一种咪唑并哒嗪类化合物及修饰后的双亲功能分子及其应用的制作方法
本发明涉及医药
技术领域:
,具体是指一种咪唑并哒嗪类化合物及修饰后的双亲功能分子,制备及其应用。
背景技术:
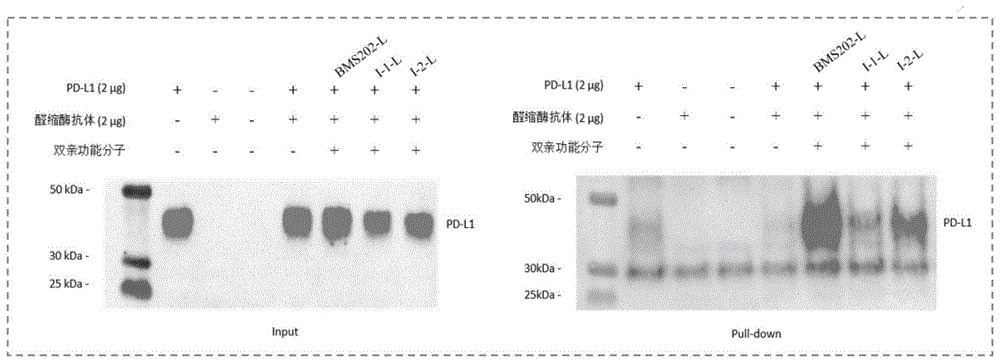
:免疫疗法已被广泛用于癌症的治疗,目前正在彻底改变各种类型癌症的治疗。癌症免疫治疗是癌症治疗中发展最快的领域之一,其目的是诱导免疫系统对抗癌症肿瘤,随着肿瘤免疫治疗的不断成功,大量的蛋白质/受体正被研究为免疫肿瘤治疗的潜在靶点,其中,程序死亡分子(programmeddeath-1,pd-l)/pd1配体(pd1ligand,pd-l1)是免疫肿瘤学领域最有前途的靶点之一。pd-l1是一种33kda的1型跨膜蛋白,pd-1/pd-l1途径在调节免疫系统中发挥重要作用,特别是劫持cd28/mhc的激活状态,通过调节t淋巴细胞增殖、细胞因子的产生和细胞毒活性负向调节免疫应答。研究表明,肿瘤微环境可诱导浸润的t细胞过表达pd-1蛋白,而pd-1的配体pd-l1在肿瘤细胞中过表达,pd-1与其配体pd-l1结合导致癌症患者t细胞功能下调,从而抑制抗肿瘤免疫,导致细t胞衰竭。pd-1/pd-l1抑制剂可以阻断pd-1与pd-l1的结合,干扰负调控机制,增强t细胞活性,从而增强免疫应答,因而达到免疫肿瘤的治疗。pd-1/pd-l1相互作用已成为一种有前景的癌症治疗靶点。批准单克隆抗体抑制pd-1/pd-l1相互作用已证明pd-1/pd-l1抑制剂的临床意义,因此,发展pd-l1的抑制剂对癌症的治疗的发展具有重要的意义。dna编码化合物(dnaencodedlibrary,del)通过组合化学,聚合酶链式反应(polymerasechainreaction,pcr)与高通量二代测序(nextgenerationsequencing,ngs)的应用可以高效推断化合物的分子组成。因此,del正逐渐成为新型苗头化合物发现的重要手段。此外可以连接蛋白和抗体的亲双功能小分子近十年发展迅速,有可能解决生物医学研究中的许多突出问题,并在未来转向治疗应用。因此基于del筛选技术得到的潜在的可以和pd-l1结合的苗头化合物分子与抗体识别的分子进行融合修饰,开发可以和pd-l1和抗体结合的异双亲功能分子,使得其靶向特异蛋白的能力增强,对临床治疗具有重要意义。技术实现要素:本发明的目的是为了克服上述现有技术中的缺失,提供一种根据del筛选后得到具有较佳的pd-l1抑制活性、在针对免疫抑制所引起的各种相关疾病具有预防或治疗作用的咪唑并哒嗪类化合物;以及修饰后可以连接pd-l1和醛缩酶抗体的双亲功能分子,使其靶向特异蛋白的能力增强,提高抗体识别靶点的能力。为了实现上述目的,本发明一方面提供了一种咪唑并哒嗪类化合物,其结构式如通式(ⅰ)所示,其中,通式(ⅰ)中,n为0-3;x为o或nh;r1为氢、取代或未取代的c1-6烷基;r2为氢、取代或未取代的c1-6烷基、烷氧基、烷硫基、卤代烷基、卤代烷氧基、氨基、c3-8环烷基、杂环烷基、取代或未取代的苯基、五元或六元单环杂芳基、取代或未取代的8-12元稠环芳基、取代或未取代的8-12元稠环杂芳基的一种或多种;其中,用于取代c1-6烷基、苯基、芳基或杂芳基的取代基为一个或多个,且独立的选自卤素、氨基、氰基、羟基、烷基、烷氧基、卤代烷基、卤代烷氧基、芳基或杂芳基中的一种或多种;r3为咪唑并哒嗪环上的取代基,r3的数目为0~3个,且独立地选自以下取代基一种或多种的组合:f、cl、br、i、c1-6烷基、氰基、硝基、甲氧基。具体的,所述通式(ⅰ)中,n所在的含氮脂肪环选自杂氮环丁烷、四氢吡咯环、哌啶环、高哌啶环中的任意一种。具体的,所述通式(ⅰ)中,1位和/或2位为手性中心。两处手性中心可分别独立地为r构型或者为s构型。具体的,所述r1可选自甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、异戊基、叔戊基中的任意一种。