混杂负载型催化剂和使用该催化剂制备聚烯烃的方法与流程
1.相关申请的交叉引用
2.本申请基于2019年9月27日递交的韩国专利申请10
‑
2019
‑
0120102和10
‑
2019
‑
0120103号和2020年9月24日递交的韩国专利申请10
‑
2020
‑
0124243号,并要求其优先权,这些专利申请的公开内容通过引用在此完整并入。
3.本发明涉及新型混杂负载型催化剂,以及使用该催化剂制备聚烯烃的方法。
背景技术:
4.烯烃聚合催化剂体系可分为ziegler
‑
natta和茂金属催化剂,并且已经根据它们的特性开发了这些高活性催化剂体系。ziegler
‑
natta催化剂自1950年代开发以来已广泛应用于商业工艺。然而,由于ziegler
‑
natta催化剂是其中混合有多个活性位点的多活性位点催化剂,因此其具有所得聚合物的分子量分布宽的特征。另外,由于共聚单体的组成分布不均匀,因此存在难以获得所需物理性质的问题。特别是,由于宽分子量分布,物理性质可能因具有相对较低分子量的聚合物链而劣化。
5.同时,茂金属催化剂包括以茂金属化合物为主要成分的主催化剂和以铝为主要成分的有机金属化合物助催化剂。所得聚合物的立构规整性、共聚特性、分子量、结晶度等可以通过改变催化剂的配体结构和聚合条件来控制。
6.美国专利5,032,562号公开了通过在一种载体上负载两种不同的过渡金属催化剂来制备聚合催化剂的方法。此催化剂通过在一种载体上负载产生高分子量的钛(ti)类ziegler
‑
natta催化剂和产生低分子量的锆(zr)类茂金属催化剂而制成,并形成双峰的分子量分布。此催化剂的缺点在于,由于助催化剂而造成负载过程复杂且聚合物的形态差。
7.美国专利5,525,678号公开了使用催化剂体系进行烯烃聚合的方法,其中,茂金属化合物和非茂金属化合物同时负载在载体上以实现高分子量聚合物和低分子量聚合物的同时聚合。然而,缺点在于茂金属化合物和非茂金属化合物必须分开负载,并且必须用各种化合物预处理载体以进行负载。
8.美国专利5,914,289号公开了使用分别负载在载体上的茂金属催化剂控制聚合物的分子量和分子量分布的方法。然而,需要大量的溶剂和大段的时间来制备负载型催化剂,并且将茂金属催化剂负载在各个载体上的过程是繁琐的。
9.而且,根据现有技术,存在的缺点在于难以有效地制备聚烯烃,特别是同时满足所需的密度水平和窄分子量分布的乙烯(共)聚合物。
10.同时,氯化聚乙烯(cpe)是通过将聚乙烯中的一部分氢用氯取代而获得的产品,并且用作聚氯乙烯(pvc)的冲击改性剂或交联以制造电缆护套或橡胶软管。
11.氯化聚乙烯在通过过氧化物类交联剂热交联的结构中用作电缆护套的材料。为了防止电缆弯曲时对护套的损坏,氯化聚乙烯在交联的混配料中必须具有出色的拉伸强度。
12.在pvc混配料产品的情况下,混配料的强度会根据氯化聚烯烃的性能而变化。在目前公知的通用氯化聚乙烯的情况下,由于应用了使用ziegler
‑
natta催化剂制备的聚乙烯,
因此聚乙烯中氯分布的均匀性由于宽分子量分布而降低。还有一个缺点是当与pvc混炼时冲击强度不足。
13.近来,为了提高电缆用氯化聚烯烃混配料的拉伸强度,将使用茂金属催化剂制备的高密度聚乙烯(hdpe)氯化以产生氯化聚乙烯,然后对其添加交联剂以制备混配料。
14.一般而言,氯化聚乙烯的门尼粘度(mv)越高,混配料的门尼粘度越高,混配料物的拉伸强度越高,但存在压缩期间的加工性降低的问题。
15.因此,需要制备在具有相似的门尼粘度的同时能够提高混配料的拉伸强度而不降低加工性的高密度聚乙烯,并需要开发用于该聚乙烯的催化剂。
技术实现要素:
16.技术问题
17.提供了一种混杂负载型催化剂,其能够制备聚烯烃,特别是分子结构被优化以提高氯化聚烯烃混配料的拉伸强度的高密度聚乙烯。
18.还提供了使用上述混杂负载型催化剂制备能够提高氯化聚烯烃混配料的拉伸强度的聚烯烃的方法。
19.技术方案
20.根据本发明的一个实施方式,提供了一种混杂负载型催化剂,其包括:由下述化学式1表示的一种或多种第一过渡金属化合物;由下述化学式2表示的一种或多种第二过渡金属化合物;和负载所述第一和第二过渡金属化合物的载体:
21.[化学式1]
[0022]
(cp1r
11
)
m
(cp2r
12
)m1(z1)3‑
m
[0023]
在化学式1中,
[0024]
m1是第4族的过渡金属;
[0025]
cp1和cp2彼此相同或不同,并且各自独立地为选自由以下基团组成的组中的任一种:环戊二烯基、茚基、4,5,6,7
‑
四氢
‑1‑
茚基和芴基,它们不具有取代基或取代有c1‑
20
烃;
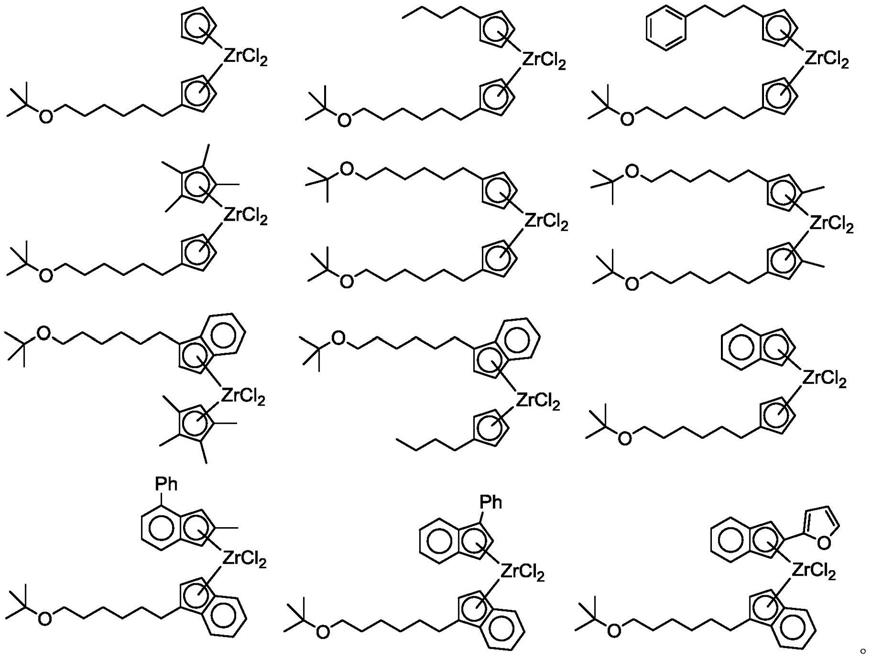
[0026]
r
11
和r
12
彼此相同或不同,并且各自独立地为氢、c1‑
20
烷基、c1‑
20
烷氧基、c2‑
20
烷氧基烷基、c6‑
20
芳基、c6‑
20
芳氧基、c2‑
20
烯基、c7‑
40
烷基芳基、c7‑
40
芳基烷基、c8‑
40
芳基烯基、c2‑
20
炔基或包含选自由n、o和s组成的组中的一个或多个杂原子的c2‑
20
杂芳基;
[0027]
z1是卤素、c1‑
20
烷基、c2‑
20
烯基、c7‑
40
烷基芳基、c7‑
40
芳基烷基、c6‑
20
芳基、具有取代基或不具有取代基的c1‑
20
次烷基、具有取代基或不具有取代基的氨基、c2‑
20
烷基烷氧基或c7‑
40
芳基烷氧基;并且
[0028]
m为1或0;
[0029]
[化学式2]
[0030][0031]
在化学式2中,
[0032]
a为碳或硅,
[0033]
m2为第4族的过渡金属,
[0034]
r
21
为取代有c1‑
20
烷基的c6‑
20
芳基,
[0035]
r
22
为c3‑
20
支化烷基,
[0036]
r
23
至r
25
各自独立地为c1‑
20
烷基,
[0037]
z
21
和z
22
各自独立地为卤素或c1‑
10
烷基,并且
[0038]
n为1至10的整数。
[0039]
本发明提供了制备聚烯烃的方法,所述方法包括在包含混杂负载型催化剂的催化剂组合物存在下聚合烯烃单体的步骤。
[0040]
在本公开中,术语“第一”和“第二”等用于描述各种成分,这些术语仅用于将某一成分与其它成分区分开。
[0041]
本文所使用的术语仅出于描述具体实施方式的目的,并不旨在限制本发明。除非上下文另外明确指出,否则单数形式也旨在包括复数形式。将进一步理解的是,术语“包括”、“具有”或“含有”在本说明书中使用时表明了存在指定的特征、数目、步骤、成分或其组合,但并不排除存在或添加一个或多个其它特征、数目、步骤、成分或其组合。
[0042]
由于可以对本发明进行各种修改并具有各种形式,因此将通过实例示出本发明的具体实施方式并将对其进行详细描述。然而,无意将本发明限制为所公开的特定形式,并且应当理解,本发明包括在本发明的构思和技术范围内的所有修改、等同形式和替换形式。
[0043]
下文中将更详细地描述本发明的一个特定实施方式的混杂负载型催化剂以及使用该催化剂制备聚烯烃的方法。
[0044]
混杂负载型催化剂
[0045]
用于橡胶软管或电缆护套的氯化聚烯烃需要高拉伸强度。可以通过提高氯化聚烯烃的门尼粘度或混配料的门尼粘度来提高氯化聚烯烃的拉伸强度。然而,在此情况下,存在挤出加工性变差的问题。为了解决此问题,有必要优化应用于氯化聚烯烃的聚烯烃(具体而言高密度聚乙烯)的分子结构。
[0046]
在本发明中,发现了当组合使用具有特定结构的两种过渡金属化合物时,制得的聚烯烃具有分子量分布曲线中形成高分子量尾的结构以及最小化的低分子含量,因此在氯化聚烯烃的制备期间可提高交联度,使得拉伸强度提高,从而完成本发明。
[0047]
具体地,本发明的一个实施方式的混杂负载型催化剂包括由化学式1表示的一种或多种第一过渡金属化合物;由化学式2表示的一种或多种第二过渡金属化合物;和负载所
述第一和第二过渡金属化合物的载体。
[0048]
在混杂负载型催化剂中,将如下更详细地描述化学式1和2的取代基。
[0049]
c1‑
20
