微凝胶的制备方法的一种优选方案,其中:所述剪切混合,在5000~10000rpm下剪切混合1~5min。
19.作为本发明水不溶性温度ph双敏感微凝胶的制备方法的一种优选方案,其中:所述紫外光照交联,光照时间为5~40min。
20.作为本发明水不溶性温度ph双敏感微凝胶的制备方法的一种优选方案,其中:所述洗涤,通过乙醇洗涤1~5次。
21.本发明的另一个目的是提供一种水不溶性温度ph双敏感微凝胶,所述水不溶性温度ph双敏感微凝胶由上述任一所述的水不溶性温度ph双敏感微凝胶的制备方法所制备得到。
22.作为本发明水不溶性温度ph双敏感微凝胶的一种优选方案,其中:所述温敏性水凝胶单体包括n
‑
异丙基丙烯酰胺、乙二醇、壳聚糖、黄原胶中的一种或多种;
23.所述ph敏感性水凝胶单体包括甲基丙烯酸、丙烯酸、甲基丙烯酸二甲氨基乙酯中的一种或多种;
24.所述交联剂包括n,n
‑
亚甲基双丙烯酸胺、乙二醇二甲基丙烯酸酯中的一种或多种;
25.所述油相包括2
‑
辛基十二烷醇、液体石蜡、角鲨烷、辛酸癸酸甘油三酯中的一种或多种;
26.所述高分子聚合物包括纤维素、壳多糖、支链淀粉、乙基纤维素中的一种或多种;
27.所述引发剂包括二苯甲酮、偶氮二异丁脒盐酸盐、安息香乙醚中的一种或多种。
28.与现有技术相比,本发明具有如下有益效果:本发明将一种温度敏感性和一种ph敏感性凝胶共聚,通过光引发聚合和乳液聚合的手段制备温度/ph双敏感型微凝胶,在不添加其他表面活性剂的情况下,在微凝胶表层附着一层乙基纤维素薄膜,使得制备出的微凝胶具有水不溶性,大幅提升微凝胶的缓释效果和其在油性环境下的应用。并且,所获得的微凝胶缓释效果可借助水相质量分数、微凝胶洗涤次数等条件进行宽范围和高效调控,有望应用于生物缓释和其他特殊性质化妆品产品。
附图说明
29.为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:
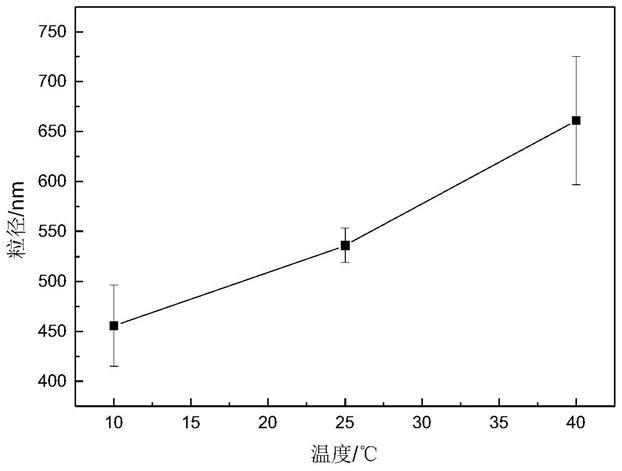
30.图1为实施例2制备的微凝胶在不同温度条件下的粒径分布。
31.图2为实施例2制备的微凝胶在不同ph条件下的粒径分布。
32.图3为实施例2制备的微凝胶在不同ph条件下的包载释放结果。
33.图4为光学显微镜观察交联后乳液形貌;其中,(a)为实施例1制备出乳液固化后乳液分散在乙醇中得到的光学显微镜观察图像;(b)为实施例2制备出乳液固化后乳液分散在乙醇中得到的光学显微镜观察图像;(c)为实施例3制备出乳液固化后乳液分散在乙醇中得到的光学显微镜观察图像。
34.图5为扫描电子显微镜观察微胶囊外观图;其中,(a)为实施例1制备的微胶囊外观图;(b)为实施例2制备的微胶囊外观图;(c)为实施例3制备的微胶囊外观图。
35.图6为微凝胶光学接触角测量结果图;其中,(a)为实施例1制备的微凝胶光学接触角测量结果图;(b)为实施例2制备的微凝胶光学接触角测量结果图;(c)为实施例3制备的微凝胶光学接触角测量结果图。
36.图7为洗涤次数不同所制得微胶囊包载释放结果对比图。
具体实施方式
37.为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合说明书实施例对本发明的具体实施方式做详细的说明。
38.在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
39.其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
40.实施例1