较佳地,所述的化合物咪唑并哒嗪类包括如下化合物:本发明另一方面还提供一种咪唑并哒嗪类化合物修饰后的双亲功能分子,其结构式如通式(ⅰ)所示,其中,通式(ⅰ)中,n为0-3;x为o或nh;r1为式(ⅱ)所示,其中式(ⅱ)中的m1是选自1-6的任意整数,由1-6个乙氧基组成的聚乙二醇链;m2是选自1-6的任意整数,由1-6个乙氧基组成的聚乙二醇链;r2为氢、取代或未取代的c1-6烷基、烷氧基、烷硫基、卤代烷基、卤代烷氧基、氨基、c3-8环烷基、杂环烷基、取代或未取代的苯基、五元或六元单环杂芳基、取代或未取代的8-12元稠环芳基、取代或未取代的8-12元稠环杂芳基的一种或多种;其中,用于取代c1-6烷基、苯基、芳基或杂芳基的取代基为一个或多个,且独立地选自卤素、氨基、氰基、羟基、烷基、烷氧基、卤代烷基、卤代烷氧基、芳基或杂芳基中的任意一种或多种;r3为咪唑并哒嗪环上取代基,r3的数目为1~3个,且独立地选自以下取代基中的任意一种或多种:f、cl、br、i、c1-6烷基、氰基、硝基、甲氧基。具体的,所述通式(ⅰ)中,n所在的含氮脂肪环选自杂氮环丁烷、四氢吡咯环、哌啶环、高哌啶环中的任意一种。具体的,所述通式(ⅰ)中,1位和/或2位为手性中心。两处手性中心可分别独立地为r构型或者为s构型。较佳地,所述的双亲功能分子包括如下化合物:本发明提供的所述的咪唑并哒嗪类化合物及修饰后的双亲功能分子,是通过以下方法得到:针对靶点蛋白进行dna编码化合物库亲和筛选,根据筛选所富集的dna编码化合物按照其计算值及测序拷贝数由高到低进行排序,确定潜在的dna编码苗头化合物的分子序列信息及设计dna标签去除化合物的合成路径;利用衔接体偶联二酮分子,最终形成双端分别包含dna编码化合物来源的苗头化合物和二酮分子的双亲功能分子。本发明提供了一种所述的咪唑并哒嗪类化合物及双亲功能分子在制备pd-l1抑制剂药物的用途。本发明提供了一种所述的咪唑并哒嗪类化合物及双亲功能分子在制备治疗pd-l1介导的疾病的药物的用途。较佳地,所述的pd-l1介导的疾病包括感染、癌症、或自身免疫性疾病。较佳地,所述的癌症为骨癌、肺癌、胃癌、结肠癌、膜腺癌、乳腺癌、前列腺癌、肺癌、脑癌、卵巢癌、膀肮癌、子宫颈癌、辜丸癌、肾癌、头颈癌、淋巴癌、白血病和皮肤癌中的一种或多种;所述的感染为皮肤感染、胃肠道感染、泌尿生殖系统感染、系统性感染、或由流感、丙型肝炎病毒、人类乳头状瘤病毒、巨细胞病毒、爱泼斯坦巴尔病毒、脊髓灰质炎病毒、水症带状殖彦病毒、柯萨奇病毒和人类免疫缺陷病毒中的一种或多种引起的病毒感染;所述的自身免疫性疾病为类风湿性关节炎、全身性红斑狼疮、混合性结缔组织病、系统硬皮病、皮肌炎、结节性脉管炎、肾病、内分泌相关疾病、肝病、银屑病和由于感染引起的自身免疫反应中的一种或多种。本发明提供了一种所述的咪唑并哒嗪类修饰的双亲功能分子化合物用于连接醛缩酶抗体的用途。本发明提供了一种药物组合物,所述的药物组合物包括治疗有效量的活性组分以及药学上可接受的辅料;所述的活性组分包括本发明所述的咪唑并哒嗪类化合物或咪唑并哒嗪类修饰的双亲功能分子、其异构体、前药、稳定的同位素衍生物或药学上可接受的盐。较佳地,所述的活性组分还包括用于癌症、病毒感染或自身免疫疾病的治疗剂。所述式ⅰ化合物、其异构体、前药、稳定的同位素衍生物或药学上可接受的盐,或所述药物组合物和一种或多种其它种类的用于治疗癌症的治疗剂和/或治疗方法联合使用;所述其它种类的用于治疗癌症的治疗剂和/或治疗方法为微管蛋白抑制剂、烷化剂、拓扑酶ⅰ/ⅰⅰ抑制剂、铂类化合物、抗代谢类药物、激素和激素类似物、信号转导通路抑制剂、血管生成抑制剂、靶向治疗、免疫治疗剂、促凋亡剂、细胞周期信号通路抑制剂和放疗中的一种或多种。较佳地,所述的药物组合物的剂型为胶囊、微囊、片剂、颗粒剂、丸剂、分散粉末、液体制剂、煎膏剂、悬浮剂、糖浆剂、凝胶剂、气雾剂、贴剂、脂质体、口服液、静脉注射液或肌肉注射液。较佳地,所述的咪唑并哒嗪类化合物、其异构体、前药、稳定的同位素衍生物或药学上可接受的盐在所述的药物组合物中的剂量为0.05mg/kg~90mg/kg。较佳地,所述的咪唑并哒嗪类修饰的双亲功能分子连接醛缩酶抗体使得其靶向特异蛋白的能力增强,从而提高抗体识别靶点的能力。实验结果表明,本发明提供的咪唑并哒嗪类化合物具有抑制pd-l1的作用,表明本发明提供的这种咪唑并哒嗪类化合物可治疗、缓解和/或预防pd-l1介导的相关疾病包括:癌症、病毒或其它感染或自身免疫性疾病中的应用。