烷基可包括直链、支化或环状烷基,具体而言,甲基(me)、乙基(et)、丙基(pr)、异丙基、正丁基(n
‑
bu)、叔丁基(t
‑
bu)、戊基(pt)、己基(hx)、庚基、辛基、环丁基、环戊基或环己基等,但不限于此。
[0050]
c1‑
20
亚烷基可包括直链或支化亚烷基,特别是亚甲基、亚乙基、亚丙基、亚丁基、亚戊基或亚己基等,但不限于此。
[0051]
c4‑
20
环烷基是指上述烷基中的环状烷基,具体地可包括环丁基、环戊基或环己基等,但不限于此。
[0052]
c2‑
20
烯基可包括直链或支化烯基,特别是烯丙基、乙烯基、丙烯基、丁烯基或戊烯基等,但不限于此。
[0053]
c6‑
20
芳基可包括单环或稠环芳基,具体而言,苯基、联苯基、萘基、菲基或芴基等,但不限于此。
[0054]
c1‑
20
烷氧基可包括甲氧基、乙氧基、苯氧基或环己氧基等,但不限于此。
[0055]
c2‑
20
烷氧基烷基可以是如上所述的烷基的一个或多个氢被烷氧基取代的官能团,具体可包括烷氧基烷基,例如甲氧基甲基、甲氧基乙基、乙氧基甲基、异丙氧基甲基、异丙氧基乙基、异丙氧基己基、叔丁氧基甲基、叔丁氧基乙基或叔丁氧基己基等;或者芳氧基烷基,例如苯氧基己基等,但不限于此。
[0056]
c1‑
20
烷基甲硅烷基或c1‑
20
烷氧基甲硅烷基是
‑
sih3的1至3个氢被1至3个如上所述的烷基或烷氧基取代的官能团,具体可包括烷基甲硅烷基,例如甲基甲硅烷基、二甲基甲硅烷基、三甲基甲硅烷基、二甲基乙基甲硅烷基、二乙基甲基甲硅烷基或二甲基丙基甲硅烷基等;烷氧基甲硅烷基,例如甲氧基甲硅烷基、二甲氧基甲硅烷基、三甲氧基甲硅烷基或二甲氧基乙氧基甲硅烷基等;烷氧基烷基甲硅烷基,例如甲氧基二甲基甲硅烷基、二乙氧基甲基甲硅烷基或二甲氧基丙基甲硅烷基等,但不限于此。
[0057]
c1‑
20
甲硅烷基烷基是如上所述的烷基的至少一个氢被甲硅烷基取代的官能团,具体可包括
‑
ch2‑
sih3、甲基甲硅烷基甲基或二甲基乙氧基甲硅烷基丙基等,但不限于此。
[0058]
卤素可以是氟(f)、氯(cl)、溴(br)或碘(i)。
[0059]
磺酸根基团具有
‑
o
‑
so2‑
r’的结构,其中r’可以是c1‑
20
烷基。具体地,c1‑
20
磺酸根基团可包括甲磺酸根基团或苯磺酸根基团等,但不限于此。
[0060]
杂芳基是包含n、o和s中的一个或多个作为杂原子的c2‑
20
杂芳基,其具体实例可包括氧杂蒽、硫杂蒽、噻吩、呋喃、吡咯、咪唑、噻唑、噁唑、噁二唑、三唑、吡啶基、联吡啶基、嘧啶基、三嗪、吖啶基、哒嗪、吡嗪基、喹啉基、喹唑啉、喹喔啉基、酞嗪基、吡啶并嘧啶基、吡啶并吡嗪基、吡嗪并吡嗪基、异喹啉、吲哚、咔唑、苯并噁唑、苯并咪唑、苯并噻唑、苯并咔唑、苯并噻吩、二苯并噻吩、苯并呋喃基、菲咯啉、异噁唑基、噻二唑基、吩噻嗪基或二苯并呋喃基等,但不限于此。
[0061]
在展现与所需效果相同或相似效果的范围内,上述取代基可以可选地取代有选自由以下基团组成的组中的一个或多个取代基:羟基;卤素;烷基、烯基、芳基或烷氧基;包含第14至16族杂原子中的一个或多个杂原子的烷基、烯基、芳基或烷氧基;甲硅烷基;烷基甲硅烷基或烷氧基甲硅烷基;膦;磷化物;磺酸根;和砜。
[0062]
此外,第4族的过渡金属可包括钛(ti)、锆(zr)或铪(hf)等,但不限于此。
[0063]
在本发明的一个实施方式的混杂负载型催化剂中,化学式1表示的第一过渡金属化合物可容易地制备具有高聚合活性的低分子量聚合物,第二过渡金属化合物可容易地制备超高分子量聚合物。因此,通过控制混杂负载型催化剂中的第一和第二过渡金属化合物的混合比,可将制备的聚合物中的低分子含量最小化,由于第二过渡金属化合物的超高分子量特性可以增加分子量分布,并且可以容易地控制粘度。因此,当使用由此制备的聚合物制备氯化聚烯烃时,可提高交联效率,从而提高拉伸强度。
[0064]
特别地,由化学式1表示的第一过渡金属化合物是含有cp1和cp2的配体的非交联化合物。cp1和cp2的配体可彼此相同或不同,各自独立地为选自由环戊二烯基、茚基、4,5,6,7
‑
四氢
‑1‑
茚基和芴基组成的组中的任一种。这些配体可以取代有一种或多种、或一种至三种c1‑
20
烃,更特别是c1‑
10
烷基。如所述的,cp1和cp2的配体可具有可充当路易斯碱的非共享电子对,从而展现出高聚合活性。特别是,当cp1和cp2的配体是具有相对较低位阻的环戊二烯基时,它们可展现出高聚合活性和低氢反应性,从而聚合得到具有高活性的低分子量烯烃聚合物。
[0065]
此外,根据取代的官能团的种类,cp1和cp2的配体可通过控制位阻的影响而容易地控制例如要制备的烯烃聚合物特性,如化学结构、分子量、分子量分布、机械性能、透明度等。具体地,cp1和cp2的配体可分别取代有r
11
和r
12
。就此而言,r
11
和r
12
彼此相同或不同,各自独立地为氢、c1‑
20
烷基、c2‑
20
烷氧基烷基、c6‑
20
芳基、c7‑
20
芳基烷基、呋喃基或噻吩基,更具体地为:c1‑
10
烷基,例如正丁基等;c2‑
10
烷氧基烷基,例如叔丁氧基己基等;c6‑
20
芳基,例如苯基等;c7‑
20
芳基烷基,例如苯基丁基等;呋喃基或噻吩基。就优异的催化活性而言,r
11
和r
12
为如上定义的取代基,其中r
11
和r
12
中的至少一个可以为c2‑
20
烷氧基烷基或c2‑
10
烷氧基烷基。
[0066]
此外,m1(z1)3‑
m
存在于cp1和cp2的配体之间,并且m1(z1)3‑
m
可能会影响金属络合物的储存稳定性。为了更有效地确保所述效果,z1可各自独立地为卤素或c1‑
20
烷基,更具体而言,各自独立地为f、cl、br或i。此外,m1可以为ti、zr或hf,更特别是zr或hf,还更特别是zr。
[0067]
更特别地,在第一过渡金属化合物中,m1是ti、zr或hf,cp1和cp2彼此相同或不同,并且各自独立地为选自由以下基团组成的组中的任一种:环戊二烯基、茚基、4,5,6,7
‑
四氢
‑1‑
茚基和芴基,它们可不具有取代基或取代有c1‑
20
烃;r
11
和r
12
各自独立地为氢、c1‑
20
烷基、c2‑
20
烷氧基烷基、c6‑
20
芳基、c7‑
20
芳基烷基、呋喃基或噻吩基,其中r
11
和r
12
中的至少一个是c2‑
20
烷氧基烷基;z1是卤素。
[0068]
化学式1表示的第一过渡金属化合物可以例如是下述结构式中任一个表示的化合物,但不限于此:
[0069][0070]
此外,第一过渡金属化合物可以是如下化合物,其中,在化学式1中,m1为zr;cp1和cp2各自独立地为未取代的环戊二烯基,或取代有至少一个c1‑
10
烷基、如甲基的环戊二烯基;r
11
和r
12
各自独立地为氢、c1‑
20
烷基、c2‑
20
烷氧基烷基、c7‑
20
芳基或c7‑
20
芳基烷基,其中r
11
和/或r
12
是烷氧基烷基,更特别是c2‑
10
烷氧基烷基,还更特别是叔丁氧基己基;z1是卤素基团;并且m是1。
[0071]
化学式1表示的第一过渡金属化合物可通过应用已知的反应来合成。特别是,通过各种合成方法制备配体化合物,然后通过加入金属前体化合物进行金属化。然而,本公开不限于此,合成方法可参见实施例。
[0072]
同时,在混杂负载型催化剂中,化学式2表示的第二过渡金属化合物形成茚衍生物和胺衍生物通过桥化合物交联的配体结构,并具有能够在配体结构中充当路易斯碱的非共价电子对,从而展现出高聚合活性。特别是,催化剂通过具有结构上稳定且电子富集的茚结构而可展现出高催化活性,并且通过在桥基中包含系链基团而可展现出优异的负载稳定性。
[0073]
另外,第二过渡金属化合物在茚结构的2位取代有具有支化结构的官能团(r
22
),并且胺衍生物的氮原子得到生长的聚合物链中的β
‑
氢通过氢键稳定,从而制备具有中等或高分子量的聚合物。另外,要制备的聚合物具有窄分子量分布,从而展现出优异的机械性能。特别是,r
22
可以是c3‑
12
或c3‑6支化烷基,例如异丙基、异丁基或已戊基等,并且可以是异丙基,这在位阻效应方面更加有利。