41.(1)将质量分数为10%的温敏性水凝胶单体n
‑
异丙基丙烯酰胺、质量分数为5%ph敏感性水凝胶单体甲基丙烯酸二甲氨基乙酯、质量分数为2%的交联剂n,n
‑
亚甲基双丙烯酸胺置于去离子水中,涡旋至完全溶解;为了模拟包载释放实验,本实施例在其中还加入了一定量的染色剂,具体的是加入质量分数为5%的甲基蓝染色剂,随之涡旋至完全溶解;
42.(2)将质量分数为0.5%的乙基纤维素置于2
‑
正辛基
‑1‑
十二烷醇的油相中,加热搅拌至完全溶解;
43.(3)将质量分数为0.5%的引发剂偶氮二异丁脒盐酸盐置于去离子水中,涡旋至完全溶解;
44.(4)将步骤(2)所得油相溶剂通入氮气,至完全排除氧气;
45.(5)将步骤(3)所得引发剂溶液加入步骤(1)所得水相溶液中,涡旋至混合均匀;
46.(6)将步骤(5)所得混合溶液按照油水比为3:1加入步骤(2)所得油相溶液中,采用fm200高速剪切机在8000rpm下剪切混合2min,得到乳液;
47.(7)将步骤(6)所得乳液置于紫外交联仪中,光照交联40min;
48.(8)将步骤(7)所得微凝胶由乙醇充分洗涤2次,置于真空冷冻干燥剂中充分干燥,得到微凝胶样品1。
49.实施例2
50.(1)将质量分数为10%的温敏性水凝胶单体n
‑
异丙基丙烯酰胺、质量分数为5%ph敏感性水凝胶单体甲基丙烯酸二甲氨基乙酯、质量分数为2%的交联剂n,n
‑
亚甲基双丙烯酸胺置于去离子水中,涡旋至完全溶解;为了模拟包载释放实验,本实施例在其中还加入了一定量的染色剂,具体的是加入质量分数为5%的甲基蓝染色剂,随之涡旋至完全溶解;
51.(2)将质量分数为5%的乙基纤维素置于2
‑
正辛基
‑1‑
十二烷醇的油相中,加热搅拌至完全溶解;
52.(3)将质量分数为0.5%的引发剂偶氮二异丁脒盐酸盐置于去离子水中,涡旋至完全溶解;
53.(4)将步骤(2)所得油相溶剂通入氮气,至完全排除氧气;
54.(5)将步骤(3)所得引发剂溶液加入步骤(1)所得水相溶液中,涡旋至混合均匀;
55.(6)将步骤(5)所得混合溶液按照油水比为3:1加入步骤(2)所得油相溶液中,采用fm200高速剪切机在8000rpm下剪切混合2min,得到乳液;
56.(7)将步骤(6)所得乳液置于紫外交联仪中,光照交联40min;
57.(8)将步骤(7)所得微凝胶由乙醇充分洗涤2次,置于真空冷冻干燥剂中充分干燥,得到微凝胶样品2。
58.图1为微凝胶样品2在不同温度下的粒径分布;由图1可知,所制备的微凝胶存在温度敏感性。
59.图2为微凝胶样品2在不同ph条件下的粒径分布;由图2可知,所制备的微凝胶存在ph敏感性。
60.对微凝胶样品2进行模拟包载释放实验,测试方法为:取1mg包载冻干样品,溶解于1ml不同ph的pbs溶液中(本次试验分别在ph为2、7.4、10的条件下进行,环境温度为恒温24℃),装入透析袋,置于99ml相同ph环境的pbs溶液中透析,并保持磁力搅拌,每隔一定时间取1ml溶液并加回1ml相同pbs溶液。测量吸光度计算累计释放量。
61.图3为微凝胶样品2在不同ph条件下微凝胶包载释放效果;由图3可知,所制备的微凝胶存在ph敏感性,ph不同影响微凝胶粒径大小,进而对释放效果产生影响。
62.实施例3
63.(1)将质量分数为10%的温敏性水凝胶单体n
‑
异丙基丙烯酰胺、质量分数为5%ph敏感性水凝胶单体甲基丙烯酸二甲氨基乙酯、质量分数为2%的交联剂n,n
‑
亚甲基双丙烯酸胺置于去离子水中,涡旋至完全溶解;为了模拟包载释放实验,本实施例在其中还加入了一定量的染色剂,具体的是加入质量分数为5%的甲基蓝染色剂,随之涡旋至完全溶解;