实验结果表明,本发明提供的咪唑并哒嗪类化合物修饰后的双亲功能分子,对pd-l1有较佳的结合相互作用且能对醛缩酶抗体进行类共价修饰连接,所形成的双亲功能分子连接醛缩酶抗体使得其靶向特异蛋白的能力增强,从而提高抗体识别靶点的能力。附图说明图1是本发明实施例9的双亲功能分子与pd-l1蛋白和醛缩酶抗体体外相互作用结果示意图。图2是本发明实施例9的双亲功能分子与pd-l1蛋白和醛缩酶抗体在细胞膜表面相互作用结果示意图。具体实施方式下面将对本发明的技术方案进行清楚、完整的描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例1(2-甲基-[1,1'-联苯]-3-基)甲基((r)-1-(2-((((r)-1-(甲基氨基)-1-氧代-3-(噻唑-4-基)丙-2-基氨基甲酰基)咪唑并[1,2-b]哒嗪-6-基)吡咯烷-3-基氨基甲酸酯(ⅰ-1)的合成。步骤16-氯咪唑并[1,2-b]哒嗪-2-羧酸乙酯(1.1)的合成称取6-氯哒嗪-3-胺(50.0g,385mmol,1.0eq)溶于450ml二氧六环后,加入碳酸氢钠(48.6g,578mmol,22.52ml,1.5eq)和3-溴丙酮乙酸乙酯(82.7g,424mmol,53.0ml,1.1eq)。加毕反应体系升温至100℃反应3小时。tlc(二氯甲烷:甲醇=10:1,碘显色)监控原料消失,并有一个新的主点被发现,结束反应。反应体系过滤,滤液加700ml水,有机层水洗3次(500ml*3),1000ml饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩,得黑胶状粗品1.1,46.0g。步骤2(r)-6-(3-(((叔丁氧羰基羰基)氨基)吡咯烷-1-基)咪唑并[1,2-b]哒嗪-2-羧酸乙酯(1.2)的合成将1.1(25.0g,101mmol,1.0eq)溶于400mln,n-二甲基甲酰胺中,加入碳酸钾(42.2g,305mmol,3.0eq)和(r)-3-叔丁氧羰基氨基吡咯烷(18.9g,101mmol,1.0eq)。加毕反应体系升温至110℃反应12小时。tlc(石油醚:乙酸乙酯=0:1,碘显色)监控原料消失,并有一个新的主点被发现,结束反应。加800ml水,用乙酸乙酯萃取3次(300ml*3),有机层饱和食盐水洗三次(500ml*3),无水硫酸钠干燥,过滤,浓缩,经硅胶柱层析(石油醚:乙酸乙酯=1:0到0:1)纯化,得黑色胶状物1.3,19.0g,产率为44.8%。步骤3(r)-6-(3-氨基吡咯烷-1-基)咪唑并[1,2-b]哒嗪-2-羧酸盐酸盐(1.3)的合成将1.2(9.0g,21.6mmol,1.0eq)溶于100ml二氯甲烷后,加入盐酸/二氧六环溶液(4m,27.0ml,5.0eq)。加毕反应体系25℃反应1小时。lc-ms监测显示1.2消失,目标产物的m/z被检测到,结束反应。反应液浓缩,得棕色固体状粗品1.3,7.50g。步骤4(2-甲基-[[1,1'-联苯]-3-基)甲基氯碳酸盐(1.6)的合成称取(2-甲基-[1,1'-联苯]-3-基)甲醇(10.0g,50.4mmol,1.0eq)溶于100ml二氯甲烷中,加入三乙醇胺(5.61g,55.48mmol,7.72ml,1.1eq),然后三光气(6.16g,20.7mmol,0.41eq)的二氯甲烷溶液(50ml)在-20℃下逐滴加入,30分钟加毕,转至0℃反应1.5小时。tlc(石油醚:乙酸乙酯=10:1)监控原料消失,并有一个新点出现,结束反应。于反应体系中加入300ml正己烷搅拌10分钟后,过滤,滤饼用二氯甲烷:正己烷(1:2.2)共64ml混合溶液洗三次。滤液浓缩,得白色固体状粗品1.6,12.0g。步骤5(r)-6-(3-((((((2-甲基-[1,1'-联苯]-3-基)甲氧基)羰基)氨基)吡咯烷-1-基)咪唑[1,2-b]哒嗪-2-羧酸盐(1.4)的合成将1.3(6.50g,18.6mmol,1.0eq)溶于65ml四氢呋喃和26ml水中,加入碳酸氢钾(4.70g,56.0mmol,2.18ml,3.0eq),而后在0℃下滴加1.6(9.73g,37.33mmol,2.0eq)的四氢呋喃溶液65ml。