[0074]
另外,茚结构通过在4位键合r
21
,特别是取代有一个或多个、或者一个或两个c1‑
20
烷基的c6‑
20
芳基而具有能够供应足够的电子的诱导效应,从而展现出更高的催化活性。更特别地,在化学式2中,r
21
可以是取代有一个或两个c3‑6支化烷基的苯基,例如4
‑
叔丁基苯基
和3,5
‑
二叔丁基苯基。
[0075]
另外,化学式2中的与n结合的r
23
可以是c1‑
20
直链或支化烷基,更特别是c3‑
12
或c3‑6支化烷基,例如叔丁基。当r
23
具有支化结构时,过渡金属化合物在空间上是稳定的,并且催化剂通过供电子效应而稳定化,从而展现出更高的催化活性。
[0076]
更特别地,在化学式2中,r
21
是取代有一个或两个c3‑6支化烷基的苯基,r
22
和r
23
各自独立地为c3‑6支化烷基。还更特别地,r
22
可以是异丙基。
[0077]
另外,在化学式2中,桥基包括能够将载体与r
24
的官能团系缚在一起的
‑
(ch2)n
‑
o
‑
r
25
的系链基团。因此,可以展现出优异的负载稳定性,并且保持优异的催化活性以制备具有高分子量的聚合物。
[0078]
特别地,r
24
可以是c1‑
12
或c1‑6直链或支化烷基。更特别地,其可以是c1‑4直链烷基或甲基以提高溶解性,使得负载效率提高。
[0079]
另外,系链基团中的r
25
可以是c1‑
12
或c1‑6直链或支化烷基,更特别地,其可以是c3‑6支化烷基或叔丁基。当系链基团具有支化结构如叔丁基时,其可与载体容易地分离和结合,从而展现出优异的负载稳定性。
[0080]
另外,系链基团中的n可具体是3至8或4至6,并且上述范围内的系链基团可具有适当的长度,从而稳定地展现出催化活性以及优异的负载稳定性。
[0081]
在桥基中,a可以是硅(si)。
[0082]
更特别地,在化学式2中,a是硅,r
25
是c3‑6支化烷基,并且n可以是4至6的整数。
[0083]
另外,化学式2的第二过渡金属化合物可包括第4族过渡金属、如钛(ti)、锆(zr)和铪(hf)作为中央金属(m2)。当过渡金属化合物含有ti作为中央金属时,催化剂通过增加结构开放性而展现出比含有其它第4族过渡金属(例如zr和hf)的情况更优异的聚合活性,并且通过供电子效应而稳定化,从而产生具有高分子量的聚合物。
[0084]
在化学式2中,z
21
和z
22
可各自独立地为卤素,例如氯;或c1‑4烷基,例如甲基。更特别地,z
21
和z
22
都可以是甲基,在此情况下,可展现出比z
21
和z
22
是卤素的情况更好的催化活性。
[0085]
更特别地,在化学式2中,m2是钛,z
21
和z
22
可各自独立地为c1‑4烷基。
[0086]
更特别地,化学式2的化合物可以是如下化合物,其中a是硅;m2是钛;r
21
是取代有一个或多个c3‑
10
支化烷基、如叔丁基的苯基;r
22
是c3‑6支化烷基,例如异丙基;r
23
是c3‑6支化烷基,例如叔丁基;r
24
是c1‑4直链烷基,例如甲基;r
25
是c3‑6支化烷基,例如叔丁基;z
21
和z
22
各自独立地为c1‑4烷基,例如甲基;n为4至6的整数。
[0087]
化学式2表示的第二过渡金属化合物可以是下述结构式之一表示的化合物,但不限于此:
[0088][0089]
上述第二过渡金属化合物可通过以下方式制备:将下述化学式3的配体化合物锂化,然后与含有第4族过渡金属的卤化物反应:
[0090]
[化学式3]
[0091][0092]
在化学式3中,a、r
21
至r
25
和n与上面定义的相同。
[0093]
下述反应方案1表示根据本发明的一个实施方式制备化学式2的第二过渡金属化合物的过程。下述反应方案1仅是用于解释本发明的一个实例,但本发明不限于此:
[0094]
[反应方案1]
[0095][0096]
在反应方案1中,a、m2、r
21
至r
25
、z
21
、z
22
和n与上面定义的相同,x1和x2各自独立地为卤素。
[0097]
如反应方案1中那样,化学式2的化合物(2)可通过以下方式制备:将化学式3的配体化合物(3)与烷基锂、如正丁基锂(nbl)反应以进行锂化,然后与含有第4族过渡金属的卤化物(4)、如ticl4反应。另外,当化学式2的化合物(2)中的x1和x2各自为c1‑
10
烷基时,可在锂化后额外添加用于使金属m烷基化的烷基化试剂,例如mmb(甲基溴化镁)。
[0098]
另外,用于制备化学式2的化合物(2)的配体化合物(3)可通过如下面反应方案2中的合成过程制备。反应方案2仅是用于解释本发明的一个实例,但本发明不限于此。
[0099]
[反应方案2]
[0100][0101]
在反应方案2中,a、r
21
至r
24
和n与上面定义的相同,x3和x4各自独立地为卤素。
[0102]
参见反应方案2,制备配体化合物(3)可包括以下步骤:将作为cp单元的茚类化合物(5)与烷基锂、如正丁基锂(nbl)反应以进行锂化;将所得反应物与用于提供系链基团的原料(6)反应以制备系链基团与茚结构结合的化合物(7);以及将化合物(7)与具有r3的取代基的伯胺(8)(例如t
‑
bunh2)反应。
[0103]
各步骤中的反应可通过应用已知反应进行,更详细的合成方法可参见后述的制备例。
[0104]
如上所述,包含第一和第二过渡金属化合物的混杂负载型催化剂可有效地提供聚烯烃,特别是由于制备氯化聚烯烃和氯化混配料时的交联度增加而能够提高拉伸强度的高密度聚乙烯,因为所述聚烯烃通过在分子量分布曲线中形成高分子量尾而具有最小化的低分子量含量和宽分子量分布。另外,上述效果可通过控制混杂负载型催化剂中的第一和第二过渡金属化合物的混合比而进一步增强。具体而言,第一和第二过渡金属化合物的混合摩尔比可以为1:3至3:1或1:1.5至2:1。
[0105]
此外,根据第一和第二过渡金属化合物的组合,通过优化混合比,催化活性可以提高,并且制备的聚乙烯的物理性质可进一步改善。例如,在第二过渡金属化合物的化学式2中,当r
21
是取代有一个c3‑
10
支化烷基的苯基时,优选以1:1.1至1:3或1:1.2至1:1.5的重量比包含第一过渡金属化合物和第二过渡金属化合物,在第二过渡金属化合物的化学式2中,当r
21
是取代有两个以上或两个c3‑
10
支化烷基的苯基时,优选以1:1至3:1或1.5:1至2:1的重量比包含第一过渡金属化合物和第二过渡金属化合物。
[0106]
另外,在混杂负载型催化剂中,包含的第一和第二过渡金属化合物为负载在载体上的负载型催化剂的形式。当过渡金属化合物以负载型催化剂的形式使用时,可以进一步改善要制备的聚乙烯的形态和物理性质,并且其可合适地用于浆液聚合、本体聚合和气相聚合。
[0107]
特别地,载体可包括二氧化硅、氧化铝、氧化镁或其混合物。此外,优选表面上具有高度反应性的羟基、硅烷醇基或硅氧烷基的载体。为此,可使用通过煅烧表面改性的载体或通过干燥而从表面去除了水分的载体。例如,可使用通过煅烧硅胶制备的二氧化硅、高温干燥的二氧化硅、二氧化硅
‑
氧化铝和二氧化硅
‑
氧化镁。另外,载体可包括氧化物、碳酸盐、硫酸盐和硝酸盐,例如na2o、k2co3、baso4和mg(no3)2等。
[0108]
优选将载体在200至600℃下,更优选在250至600℃下煅烧或干燥。当温度低于200℃时,载体含过多的水分,使得表面上的水分可能与助催化剂反应。另外,由于过量的羟基,助催化剂的负载率可能相对较高,但这需要大量的助催化剂。当温度高于600℃时,载体表面上的孔可彼此结合而减小表面积,并且许多羟基或硅烷醇基会从表面失去,仅留下硅氧
烷基。因此,与助催化剂的反应位点可能会减少,这不是优选的。
[0109]
羟基的量可通过制备方法、制备条件或干燥条件(例如温度、时间、真空或喷雾干燥载体)来控制。当羟基的量过低时,与助催化剂的反应位点可能不足。当羟基的量过高时,除了存在于载体颗粒表面上的羟基以外,还可能由水分引起,这是不希望的。例如,表面上羟基的量可以为0.1至10mmol/g或0.5至5mmol/g。
[0110]