64.(2)将质量分数为10%的乙基纤维素置于2
‑
正辛基
‑1‑
十二烷醇的油相中,加热搅拌至完全溶解;
65.(3)将质量分数为0.5%的引发剂偶氮二异丁脒盐酸盐置于去离子水中,涡旋至完
全溶解;
66.(4)将步骤(2)所得油相溶剂通入氮气,至完全排除氧气;
67.(5)将步骤(3)所得引发剂溶液加入步骤(1)所得水相溶液中,涡旋至混合均匀;
68.(6)将步骤(5)所得混合溶液按照油水比为3:1加入步骤(2)所得油相溶液中,采用fm200高速剪切机在8000rpm下剪切混合2min,得到乳液;
69.(7)将步骤(6)所得乳液置于紫外交联仪中,光照交联40min;
70.(8)将步骤(7)所得微凝胶由乙醇充分洗涤2次,置于真空冷冻干燥剂中充分干燥,得到微凝胶样品3。
71.图4为实施例1~3中添加不同质量分数乙基纤维素时,制备出乳液固化后乳液分散在乙醇中得到的光学显微镜观察图像。从图4可知,实施例1~3中采用0.5~10%乙基纤维素制得的微粒随乙基纤维素含量的增加变小,并且当乙基纤维素含量过少时,粒子出现黏连,破碎等现象。
72.扫描电子显微镜检测:将微凝胶粒子冷冻干燥后样品置于硅片上,喷金后通过场致发射扫描电子显微镜(fesem,jsm 7401f,jeol,japan)观察微凝胶粒子形态,见图5。从图5可知,实施例1~3中采用0.5~10%乙基纤维素制得的微粒形貌随乙基纤维素含量的不同有较大改变。当乙基纤维素含量过少(0.5%)时,粒子冷冻干燥后皱缩明显,不能保证球形形貌,甚至出现破碎;当乙基纤维素含量过多(10%)时,粒子之间不能保证分散,黏连现象明显,甚至出现团聚。
73.图6为对所得微凝胶光学接触角测量结果。从图6可知,样品1~3压片后,其静态接触角随加入乙基纤维素含量的增加逐步增大,可以证明乙基纤维素的加入确实可以使制得的粒子呈现水不溶性。
74.实施例4
75.(1)将质量分数为10%的温敏性水凝胶单体n
‑
异丙基丙烯酰胺、质量分数为5%ph敏感性水凝胶单体甲基丙烯酸二甲氨基乙酯、质量分数为2%的交联剂n,n
‑
亚甲基双丙烯酸胺置于去离子水中,涡旋至完全溶解;为了模拟包载释放实验,本实施例在其中还加入了一定量的染色剂,具体的是加入质量分数为5%的甲基蓝染色剂,随之涡旋至完全溶解;
76.(2)将质量分数为5%的乙基纤维素置于2
‑
正辛基
‑1‑
十二烷醇的油相中,加热搅拌至完全溶解;
77.(3)将质量分数为0.5%的引发剂偶氮二异丁脒盐酸盐置于去离子水中,涡旋至完全溶解;
78.(4)将步骤(2)所得油相溶剂通入氮气,至完全排除氧气;
79.(5)将步骤(3)所得引发剂溶液加入步骤(1)所得水相溶液中,涡旋至混合均匀;
80.(6)将步骤(5)所得混合溶液按照油水比为3:1加入步骤(2)所得油相溶液中,采用fm200高速剪切机在8000rpm下剪切混合2min,得到乳液;
81.(7)将步骤(6)所得乳液置于紫外交联仪中,光照交联40min;
82.(8)将步骤(7)所得微胶囊由乙醇充分洗涤1次,置于真空冷冻干燥剂中充分干燥,得到微胶囊样品4。
83.实施例5
84.(1)将质量分数为10%的温敏性水凝胶单体n
‑
异丙基丙烯酰胺、质量分数为5%ph
敏感性水凝胶单体甲基丙烯酸二甲氨基乙酯、质量分数为2%的交联剂n,n
‑
亚甲基双丙烯酸胺置于去离子水中,涡旋至完全溶解;为了模拟包载释放实验,本实施例在其中还加入了一定量的染色剂,具体的是加入质量分数为5%的甲基蓝染色剂,随之涡旋至完全溶解;
85.(2)将质量分数为5%的乙基纤维素置于2