加毕反应体系升温至25℃反应2小时。lc-ms监测显示1.3消失,目标产物的m/z被检测到,结束反应。加300ml水,用乙酸乙酯萃取3次(200ml*3),有机层用500ml饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩,经prep-hplc(柱:phenomenexlunac18250mm*100mm*10um;流动相:[水-(0.05%盐酸)-乙腈];b%:36-66乙腈%,25min)纯化,得黄色固体1.5,5.12g,产率为55.1%。步骤6(r)-6-(3-((((((2-甲基-[1,1'-联苯]-3-基)甲氧基)羰基)氨基)吡咯烷-1-基)咪唑[1,2-b]哒嗪-2-羧酸(1.5)的合成将1.4(3.10g,6.16mmol,1.0eq)溶于25ml乙醇和10ml水中,加入氢氧化锂-一水合物(774mg,18.4mmol,3.0eq),而后反应体系于25℃反应12小时。tlc(石油醚:乙酸乙酯=0:1)监控原料消失,并有一个新点出现,结束反应。反应体系浓缩除去乙醇后,加30ml水,加1m盐酸调ph至6~7。白色固体析出,过滤,滤饼干燥得棕色固体1.5,1.51g,产率为86.5%。步骤7(r)-2-((叔丁氧基羰基)氨基)-3-(噻唑-4-基)丙酸甲酯盐酸盐(1.7)的合成称取(r)-2-((叔丁氧基羰基)氨基)-3-(噻唑-4-基)丙酸(1.0g,3.67mmol,1.0eq)溶于盐酸甲醇(4m,20ml,21.7eq)溶液中,而后反应体系升温至80℃反应12小时。lc-ms监测显示原料消失,目标产物的m/z被检测到,结束反应。反应物浓缩得黄色固体状粗品1.7,912mg。步骤8化合物i-1的合成将1.5(0.69g,1.46mmol,1.0eq)溶于10ml二氯甲烷中,加入n,n-二异丙基乙胺(378mg,2.93mmol,509ul,2.0eq),1-羟基苯并三唑(296mg,2.20mmol,1.5eq)和1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(420mg,2.20mmol,1.5eq),该反应混合物15℃下反应15分钟。而后加入1.6(325mg,1.46mmol,1.0eq),于15℃下再反应2小时。lc-ms监测显示1.5消失,目标产物的m/z被检测到,结束反应。反应液浓缩,经prep-hplc(柱:watersvinridissilica2-epobd50*150mm*5um,流动相:[正庚烷-乙醇(0.1%氨水)];b%:25-65%乙醇,10min)纯化,得黄色固体i-1,510mg,产率为54.6%。1hnmr(400mhz,cdcl3)δppm8.78(d,j=1.6hz,1h),8.25-8.06(m,2h),7.61(d,j=10.0hz,1h),7.45-7.35(m,2h),7.34-7.30(m,2h),7.29-7.24(m,1h),7.23-7.15(m,2h),7.14-7.10(m,1h),6.59(d,j=10.0hz,1h),5.35-5.10(m,4h),4.44(s,1h),3.80-3.75(m,1h),3.72-3.70(m,3h),3.64-3.50(m,2h),3.48-3.31(m,3h),2.37-2.26(m,1h),2.22(s,3h),2.11-1.97(m,1h).lc-ms:rt=0.921min,m/z=640.1[m+h]+.实施例2甲基nτ-苄基-nα-(6-((r)-3-(((((2-甲基-[1,1'-联苯]-3-基)甲氧基)羰基)氨基)吡咯烷-1-基)咪唑并[1,2-b]哒嗪-2-羰基)-l-组氨酸盐(ⅰ-2)的合成步骤1nτ-苄基-l-组氨酸甲酯盐酸盐(2.1)的合成称取nτ-苄基-nα-(叔丁氧羰基)-l-组氨酸(1.0g,2.90mmol,1.0eq)溶于盐酸甲醇(4m,20ml,21.7eq)溶液中,而后反应体系升温至80℃反应5小时。tlc(石油醚:乙酸乙酯=0:1)监控原料消失,并有一个新点出现,结束反应。反应物浓缩得黄色油状粗品2.1,750mg。