在上述载体中,通过煅烧二氧化硅(特别是硅胶)制备的二氧化硅在丙烯的聚合过程中几乎没有催化剂从载体表面释放,因为过渡金属化合物化学键合并负载到二氧化硅载体上。结果,当通过浆液聚合或气相聚合制备聚烯烃时,粘附在反应器壁表面上或彼此粘附的结垢现象可被最小化。
[0111]
当过渡金属化合物以负载型催化剂的形式使用时,基于载体的重量,例如1g二氧化硅,第一和第二过渡金属化合物的含量可以为10μmol以上,30μmol以上或60μmol以上,且120μmol以下,或100μmol以下。当在上述含量范围内负载时,负载型催化剂可展现出适当的活性,这就保持催化活性而言是有利的。
[0112]
具有上述构造的混杂负载型催化剂展现出优异的聚合活性,并且可制备结构被优化以提高氯化聚烯烃或氯化混配料的拉伸强度的聚烯烃。
[0113]
聚烯烃的制备
[0114]
根据本发明的另一个实施方式,提供了制备聚烯烃的方法,所述方法包括在包含上述混杂负载型催化剂的催化剂组合物存在下使烯烃单体聚合的步骤。
[0115]
在制备方法中,催化剂组合物包含上述的混杂负载型催化剂。
[0116]
混杂负载型催化剂可直接引入聚合体系中,或可溶解或稀释于c5‑
12
脂族烃溶剂(如戊烷、己烷、庚烷、壬烷、癸烷及其异构体)、芳烃溶剂(如甲苯和苯)或氯取代的烃溶剂(如二氯甲烷和氯苯),然后引入聚合体系中。在此使用的溶剂优选在通过用少量烷基铝处理除去少量水或空气(充当催化剂毒物)后使用。
[0117]
催化剂组合物还可包括助催化剂和抗静电剂中的一种或多种。
[0118]
特别地,就提高活性和工艺稳定性而言,催化剂组合物还可包括助催化剂。助催化剂可包括下述化学式9、化学式10和化学式11表示的一种或多种化合物:
[0119]
[化学式9]
[0120]
‑
[al(r
a
)
‑
o]
m
‑
[0121]
在化学式9中,
[0122]
r
a
可以彼此相同或不同,且各自独立地为:卤素;c1‑
20
烃;或卤素取代的c1‑
20
烃;并且
[0123]
m是2以上的整数;
[0124]
[化学式10]
[0125]
j(r
b
)3[0126]
在化学式10中,
[0127]
r
b
可以彼此相同或不同,且各自独立地为卤素;c1‑
20
烃;或卤素取代的c1‑
20
烃;并且
[0128]
j是铝或硼;
[0129]
[化学式11]
[0130]
[e
‑
h]
+
[zq4]
‑
或[e]
+
[zq4]
‑
[0144]
在化学式12中,r可以是c8‑
30
烷基,并且当r包括具有上述范围内的碳数的烷基时,其可以表现出通过优异的抗静电作用而减少细屑且不会引起难闻的气味的效果。
[0145]
更特别地,乙氧基化烷基胺可以是具有化学式1的化合物,其中r是c8‑
22
直链烷基,或c
10
‑
18
直链烷基,或c
13
‑
15
直链烷基,并且这些化合物可单独使用或以其中两种以上的混合物使用。其具体实例可包括n,n
‑
二(2
‑
羟乙基)十三烷基胺或n,n
‑
二(2
‑
羟乙基)十五烷基胺等,可使用市售的atmer 163
tm
(可获自croda)。
[0146]
当还包含抗静电剂时,基于100重量份的载体,例如二氧化硅,抗静电剂的含量可以为0.5重量份以上,1重量份以上,或2重量份以上,且20重量份以下,10重量份以下,或7重量份以下。
[0147]
上述助催化剂和抗静电剂可分别与上述混杂负载型催化剂组合使用,或者可在负载在混杂负载型催化剂中的载体上的状态下使用。当它们在负载在混杂负载型催化剂中的载体上的状态下使用时,催化剂组合物可通过包括如下步骤的制备方法制备:将助催化剂化合物负载在载体上,并将过渡金属化合物负载在载体上;将浆液态的抗静电剂引入负载有助催化剂和过渡金属化合物的载体中,然后热处理。就此而言,过渡金属化合物的负载可通过先负载第一过渡金属化合物然后负载第二过渡金属化合物来进行,反之亦然。具有根据这样的负载顺序确定的结构的负载型催化剂可以在聚烯烃制备过程中展现出更高的催化活性和优异的工艺稳定性。
[0148]
催化剂组合物可根据制备方法以在溶剂中的浆液或稀释液的形式使用,或者可以以与油脂的混合物混合的泥浆催化剂的形式使用。
[0149]
当催化剂组合物以在溶剂中的浆液或稀释液的形式使用时,溶剂为适合于丙烯单体聚合过程的具有5至12个碳原子的脂族烃溶剂,例如戊烷、己烷、庚烷、壬烷、癸烷及其异构体;芳烃溶剂,例如甲苯和苯;或者氯原子取代的烃溶剂,例如二氯甲烷和氯苯等,并且可使用它们中的任一种或两种以上的混合物。在此情况下,催化剂组合物还可包括上述溶剂,并且在使用前可通过用少量烷基铝处理所述溶剂而除去少量水或空气(其可能充当催化剂毒物)。
[0150]
另外,当使用诸如连续本体聚合等聚合方法时,催化剂组合物可以以与油脂混合物混合的泥浆催化剂的形式使用。在此情况下,与使用溶解或稀释在溶剂中的形式的催化剂组合物的情况相比,可进一步减少制备的均聚乙烯中所含的挥发性有机化合物的量,由此,还可减少由挥发性有机化合物造成的气味。
[0151]
另一方面,制备聚烯烃的聚合反应可在一个连续浆液聚合反应器、环形浆液反应器、气相反应器或溶液反应器中通过一种烯烃单体的均聚或两种以上单体的共聚来进行。然而,根据一个实施方式的方法,烯烃单体更优选通过浆液聚合或气相聚合聚合,以便更有效地控制分子量分布。
[0152]
特别地,聚合反应可以通过在烃类溶剂(例如脂族烃类溶剂,如己烷、丁烷或戊烷)中的浆液相聚合来进行。由于本公开的第一和第二过渡金属化合物在脂族烃类溶剂中具有优异的溶解性,因此它们被稳定地溶解并供应至反应体系,因此可以有效地进行聚合反应。
[0153]
此外,本发明的一个实施方式的制备聚烯烃的方法可在单一cstr反应器中进行。
[0154]
在聚合反应器中,聚合可以例如在惰性气体、如氮气的存在下进行。惰性气体可以通过抑制在聚合反应开始时茂金属催化剂的快速反应来起到延长催化剂中包含的茂金属
化合物的反应活性的作用。
[0155]
另外,在聚合反应期间,出于控制聚烯烃的分子量和分子量分布的目的,可使用氢气。氢气激活茂金属催化剂的惰性位点,并通过引起链转移反应来控制分子量。当在聚合反应期间进一步添加氢气时,其添加量基于烯烃单体的总体积可相当于0.1体积%以上,或0.12体积%以上,且0.2体积%以下,或0.18体积%以下。当氢气的添加量在上述范围内时,制备的聚合物的分子量减小,从而改善加工性。
[0156]
另外,聚合反应期间的温度可以为70℃至100℃或80℃至90℃。当聚合温度过低时,就聚合速率和生产率而言其是不优选的。相反,当聚合温度高于必要时,可能在反应器中引起结垢。
[0157]
另外,聚合反应期间的压力可以为6.8kg/cm2至9kg/cm2,更特别是6.8kg/cm2以上,或7.0kg/cm2以上,或8.0kg/cm2以上,且9kg/cm2以下或8.7kg/cm2以下。就防止因高分子量聚合物过量生成和优化生产率而言,聚合压力可以为6.8kg/cm2以上,而考虑到防止高压聚合条件下的副反应,则聚合压力可以为9kg/cm2以下。
[0158]
另外,可进一步使用有机溶剂作为聚合反应中的反应介质或稀释剂。例如,考虑到烯烃单体的含量,这种有机溶剂可以按足以适当地进行浆液相聚合的量使用。
[0159]
例如,在聚合反应期间可以可选地添加三烷基铝,例如三乙基铝。
[0160]
当聚合反应器中存在水分或杂质时,一部分催化剂分解。三烷基铝充当清除剂,以预先捕获反应器中的水分或杂质或单体中包含的水分。因此,可使制备中所用的催化剂的活性最大化,由此可更高效地制备具有优异的物理性质、特别是窄分子量分布的均聚乙烯。特别地,在三烷基铝中,烷基是如上所定义的,特别是c1‑
20
烷基,更特别是c1‑6直链或支化烷基,例如甲基、乙基或异丁基等。
[0161]
另外,基于乙烯单体的总重量,三烷基铝(基于1m)的添加量可以为300ppm以上,或400ppm以上,且1500ppm以下,或1350ppm以下。当在该含量范围内的三烷基铝的存在下进行聚合反应时,可以更容易地制备具有优异强度特性的均聚乙烯。
[0162]
另外,烯烃单体可以是具有两个以上双键的乙烯、α
‑