‑
正辛基
‑1‑
十二烷醇的油相中,加热搅拌至完全溶解;
86.(3)将质量分数为0.5%的引发剂偶氮二异丁脒盐酸盐置于去离子水中,涡旋至完全溶解;
87.(4)将步骤(2)所得油相溶剂通入氮气,至完全排除氧气;
88.(5)将步骤(3)所得引发剂溶液加入步骤(1)所得水相溶液中,涡旋至混合均匀;
89.(6)将步骤(5)所得混合溶液按照油水比为3:1加入步骤(2)所得油相溶液中,采用fm200高速剪切机在8000rpm下剪切混合2min,得到乳液;
90.(7)将步骤(6)所得乳液置于紫外交联仪中,光照交联40min;
91.(8)将步骤(7)所得微胶囊由乙醇充分洗涤3次,置于真空冷冻干燥剂中充分干燥,得到微胶囊样品5。
92.实施例6
93.(1)将质量分数为10%的温敏性水凝胶单体n
‑
异丙基丙烯酰胺、质量分数为5%ph敏感性水凝胶单体甲基丙烯酸二甲氨基乙酯、质量分数为2%的交联剂n,n
‑
亚甲基双丙烯酸胺置于去离子水中,涡旋至完全溶解;为了模拟包载释放实验,本实施例在其中还加入了一定量的染色剂,具体的是加入质量分数为5%的甲基蓝染色剂,随之涡旋至完全溶解;
94.(2)将质量分数为5%的乙基纤维素置于2
‑
正辛基
‑1‑
十二烷醇的油相中,加热搅拌至完全溶解;
95.(3)将质量分数为0.5%的引发剂偶氮二异丁脒盐酸盐置于去离子水中,涡旋至完全溶解;
96.(4)将步骤(2)所得油相溶剂通入氮气,至完全排除氧气;
97.(5)将步骤(3)所得引发剂溶液加入步骤(1)所得水相溶液中,涡旋至混合均匀;
98.(6)将步骤(5)所得混合溶液按照油水比为3:1加入步骤(2)所得油相溶液中,采用fm200高速剪切机在8000rpm下剪切混合2min,得到乳液;
99.(7)将步骤(6)所得乳液置于紫外交联仪中,光照交联40min;
100.(8)将步骤(7)所得微胶囊由乙醇充分洗涤4次,置于真空冷冻干燥剂中充分干燥,得到微胶囊样品6。
101.对微凝胶样品2、4、5、6进行模拟包载释放实验,测试方法为:取1mg包载冻干样品,溶解于1ml ph为7.5的pbs溶液中,环境温度为恒温24℃,装入透析袋,置于99ml相同ph环境的pbs溶液中透析,并保持磁力搅拌,每隔一定时间取1ml溶液并加回1ml相同pbs溶液。测量吸光度计算累计释放量。
102.图7为微凝胶样品2、4、5、6采用微胶囊洗涤次数不同所制得微胶囊包载释放效果。由图7可知,随着洗涤次数的增加,微胶囊的包载释放效果变快,微凝胶的缓释效果可通过洗涤次数进行调节。
103.本发明获得了一种水不溶性温度/ph双重敏感微凝胶,该凝胶可通过温度和ph两种刺激下分别引起粒径变化,乙基纤维素在制备乳液过程中代替了表面活性剂,通过物理
吸附,附着在凝胶液滴表面上进行稳定,还可以在微凝胶表面形成一层疏水薄膜,使其具有水不溶性。大幅提高微凝胶的缓释效果和其在油性环境下的应用。
104.本发明提供了一种以前从未报道过的由温敏性水凝胶单体、ph敏感性水凝胶单体共聚得到的水不溶性多重响应性微凝胶,该微凝胶在制备过程中没有加入常用表面活性剂,而是利用生物友好型高分子聚合物乙基纤维素在液滴表面的附着进行乳化,通过光照交联进行固化。此时乙基纤维素不仅能起到乳化作用,还能附着在微凝胶表面,对微凝胶表面进行修饰,使其获得一层疏水层,进一步增强微凝胶缓释效果和扩大其微凝胶的利用面。此外,微凝胶的缓释效果可通过洗涤次数进行调节。
105.应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1