步骤2化合物i-2的合成将2.1(500mg,1.06mmol,1.0eq),n,n-二甲基甲酰胺(7.75mg,106umol,8.16ul,0.10eq)和三乙醇胺(322mg,3.18mmol,443ul,3.0eq)溶于10ml二氯甲烷中,0℃下加入草酰氯(162mg,1.27mmol,111ul,1.20eq),搅拌30分钟。然后加入2.1(337mg,1.06mmol,1.0eq)的二氯甲烷溶液(5ml),反应体系升至25℃搅拌2小时。lc-ms监测显示2.1消失,目标产物的m/z被检测到,结束反应。反应液加20ml水,用乙酸乙酯萃取三次(20ml*3),有机相饱和食盐水洗两次(50ml*2),无水硫酸钠干燥,过滤,浓缩。浓缩物经prep-hplc(柱:welchultimatexb-diol250*50*10um,流动相:[正庚烷-乙醇(0.1%氨水)];b%:25-65%乙醇,10min)纯化,得黄色固体i-2,450mg,产率为54.8%。1hnmr(400mhz,cdcl3)δppm8.37(d,j=8.4hz,1h),8.12(s,1h),7.61-7.58(m,1h),7.56(s,1h),7.48-7.45(m,2h),7.41-7.39(m,3h),7.37-7.32(m,4h),7.31-7.29(m,2h),7.22-7.11(m,3h),6.72(s,1h),6.58(d,j=10.0hz,1h),5.22(s,2h),5.08-5.03(m,4h),4.44(s,1h),3.77-3.74(m,1h),3.66(s,3h),3.44-3.42(m,1h),3.20-3.16(m,2h),3.05-3.03(m,1h),2.34-2.32(m,1h),2.23(s,3h),2.07-2.05(m,1h).lc-ms:rt=0.734min,m/z=713.3[m+h]+.实施例3(r)-2-(6-((r)-3-(((((2-甲基-[1,1'-联苯]-3-基]甲氧基)羰基)氨基)吡咯烷-1-基)咪唑[1,2-b]哒嗪-2-羧酰胺基)-3-(噻唑-4-基)丙酸(i-1-a)的合成将i-1(510mg,786umol,1.0eq)溶于5ml甲醇和2ml水中,加入氢氧化锂-一水合物(65.9mg,1.57mmol,2.0eq),而后反应体系于15℃反应2小时。tlc(二氯甲烷:甲醇=10:1,碘显色)监控原料消失,并有一个新点出现,结束反应。反应体系加30ml水,用乙酸乙酯萃取两次(15ml*2)。然后水层加1m盐酸调ph至5~6,加乙酸乙酯萃取3次(20ml*3),有机层20ml饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩,得白色固体i-1-a,390mg,产率为76.3%。1hnmr(400mhz,dmso-d6)δppm9.04(d,j=1.6hz,1h),8.30(d,j=8.0hz,1h),8.15(s,1h),7.83(d,j=10.0hz,1h),7.75-7.60(m,1h),7.49-7.35(m,5h),7.30-7.20(m,3h),7.19-7.11(m,1h),7.00-6.90(m,1h),5.11(s,2h),4.82-4.77(m,1h),4.28-4.15(m,1h),3.70-3.65(m,1h),3.62-3.47(m,2h),3.42-3.35(m,3h),2.25-2.17(m,1h),2.15(s,3h),2.00-1.90(m,1h).lc-ms:rt=0.942min,m/z=626.3[m+h]+.实施例4nτ-苄基-nα-(6-((r)-3-(((((2-甲基-[1,1'-联苯]-3-基)甲氧基)羰基)氨基)吡咯烷-1-基)咪唑[1,2-b]哒嗪-2-羰基)-l-组氨酸(i-2-a)的合成参照化合物i-1-a的合成方法,得白色固体i-2-a,350mg,产率为82.5%。