烯烃、环状烯烃、二烯烯烃或三烯烯烃。
[0163]
烯烃单体的具体实例可包括乙烯、丙烯、1
‑
丁烯、1
‑
戊烯、4
‑
甲基
‑1‑
戊烯、1
‑
己烯、1
‑
庚烯、1
‑
辛烯、1
‑
癸烯、1
‑
十一碳烯、1
‑
十二碳烯、1
‑
十四碳烯、1
‑
十六碳烯、1
‑
二十碳烯、降冰片烯、降冰片二烯、亚乙基降冰片烯、苯基降冰片烯、乙烯基降冰片烯、二环戊二烯、1,4
‑
丁二烯、1,5
‑
戊二烯、1,6
‑
己二烯、苯乙烯、α
‑
甲基苯乙烯、二乙烯基苯或3
‑
氯甲基苯乙烯等,并且可通过将所述单体中的两种以上混合进行共聚。
[0164]
由上述制备方法制备的聚烯烃在通过凝胶渗透色谱法(gpc)分析时展现出多峰分子量分布,并具有宽分子量分布以及最小化的低分子含量。在由聚烯烃的凝胶渗透色谱法分析而得的分子量分布曲线中,相对于分布曲线的总面积,log mw为3.5以下的区域所占的面积比为1.4%以下,分子量分布为6至15。
[0165]
具体而言,对于聚烯烃,当通过在x轴上绘制重均分子量(mw)的log值(log mw)并在y轴上绘制相对于该log值的分子量分布(dwt/dlog mw)而获得分子量分布时,面积比,即log mw为3.5以下(log mw≤3.5)的区域相对于总面积所占的分数为1.4%以下,或1.2%以下,或1.05%以下,或1%以下。log mw为3.5以下的区域所占的面积比是指聚烯烃中具有
10
3.5
g/mol以下的重均分子量(mw)的低分子含量。在使用本发明的混杂负载型催化剂制备的聚烯烃中,低分子含量可如上所述最小化,从而实现优异的交联性。
[0166]
然而,由于聚烯烃中的低分子含量较小,就交联度而言更有利。然而,当低分子含量过低且高分子含量相对过高时,可以理解的是,由于在氯化聚乙烯混配料制备期间的门尼粘度增加,加工性可能降低。因此,考虑到使用氯化聚乙烯制造电缆期间的适当的门尼粘度和加工性改善,log mw为3.5以下的区域的分数可以为0.1%以上,0.5%以上,或0.8%以上。
[0167]
聚烯烃根据分子量分布曲线中形成的高分子量尾而具有宽分子量分布(pdi),以及最小化的低分子含量。特别是,聚烯烃的pdi为6以上或6.1以上,且15以下或12以下。
[0168]
同时,在本发明中,聚烯烃的分子量分布(pdi,多分散性指数)可通过以下方式确定:测量聚烯烃的重均分子量(mw)和数均分子量(mn),然后用重均分子量除以数均分子量。
[0169]
此外,聚烯烃中的低分子含量、重均分子量和数均分子量可通过凝胶渗透色谱法(gpc,由water制造)分析来测量,并且具体测量方法与下面的实验例中描述的相同。
[0170]
此外,除了上述低分子含量和分子量分布特性之外,聚烯烃可满足下述条件(i)至(viii)中的一个或多个、两个以上或全部:
[0171]
(i)根据gpc分析,分子量分布曲线中的log mw超过3.5且为4.0以下(3.5<log mw≤4.0)的分数:1%至5%;
[0172]
(ii)根据gpc分析,分子量分布曲线中的log mw为4.0以下的分数:6%以下;
[0173]
(iii)重均分子量:185,000g/mol至500,000g/mol;
[0174]
(iv)在500rad/s的频率下测得的复数粘度:790pa
·
s至810pa
·
s;
[0175]
(v)熔体流动速率比(mfrr,mi
21.6
/mi5):10至25;
[0176]
(vi)熔融指数(mi
5.0
,条件e,190℃,在5.0kg的负荷下):0.5g/10min至3g/10min;
[0177]
(vii)根据astm d
‑
1505测得的密度:0.94g/cm3至0.96g/cm3;
[0178]
(viii)mdr扭矩(m
h
‑
m
l
):7.5nm至12nm。
[0179]
特别地,在聚烯烃中,降低交联效率的log mw超过3.5且为4.0以下(3.5<log mw≤4.0)的分数(即重均分子量(mw)超过10
3.5
g/mol且为10
4.0
g/mol以下的低分子含量)可以为5%以下,或4.8%以下,或4.5%以下,且1%以上,或3%以上,并且log mw为4.0以下的分数(即重均分子量(mw)为10
4.0
g/mol以下的低分子含量)可以为6%以下,更特别是5.5%以下。
[0180]
在聚烯烃中,当低分子含量,特别是mw为10
4.0
g/mol以下的低分子含量较高时,低分子成分熔化,因此流动性增加,这可能会阻塞聚烯烃颗粒的孔并降低氯化生产率。相比之下,上述聚烯烃可表现出优异的提高交联度的效果,而无需担心氯化生产率的降低。
[0181]
此外,聚烯烃具有高重均分子量(mw),特别是mw为185,000g/mol以上,或190,000g/mol以上,且500,000g/mol以下,或350,000g/mol以下。当聚烯烃具有上述范围的mw和pdi时,其可在优异的机械性能和改善的加工性之间展现出良好的平衡。特别是,氯化后的聚烯烃的分子量没有太大差异,因此氯可以均匀取代。
[0182]
此外,在500rad/s的频率下测得的聚烯烃的复数粘度(η*(ω500))为790pa
·
s以上且810pa
·
s以下,因此其适合于制备用于电线或电缆的氯化聚烯烃。
[0183]
在本发明中,可使用ares(advanced rheometric expansion system)在190℃的温度和0.05rad/s的频率(ω)下测量聚烯烃的复数粘度,其具体测量方法与下面的实验例
中描述的相同。
[0184]
此外,聚烯烃的熔体流动速率比为25以下,或20以下,或18以下,且10以上,或10.3以上,并且熔融指数(mi
5.0
,条件e,190℃,在5.0kg的负荷下)为0.5g/10min以上,0.8g/10min以上,或1g/10min以上,且3g/10min以下,或2.5g/10min以下。当聚烯烃具有上述范围内的熔体流动速率比和熔融指数时,可以适当地控制门尼粘度而不会使氯化聚烯烃制备期间的物理性质劣化,从而提高加工性。
[0185]
在本发明中,聚烯烃的熔融指数(mi
5.0
)可根据astm d1238(条件e,190℃,在5.0kg的负荷下)测量。此外,可通过用mfr
21.6
除以mfr5计算熔体流动速率比(mfrr,21.6/5),mfr
21.6
可以根据astm d 1238在21.6kg的负荷下于190℃的温度测量,并且mfr5可以根据astm d 1238在5kg的负荷下于190℃的温度测量。
[0186]
此外,聚烯烃展现出0.94g/cm3以上或0.945g/cm3以上且0.96g/cm3以下或0.955g/cm3以下的高密度。这意味着聚烯烃具有高含量的晶体结构且致密,表明在氯化过程中晶体结构几乎不变。在本发明中,可通过依照astm d
‑
1505的方法测量聚烯烃的密度。
[0187]
此外,聚烯烃展现出7.5nm以上、或8nm以上或8.5nm以上且12nm以下或11.5nm以下的mdr扭矩(m
h
‑
m
l
)。当聚烯烃具有上述范围内的mdr扭矩时,其可展现出高交联度和优异的机械性能。
[0188]
聚烯烃的mdr扭矩(m
h
‑
m
l
)是指交联度。高交联度意味着施用相同交联剂时的高m
h
‑
m
l
和优异的交联效率。聚烯烃的mdr扭矩可利用诸如mdr(动模流变仪)来测量,并且可通过在180℃和10min的条件下测量m
h
值和m
l
值然后从m
h
值中减去m
l
值来确定。在此,m
h
表示在完全固化时测得的最大硫化扭矩,而m
l
表示最小硫化扭矩。其具体测量方法与下面的实验例中描述的相同。
[0189]
聚乙烯可以是不含共聚单体的烯烃均聚物,例如乙烯均聚物。例如,当聚烯烃是乙烯均聚物、优选满足上述密度条件的高密度聚乙烯(hdpe)时,则可更适当地满足上述物理性质。高密度聚乙烯具有优异的软化点、刚性、强度和电绝缘性,因此可用于各种容器、包装膜、纤维、管材、填料或绝缘材料等。
[0190]
如上所述,使用本发明的混杂负载型催化剂制备的聚烯烃具有优化的分子结构(例如宽分子量分布)以及最小化的低分子含量。因此,可使氯化聚烯烃制备期间的交联度增加,由此可显著提高拉伸强度。因此,聚烯烃可特别适用于制备电线或电缆用氯化聚烯烃。
[0191]
根据本发明的另一个实施方式,提供了制备氯化聚烯烃的方法,所述方法包括通过用氯进行处理而使由上述方法制备的聚烯烃氯化的步骤。
[0192]
有益效果
[0193]
本发明的混杂负载型催化剂包含两种以上具有特定化学结构的过渡金属化合物,从而制备聚烯烃,特备是分子结构被优化以提高氯化聚烯烃混配料的拉伸强度的高密度聚乙烯。
具体实施方式
[0194]
将参考以下示例性实施方式更详细地描述本发明。然而,这些示例性实施方式仅出于说明性目的,并且本发明的范围不旨在由以下示例性实施方式限制。
[0195]
<第一过渡金属化合物的制备>
[0196]
合成例1:制备[tert
‑
bu
‑
o
‑
(ch2)6‑
c5h4]2zrcl2[0197]
使用6
‑
氯己醇通过tetrahedron lett.2951(1988)中所示的方法制备叔丁基
‑
o
‑
(ch2)6‑
cl,并将其与nacp反应,从而获得叔丁基
‑
o
‑
(ch2)6‑
c5h5(产率60%,沸点80℃/0.1mmhg)。
[0198]
另外,在
‑
78℃将叔丁基
‑
o
‑
(ch2)6‑
c5h5溶于thf,并且向其中缓慢添加正丁基锂(n
‑
buli)。其后,将其加热至室温并反应8小时。在
‑
78℃将如上所述合成的锂盐溶液缓慢加入zrcl4(thf)2(1.70g,4.50mmol)/thf(30ml)的悬浮液中,并在室温下进一步反应约6小时。
[0199]
真空干燥掉所有挥发物,并通过加入己烷溶剂来过滤所得油状液体材料。将过滤后的溶液真空干燥,并加入己烷以在低温下(
‑
20℃)获得沉淀物。将所得沉淀物在低温下过滤,从而获得白色固体形式的[tert
‑
bu
‑
o
‑
(ch2)6‑
c5h4]2zrcl2(产率92%)。
[0200]1h nmr(300mhz,cdcl3):6.28(t,j=2.6hz,2h),6.19(t,j=2.6hz,2h),3.31(t,6.6hz,2h),2.62(t,j=8hz),1.7
‑
1.3(m,8h),1.17(s,9h)。
[0201]
13
c nmr(cdcl3):135.09,116.66,112.28,72.42,61.52,30.66,30.61,30.14,29.18,27.58,26.00。
[0202]
<第二过渡金属化合物的制备>
[0203]
合成例2
‑1[0204]
步骤1:配体化合物的制备
[0205][0206]
将作为cp单元的4
‑
(4
‑
(叔丁基)苯基)
‑2‑
异丙基
‑
1h
‑
茚(i)(2.9g,10mmol)放入100ml舒伦克瓶中,向其中加入四氢呋喃(thf;35ml),并冷却至
‑
20℃以下。将冷却的混合溶液搅拌5分钟,然后向其中加入正丁基锂(nbl;4.2ml,2.5m己烷溶液),使其反应过夜以制备锂化cp。当加入正丁基锂时,混合溶液具有棕色。
[0207]
将作为系链硅烷的二氯(叔丁氧基己基)甲基硅烷(ii)(2.84g)放入另一个100ml舒伦克瓶中,并向其中加入mtbe(甲基叔丁基醚)(35ml)。将舒伦克瓶冷却至
‑
20℃以下,然后滴加制备的锂化cp并使其反应。当反应完成时,通过在真空下蒸馏去除反应产物中的溶剂,并通过使用己烷(hex)过滤去除所得的盐。将t
‑
bunh2(4.5ml)加入所得反应产物(iii)并使其反应,然后通过使用己烷过滤去除所得沉淀物,从而获得1
‑
(6
‑
(叔丁氧基)己基)
‑
n
‑
(叔丁基)
‑1‑
(4
‑
(4
‑
(叔丁基)苯基)
‑2‑
异丙基
‑
1h
‑
茚
‑1‑
基)
‑1‑
甲基硅烷胺的配体化合物(2a)(黄色油,5.50g,产率98%(以摩尔计))。
[0208]
nmr(400mhz,c6d6)7.72
‑
7.65(m,2h),7.59
‑
7.21(m,5h),7.05(s,1h)3.73
‑
3.65(m,1h),3.33
‑
3.21(m,3h),3.11
‑
2.85(m,1h),1.66
‑
1.51(m,3h),1.51
‑
1.34(m,4h),1.26(s,9h),1.12(s,9h),1.24
‑
1.18(m,6h),1.06(s,9h),1.04
‑
0.99(m,1h),0.64
‑
0.58(m,1h),
0.54
‑
0.49(m,1h),0.30(s,1.5h),0.19(s,1.5h)
[0209]
步骤2:过渡金属化合物的制备
[0210][0211]
将步骤1中制备的配体化合物(2a)(4.81g,8.6mmol)放入100ml舒伦克瓶中,加入甲苯(43ml),然后冷却至
‑
20℃以下。通过搅拌将溶液充分冷却5分钟,并向所得混合溶液中加入nbl(7.2ml,2.5m己烷溶液),然后锂化。确认了在锂化后,混合溶液的颜色从浅黄色变为深黄色。在锂化反应完成后,将所得反应溶液冷却至0℃,加入mmb(甲基溴化镁)(8.6ml,3m乙醚溶液),将温度立即降至
‑
20℃,并添加ticl4(8.6ml,1m甲苯溶液)。添加时,产生烟,反应溶液立即变成棕色。添加后,进行o/n搅拌,完成后,通过过滤器除去盐,得到棕色油相过渡金属化合物(1a)(棕色油,4.5g,产率82%(以摩尔计))。
[0212]
nmr(400mhz,c6d6),7.25
‑
7.75(m,8h),3.20
‑
3.36(m,2h),3.20
‑
2.64(m,4h)2.64
‑
2.74(m,1h),1.59
‑
1.71(m,4h),1.53(s,9h),1.40
‑
1.35(m,2h),1.25(s,9h),1.14(s,9h)1.12(s,6h),0.97(s,3h)0.58(s,3h),0.12(s,3h)
[0213]
合成例2
‑2[0214]
步骤1:配体化合物的制备
[0215]
将作为cp单元的4
‑
(3,5
‑
二
‑
叔丁基苯基)
‑2‑
异丙基
‑
1h
‑
茚(1.39g,4mmol)加入50ml舒伦克瓶中,向其中加入thf(13ml),并冷却至
‑
20℃以下。将冷却的混合溶液搅拌5分钟,然后向其中加入nbl(1.7ml,2.5m己烷溶液),使其反应过夜以制备锂化cp。当加入nbl时,混合溶液变为红棕色。
[0216]
将二氯(叔丁氧基己基)甲基硅烷(1.14g)加入另一个100ml舒伦克瓶中,并向其中加入thf(13ml)。在将舒伦克瓶冷却至
‑
20℃以下后,滴加上述制备的锂化cp以进行反应。当反应完成时,通过在减压下真空蒸馏去除所得反应物中的溶剂,并使用己烷(hex)过滤出所得的盐。将t
‑
bunh2(1.7ml)加入所得反应物进行反应后,使用己烷过滤出所得沉淀物,并且获得1
‑
(6
‑
(叔丁氧基)己基)
‑
n
‑
(叔丁基)
‑1‑
(4
‑
(3,5
‑
二
‑
叔丁基苯基)
‑2‑
异丙基
‑
1h
‑
茚
‑1‑
基)
‑1‑
甲基硅烷胺的配体化合物(黄色油,2.41g,产率97%(以摩尔计))。
[0217]
nmr(400mhz,c6d6),7.70
‑
7.68(m,1h),7.60
‑
7.47(m,4h),7.34
‑
7.19(m,2h),7.07(s,0.5h),6.89(s,0.5h),3.36
‑
3.21(m,4h),3.12(s,1h),2.52
‑
2.44(m,0.5h),2.00
‑
1.92(m,0.5h),1.72
‑
1.39(m,8h),1.39(s,9h),1.31(s,9h),1.23(s,3h)1.19(s,3h),1.13(s,9h)0.98(s,9h)0.32(s,1h),0.25(s,0.5h),0.2 0(s,1h),0.12(s,0.5h)
[0218]
步骤2:过渡金属化合物的制备
[0219]
将步骤1中制备的1
‑
(6
‑
(叔丁氧基)己基)
‑
n
‑
(叔丁基)
‑1‑
(4
‑
(3,5
‑
二
‑
叔丁基苯
基)
‑2‑
异丙基
‑
1h