1hnmr(400mhz,dmso-d6)δppm9.19(s,1h),8.73(s,1h),8.27(s,1h),7.85-7.83(m,1h),7.77-7.76(m,1h),7.50-7.47(m,1h),7.45-7.43(m,2h),7.40-7.38(m,2h),7.35-7.26(m,8h),7.25-7.21(m,1h),7.18-7.07(m,1h),5.36(s,2h),5.32(s,2h),4.80-4.76(m,1h),4.25(s,1h),3.74-3.73(m,1h),3.72-3.70(m,1h),3.64-3.54(m,1h),3.41-3.39(m,1h),3.38-3.23(m,2h),2.24-2.16(m,4h),1.99-1.95(m,1h).lc-ms:rt=1.017min,m/z=699.4[m+h]+.实施例5(2-甲基-[1,1'-联苯]-3-基)甲基((r)-1-(2-((((r)-37-((4-(3,5-二氧己基))苯基)氨基)-3,20,33,37-四氧-1-(噻唑-4-基)-7,10,13,16,23,26,29-庚七醇-4,19,32-三氮杂庚并烷-2-基)氨基甲酰基)咪唑并[1,2-b]哒嗪-6-基)吡咯烷-3-基氨基甲酸酯(ⅰ-1-l)的合成步骤1n1-(1-氨基-16-氧代-3,6,9,12,19,22,25-庚烷-15-氮杂庚烷-27-基)-n5-(4-(3,5-二氧己基)苯基戊二酰胺(3.2)的合成称取叔丁氧羰基保护的原料(3.1,56.0mg,66.5umol,1.0eq)溶于0.5ml二氯甲烷溶液中,而后加入盐酸/二氧六环(4m,166ul,10.0eq)溶液,反应体系于15℃反应10分钟。tlc(二氯甲烷:甲醇=10:1,碘显色)监控原料消失,并有一个新点出现,结束反应。反应物浓缩得黄色油状粗品3.2,50mg。步骤2化合物i-1-l的合成将i-1-a(40.0mg,61.5umol,1.0eq)溶解于1.5ml二氯甲烷中,加入n,n-二异丙基乙胺(23.85mg,184umol,32.1ul,2.0eq),1-羟基苯并三唑(12.46mg,92.25umol,1.5eq)和1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(17.68mg,92.25umol,1.5eq),该反应混合物15℃下反应15分钟。而后加入3.2(47.81mg,64.53umol,1.1eq),于15℃下再反应12小时。lc-ms监测显示约9%的i-1-a剩余,约55%的目标产物的m/z被检测到,结束反应。反应液加5ml水,用二氯甲烷萃取两次(20ml*3),有机相5ml饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩。浓缩物经prep-hplc(柱:watersxbridge150*25mm*5um,流动相:[水(10mm碳酸氢铵)-乙腈];b%:33-66%乙腈,9min)纯化,得黄色固体i-1-l,15.0mg,产率为17.6%。1hnmr(400mhz,dmso-d6)δppm9.78(s,1h),9.00(s,1h),8.47-8.02(m,3h),7.95-7.78(m,3h),7.73-7.65(m,1h),7.52-7.31(m,8h),7.29-7.07(m,7h),6.98-6.91(m,1h),5.11(s,2h),4.82-4.77(m,1h),4.28-4.15(m,1h),3.70-3.55(m,7h),3.54-3.45(m,22h),3.40-3.35(m,2h),3.27-3.13(m,10h),2.83-2.65(m,4h),2.34-2.23(m,5h),2.18-2.06(m,8h),2.00-1.90(m,2h),1.84-1.72(m,2h).lc-ms:rt=0.994min,m/z=1348.9[m+h]+.实施例6(2-甲基-[1,1'-联苯]-3-基)甲基((r)-1-(2-((((s)-1-(1-苄基-1h-咪唑-4-基)-37-((4-(3,5-二氧己基)苯基)氨基)-3,20,33,37-四氧-7,10,13,16,23,26,29-庚氧基-4,19,32-三氮杂庚并烷-2-基氨基甲酰基)咪唑并[1,2-b]哒嗪-6-基)吡咯烷-3-基)氨基甲酸酯(ⅰ-2-l)的合成参照化合物i-1-l的合成方法,得黄色固体i-2-l,12.0mg,产率为6.16%。