‑
茚
‑1‑
基)
‑1‑
甲基硅烷胺的配体化合物(2.4g,3.9mmol)加入100ml舒伦克瓶中,向其中加入甲苯(13ml),然后冷却至
‑
20℃以下。通过搅拌充分冷却5分钟后,向所得混合溶液中加入nbl(5.1ml,2.5m己烷溶液)以进行锂化。确认了在锂化后,混合溶液的颜色变为棕色。当锂化完成后,将所得反应溶液冷却至0℃。向其中加入nmb(13ml,3m乙醚溶液)后,将温度立即降至
‑
20℃,并添加ticl4(3.9ml,1m甲苯溶液)。添加时产生烟,反应溶液立即变成棕色。添加后,进行o/n搅拌,然后通过过滤器除去盐,从而获得过渡金属化合物(1b)(棕色油,2.16g,产率80%(以摩尔计))。
[0220][0221]
nmr(400mhz,c6d6),7.79
‑
7.76(m,2h),7.64
‑
7.47(m,5h),3.35
‑
3.21(m,2h),2.76
‑
2.49(s,2h),1.99
‑
1.91(m,4h),1.70
‑
1.60(m,4h),1.53(s,9h),1.51
‑
1.44(m,4h),1.36(s,9h),1.30(s,9h),1.20(s,6h),1.13(s,9h),0.59(s,3h),0.12(s,3h)
[0222]
比较合成例2
‑1[0223]
步骤1:配体化合物的制备
[0224]
将作为cp单元的4
‑
(4
‑
(叔丁基)苯基)
‑2‑
甲基
‑
1h
‑
茚(2.6g,10mmol)放入100ml舒伦克瓶中,向其中加入thf(35ml),并冷却至
‑
20℃以下。将冷却的混合溶液搅拌5分钟,然后向其中加入nbl(4.2ml,2.5m己烷溶液),使其反应过夜以制备锂化cp。当加入nbl时,混合溶液变为棕色。
[0225]
将作为系链硅烷的二氯(叔丁氧基己基)甲基硅烷(2.84g)放入另一个100ml舒伦克瓶中,并向其中加入mtbe(35ml)。将舒伦克瓶冷却至
‑
20℃以下,然后滴加制备的锂化cp并使其反应。当反应完成时,通过在真空下蒸馏去除反应产物中的溶剂,并通过使用己烷(hex)过滤去除所得的盐。将t
‑
bunh2(4.5ml)加入所得反应产物并使其反应,然后通过使用己烷过滤去除所得沉淀物,从而获得1
‑
(6
‑
(叔丁氧基)己基)
‑
n
‑
(叔丁基)
‑1‑
(4
‑
(4
‑
(叔丁基)苯基)
‑2‑
甲基
‑
1h
‑
茚
‑1‑
基)
‑1‑
甲基硅烷胺的配体化合物(黄色油,5.2g,产率98%(以摩尔计))。
[0226]
nmr(400mhz,c6d6)7.78
‑
7.67(m,2h),7.60
‑
7.7.19(m,5h),7.05(s,1h)3.68(s,1h),3.27
‑
3.06(m,3h),2.92
‑
2.86(m,1h),1.26
‑
1.12(m,4h),1.11(s,9h),1.09(s,9h),1.08(s,3h),1.06(s,9h),1.04
‑
0.99(m,4h),0.54
‑
0.49(m,1h),0.18(s,1.5h),0.05(s,1.5h)
[0227]
步骤2:过渡金属化合物的制备
[0228]
将步骤1中制备的配体化合物1
‑
(6
‑
(叔丁氧基)己基)
‑
n
‑
(叔丁基)
‑1‑
(4
‑
(4
‑
(叔丁基)苯基)
‑2‑
甲基
‑
1h
‑
茚
‑1‑
基)
‑1‑
甲基硅烷胺(4.6g,8.6mmol)放入100ml舒伦克瓶中,加入甲苯(43ml),然后冷却至
‑
20℃以下。通过搅拌将溶液充分冷却5分钟,并向所得混合溶
液中加入nbl(7.2ml,2.5m己烷溶液),然后锂化。确认了在锂化后,混合溶液的颜色从浅黄色变为深黄色。在锂化反应完成后,将所得反应溶液冷却至0℃,加入mmb(8.6ml,3m乙醚溶液),将温度立即降至
‑
20℃,并添加ticl4(8.6ml,1m甲苯溶液)。添加时,产生烟,反应溶液立即变成棕色。添加后,进行o/n搅拌,完成后,通过过滤器除去盐,得到具有下述结构的棕色油相过渡金属化合物(a)(棕色油,3.9g,产率74%(以摩尔计))。
[0229][0230]
nmr(400mhz,c6d6),7.75
‑
7.12(m,8h),3.30
‑
3.26(m,2h),2.68
‑
2.45(m,1h),1.76
‑
1.52(m,6h),1.50
‑
1.13(m,4h),1.25(s,9h),1.20
‑
1.15(m,2h),1.14(s,9h),1.10(s,9h)0.96(s,3h),0.58(s,3h),0.11(s,3h)
[0231]
比较合成例2
‑2[0232]
步骤1:配体化合物的制备
[0233]
将作为cp单元的2
‑
异丙基
‑4‑
苯基
‑
1h
‑
茚(0.94g,4mmol)放入50ml舒伦克瓶中,向其中加入thf(13ml),冷却至
‑
20℃以下。将冷却的混合溶液搅拌5分钟,然后向其中加入nbl(1.7ml,2.5m己烷溶液),使其反应过夜以制备锂化cp。当加入nbl时,混合溶液变为红棕色。
[0234]
将作为系链硅烷的二氯(叔丁氧基己基)甲基硅烷(1.16g)放入另一个100ml舒伦克瓶中,并向其中加入thf(13ml)。将舒伦克瓶冷却至
‑
20℃以下,然后滴加制备的锂化cp并使其反应。当反应完成后,通过在真空下蒸馏去除反应产物中的溶剂,并通过使用己烷(hex)过滤去除所得的盐。将t
‑
bunh2(1.7ml)加入所得反应产物并使其反应,然后通过使用己烷过滤去除所得沉淀物,从而获得1
‑
(6
‑
(叔丁氧基)己基)
‑
n
‑
(叔丁基)
‑1‑
(2
‑
异丙基
‑4‑
苯基
‑
1h
‑
茚
‑1‑
基)
‑1‑
甲基硅烷胺的配体化合物(黄色油,1.98g,产率98%(以摩尔计))。
[0235]
nmr(400mhz,c6d6),7.68
‑
7.63(m,2h),7.57
‑
7.41(m,2h),7.41
‑
7.16(m,4h),6.98(s,1h),3.67(s,1h),3.30
‑
3.19(m,4h),3.06
‑
2.99(m,0.5h),2.94
‑
2.87(m,0.5h),2.52
‑
2.42(m,0.5h),2.02
‑
1.92(m,0.5h),1.69
‑
1.38(m,8h),1.13(s,9h),1.05(s,6h),0.98(s,9h),0.27(s,1.5h),0.16(s,1.5h)
[0236]
步骤2:过渡金属化合物的制备
[0237]
将步骤1中制备的配体化合物1
‑
(6
‑
(叔丁氧基)己基)
‑
n
‑
(叔丁基)
‑1‑
(4
‑
(3,5
‑
二
‑
叔丁基苯基)
‑2‑
异丙基
‑
1h
‑
茚
‑1‑
基)
‑1‑
甲基硅烷胺(1.98g,3.9mmol)放入100ml舒伦克瓶中,加入甲苯(13ml),然后冷却至
‑
20℃以下。通过搅拌将溶液充分冷却5分钟,并向所得混合溶液中加入nbl(5.1ml,2.5m己烷溶液),然后锂化。确认了在锂化后,混合溶液的颜色变黄。在锂化反应完成后,将所得反应溶液冷却至0℃,加入mmb(13ml,3m乙醚溶液),将温度立即降至
‑
20℃,并添加ticl4(3.9ml,1m甲苯溶液)。添加时,产生烟,反应溶液立即变成
棕色。添加后,进行o/n搅拌,完成后,通过过滤器除去盐,得到棕色油相过渡金属化合物(b)(棕色油,1.79g,产率79%(以摩尔计))。
[0238][0239]
nmr(400mhz,c6d6),7.71
‑
7.69(m,2h),7.52
‑
7.44(m,2h),7.29
‑
7.22(m,1h),7.29
‑
7.17(s,4h),3.38
‑
3.24(m,2h),2.88
‑
2.41(m,2h),1.68
‑
1.60(m,7h),1.50(s,9h),1.30
‑
1.20(m,5h),1.19(s,6h),1.14(s,9h),0.58(s,3h),0.07(s,3h)
[0240]
比较合成例2
‑3[0241]
使用通过已知方法制备的具有下述结构的过渡金属化合物(c)。
[0242][0243]