1hnmr(400mhz,dmso-d6)δppm9.79(s,1h),8.26-8.20(m,2h),8.14-7.89(m,1h),7.85-7.83(m,3h),7.75-7.70(m,1h),7.64(s,1h),7.46-7.42(m,4h),7.38-7.30(m,2h),7.28-7.24(m,6h),7.20-7.14(m,5h),7.13-7.11(m,1h),6.88(s,1h),5.11(s,3h),4.67-4.62(m,1h),4.23(s,1h),3.68-3.62(m,2h),3.58-3.52(m,3h),3.48-3.44(m,22h),3.41-3.39(m,6h),3.32-3.30(m,2h),3.26-3.18(m,5h),2.85-2.78(m,2h),2.75-2.68(m,4h),2.30-2.27(m,5h),2.25-2.15(m,7h),2.11-2.07(m,1h),1.79-1.78(m,3h).lc-ms:rt=1.037min,m/z=1421.9[m+h]+.实施例7连接dna的咪唑并哒嗪类化合物(咪唑并哒嗪类on-dna化合物)对pd-l1的结合作用研究。针对靶蛋白pd-l1进行del库亲和筛选,从上百万化合物中筛选得到两个富集较佳的咪唑并哒嗪类on-dna化合物的分子序列信息,并按照库合成路线重新合成了on-dna化合物。并对on-dna化合物重合成,对合成后的混合物使用两种测试方法测试on-dna化合物结合作用。其中,咪唑并哒嗪类on-dna化合物重合成步骤如下:步骤一,向离心管中加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和n-羟基丁二酰亚胺的二甲基亚砜溶液,涡旋离心后向反应液中加入合成砌块1(bb1)的n,n-二甲基乙酰胺溶液,涡旋离心后在摇床中20℃反应15分钟后,加入化合物4.1,20℃摇床反应16个小时,加乙醇-80℃,1小时后离心,去除上清液,残留固体得粗品化合物4.2。步骤二,粗品化合物4.2溶于双蒸水后,加入20%的哌啶的双蒸水溶液,涡旋离心后20℃摇床中反应2个小时,加乙醇-80℃,1小时后离心,去除上清液,残留固体冻干;而后加双蒸水溶解,超滤管(ultracel-3kda,14000g,30min,20℃)纯化,收集浓缩液,得粗品化合物4.3。步骤三,于离心管中加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和n-羟基丁二酰亚胺的二甲基亚砜溶液,涡旋离心后向反应液中加入合成砌块2(bb2)的n-甲基吡咯烷酮溶液,涡旋离心后在摇床中20℃反应15分钟后,加入化合物4.3,20℃摇床反应16个小时,加乙醇-80℃,1小时后离心,去除上清液,残留固体冻干得粗品化合物4.4。步骤四,粗品化合物4.4溶于双蒸水后,加入氯化镁和醋酸钠的双蒸水溶液,涡旋离心后,90℃反应16个小时,加乙醇-80℃,1小时后离心,去除上清液,残留固体;而后加双蒸水溶解,超滤管(ultracel-3kda,14000g,30min,20℃)纯化,收集浓缩液,得粗品化合物4.5。步骤五,于离心管中加入合成砌块3(bb3)和三乙醇胺的n,n-二甲基乙酰胺溶液,涡旋离心后在摇床中20℃反应10分钟后,加入n,n'-二琥珀酰亚胺基碳酸酯的n,n-二甲基乙酰胺溶液,涡旋离心后20℃摇床反应1.5个小时,再加入粗品化合物4.5,涡旋离心后20℃摇床反应1.5个小时,加乙醇-80℃,1小时后离心,去除上清液,残留固体冻干得;用zeba脱盐柱纯化,收集原液以及三次洗液,冻干后得白色固体混合终产物。采用ltq质谱检测lcms,检测终产物i-1-dna纯度为38.54%;终产物i-2-dna纯度为43.38%。其中,使用的一种测试方法是亲和筛选-质谱分析法(as-ms)测试咪唑并哒嗪类on-dna化合物的结合效果和结构。as-ms测试步骤参照中国专利cn111965295a中提供的测试方法。测试结果如下:表1咪唑并哒嗪类on-dna化合物对pd-l1结合的as-ms测试结果由表1结果可见,咪唑并哒嗪类on-dna化合物对pd-l1有较佳的结合相互作用,并通过ms分析可推断出on-dna化合物的具体结构。其中,使用的另一种测试方法是表面等离子体共振(surfaceplasmaresonance,spr)测试咪唑并哒嗪类on-dna化合物的结合效果。测试步骤参照spr测试小分子和蛋白结合操作,将靶蛋白pd-l1蛋白偶联于芯片表面,将不同浓度的咪唑并哒嗪类on-dna化合物进样,使之流过芯片表面与偶联的pd-l1结合,记录并分析传感图谱,获取相应分子相互作用信息。测试结果如下:表2咪唑并哒嗪类on-dna化合物对pd-l1结合的spr测试结果compoundkdrmaxi-1-dna57.4μm47.3i-2-dna34.5μm58.9由表2结果可见,咪唑并哒嗪类on-dna化合物对pd-l1有较佳的结合相互作用,kd可达到几十μm水平。实施例8咪唑并哒嗪类双亲功能分子对pd-l1的结合作用研究。测试步骤参照spr测试小分子和蛋白结合操作,将靶蛋白pd-l1偶联于芯片表面,将不同浓度的咪唑并哒嗪类双亲功能分子进样,使之流过芯片表面与偶联的pd-l1结合,记录并分析传感图谱,获取相应分子相互作用信息。测试结果如下:表3咪唑并哒嗪类亲双功能分子对pd-l1结合的spr测试结果compoundkdrmaxi-1-l6.12μm25.9i-2-l5.53μm15.5由表3结果可见,咪唑并哒嗪类亲双功能分子对pd-l1有较佳的结合相互作用,kd可达到个位数μm水平。kd可越小,表明对目标的结合亲和力就越大。实施例9咪唑并哒嗪类双亲功能分子对醛缩酶抗体的亲和相互作用研究。在体外和细胞水平分别测试该咪唑并哒嗪类双亲功能分子对醛缩酶抗体的相互作用。其中,双亲功能分子与醛缩酶抗体体外相互作用实验,设置参照组1仅pd-l1蛋白组,参照组2仅醛缩酶抗体38c2组,参照组3无蛋白组,实验组1为无双亲分子组,实验组2阳性对照组(bms202-l),实验组3双功能分子组(i-1-l),实验组4双功能分子组(i-2-l)。将醛缩酶抗体38c2固载在proteing免疫磁珠上,将双亲功能分子i-1-l和i-2-l在37℃条件下,翻转孵育1小时,再加入pd-l1底物蛋白室温条件下,翻转孵育1小时。而后使用westernblot检测,胶图如图1。如图1结果可见,胶图中可以明显发现靶蛋白pd-l1和醛缩酶抗体38c2所在分子量的条带,表明亲双功能分子i-1-l和i-2-l可以在体外试验中亲和作用于靶蛋白pl-l1蛋白和醛缩酶抗体38c2。其中,双亲功能分子与醛缩酶抗体在细胞膜表面相互作用实验。pd-l1质粒转染细胞后,将醛缩酶抗体38c2溶解于细胞亲和缓冲液中,并分装至参照组和实验组,将双亲功能分子(i-1-l和i-2-l)加入实验组,37℃翻转孵育1小时。形成38c2抗体-小分子复合物。将38c2抗体-小分子复合物加入到pd-l1过表达的freestyle293细胞(悬浮),37℃孵育1小时,洗涤,离心,弃上清收集细胞。之后加入荧光标记的抗鼠源38c2的二抗常温孵育1小时,洗涤,离心,弃上清收集细胞。细胞通过bectondickinsonfacsariatmiiiflowcytometer(bdbiosciences)进行流式细胞术检测。以前向反射光(fsc)与侧向反射光(ssc)确定所分析的细胞类群,圈门后检测细胞表面荧光的表达;以平均荧光表达强度为对比值衡量参照组与实验组荧光表达的差异,进而推断细胞膜靶蛋白通过双亲功能分子的亲和作用于醛缩酶抗体38c2,并以其作为指示来指征细胞膜靶蛋白的表达与亲和作用特异性。如图2结果可见,参照仅添加醛缩酶抗体组,将双功能分子i-1-l和i-2-l加入pd-l1蛋白后的实验组相比加入前的实验组,平均荧光表达强度明显增强。表明细胞膜靶蛋白pd-l1可以通过双亲功能分子i-1-l和i-2-l的亲和作用于醛缩酶抗体38c2。综上所述,上述各实施例仅为本发明的较佳实施例而已,并不用以限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,皆应包含在本发明的保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1