比较合成例2
‑4[0244]
使用通过已知方法制备的具有下述结构的过渡金属化合物(d)。
[0245][0246]
<混杂负载型催化剂的制备>
[0247]
制备例1
[0248]
(1)制备载体
[0249]
将二氧化硅(sylopol 948
tm
,由grace davison制造)脱水,并在真空下于600℃的温度下干燥12小时。
[0250]
(2)制备混杂负载型催化剂
[0251]
在室温下将10g经干燥的二氧化硅放入玻璃反应器中,加入100ml甲苯,然后搅拌。
在将二氧化硅充分分散后,加入106ml的10重量%的甲基铝氧烷(mao)/甲苯溶液,在搅拌下于40℃和200rpm下缓慢反应14小时。反应后,停止搅拌,进行30分钟的沉降,将剩余溶液进行倾析。加入100ml甲苯并搅拌10分钟,进行30分钟的沉降,然后将剩余溶液进行倾析。加入50ml甲苯,然后将合成例1的第一过渡金属化合物0.15mmol/gsio2溶于甲苯中,并加入此溶液,使其反应1小时。在反应完成后,将合成例2
‑
1的第二过渡金属化合物0.18mmol/gsio2溶于甲苯中,并加入此溶液,在搅拌下使其反应2小时。在反应完成后,停止搅拌,分离并除去甲苯层,加入100ml正己烷,然后搅拌10分钟。在搅拌完成后,进行30分钟的沉降,然后除去剩余溶液,并在减压下除去剩余己烷,从而获得混杂负载型催化剂。
[0252]
制备例2
[0253]
以与制备例1相同的方式制备混杂负载型催化剂,不同之处在于使用合成例2
‑
2中制备的化合物代替制备例1中的第二过渡金属化合物,并以下表1中所述的量使用第一和第二过渡金属化合物。
[0254]
制备例3
[0255]
以与制备例2相同的方式制备混杂负载型催化剂,不同之处在于,在制备例2中,以下表1中所述的量使用第一和第二过渡金属化合物。
[0256]
比较制备例1
[0257]
以与制备例1相同的方式制备混杂负载型催化剂,不同之处在于,使用比较合成例2
‑
1中制备的过渡金属化合物(a)代替制备例1中的第二过渡金属化合物。
[0258]
比较制备例2
[0259]
以与制备例1相同的方式制备混杂负载型催化剂,不同之处在于,使用比较合成例2
‑
2中制备的过渡金属化合物(b)代替制备例1中的第二过渡金属化合物,并以下表1中所述的量使用第一和第二过渡金属化合物。
[0260]
比较制备例3
[0261]
以与制备例1相同的方式制备混杂负载型催化剂,不同之处在于,使用比较合成例2
‑
3中制备的过渡金属化合物(c)代替制备例1中的第二过渡金属化合物,并以下表1中所述的量使用第一和第二过渡金属化合物。
[0262]
比较制备例4
[0263]
以与制备例1相同的方式制备混杂负载型催化剂,不同之处在于,使用比较合成例2
‑
4中制备的过渡金属化合物(d)代替制备例1中的第二过渡金属化合物,并以下表1中所述的量使用第一和第二过渡金属化合物。
[0264]
<聚烯烃的制备>
[0265]
实施例1
[0266]
在下表1中所示的条件下,使用制备例1的混杂负载型催化剂(合成例1和2
‑
1的化合物)进行乙烯均聚。
[0267]
为了制备烯烃聚合物,准备2l金属合金反应器,其配备有机械搅拌器,温度可控,并可用于高压反应。将反应器在真空下于120℃干燥,冷却,并引入0.9kg纯化的正己烷,在室温下引入1350ppm的三乙基铝(1m己烷溶液),然后将15mg制备例1中制备的负载型茂金属催化剂引入反应器中。之后,将反应器的温度缓慢升至80℃,然后进行2小时的聚合过程。此时,连续注入乙烯气体以将反应器的压力保持在约9kgf/cm2。基于乙烯,以0.16体积%注入
氢气。在反应完成后,排出未反应的乙烯和氢。
[0268]
实施例2和3
[0269]
以与实施例1相同的方式制备各种聚乙烯,不同之处在于,如下表1中所示改变催化剂和聚合条件。
[0270]
比较例1
[0271]
使用利用ziegler
‑
natta催化剂制备的高密度聚乙烯(ce2080
tm
,由lg chem.制造)。
[0272]
比较例2至4
[0273]
以与实施例1相同的方式制备各种聚乙烯,不同之处在于如下表1中所示改变催化剂和聚合条件。
[0274]
表1
[0275][0276]
*在比较例4中,由于用于制备聚乙烯的第二过渡金属化合物中没有系链基团,因此10聚合期间的活性低,并且由于浸出而在反应器中发生结垢。
[0277]
实验例1
[0278]
针对用于制备本发明的混杂负载型催化剂的过渡金属化合物,评价其作为单一催化剂的活性。具体地,分别使用合成例和比较合成例中制备的过渡金属化合物作为单一催化剂来制备均聚乙烯。分别测量制备的聚合物的聚合活性、重均分子量(mw)和分子量分布(pdi)。结果在下表2中示出。
[0279]
<负载型茂金属催化剂的制备>
[0280]
将硅胶(sylopol 952x
tm
,在250℃煅烧,7g)在氩气(ar)下放入玻璃反应器中,并在室温下缓慢引入53.1ml(相当于10mmol/1g二氧化硅)的10重量%甲基铝氧烷(mao)甲苯溶
液,并在95℃搅拌12小时。反应完成后,将反应器冷却至室温,静置15分钟,并使用套管倾析出溶剂。引入50ml甲苯并搅拌1分钟,静置15分钟,并使用套管倾析出溶剂。然后,引入50ml甲苯,将60μmol(相当于60μmol/1g二氧化硅)的合成例或比较合成例中制备的各种过渡金属化合物溶于10ml甲苯中,并使用套管转移至反应器。在80℃搅拌2小时后,将反应器冷却,静置15分钟,并使用套管倾析出溶剂。引入50ml甲苯并搅拌1分钟,静置15分钟,并使用套管倾析出溶剂。将此过程重复两次。以相同的方式引入50ml己烷并搅拌1分钟,静置15分钟,并使用套管倾析出溶剂。使用套管转移3.1ml(基于100重量份的二氧化硅,抗静电剂含量=2重量份)溶于己烷的抗静电剂(atmer 163
tm
,croda)。在室温下搅拌20分钟,并将溶液转移至玻璃过滤器中以除去溶剂。在室温下真空干燥5小时,从而获得各负载型催化剂。
[0281]
<均聚乙烯的制备>
[0282]
准备600ml金属合金反应器,其配备有机械搅拌器,温度可控,并可用于高压反应。将反应器在真空下于120℃干燥,冷却,在室温下引入450ppm的三乙基铝(1m己烷溶液),然后将15mg的合成例和比较例中制备的各过渡金属化合物负载型催化剂引入反应器中。之后,将反应器的温度缓慢升至80℃,然后进行1小时的聚合过程。此时,连续注入乙烯气体以将反应器的压力保持在约9kgf/cm2。在反应完成后,排出未反应的乙烯。
[0283]
<评价>
[0284]
(1)聚合活性(kg pe/g cat
·
hr):基于单位时间(h),通过所产生的聚合物的重量(kg pe)与所用负载型催化剂的重量(g)之比来确定。
[0285]
(2)重均分子量(mw,g/mol)和分子量分布(pdi,多分散性指数):通过凝胶渗透色谱法(gpc)测量重均分子量(mw)和数均分子量(mn),并测定mw/mn之比作为分子量分布。具体而言,通过使用来自polymer laboratories的长度300mm的plgel mix
‑
b柱的waters pl
‑
gpc220仪器进行测量。此时,评价温度为160℃,使用1,2,4
‑
三氯苯作为溶剂,流速为1ml/min。样品以10mg/10ml的浓度制备,并以200μl的量供应。使用通过聚苯乙烯标样建立的校准曲线来导出mw和mn值。聚苯乙烯标样的分子量(g/mol)为2,000/10,000/30,000/70,000/200,000/700,000/2,000,000/4,000,000/10,000,000九种类型。
[0286]
表2
[0287][0288]
作为实验结果,即使作为单一催化剂使用时,用于制备本发明的混杂负载型催化剂的第一和第二过渡金属化合物也展现出优异的聚合活性。相比之下,当比较合成例2
‑
1中制备的过渡金属化合物用作单一催化剂时,其展现出0.9kg pe/g cat
·
hr的非常低的聚合活性,表明在用作混杂负载型催化剂时其催化活性也变差,并且其改善聚合物的物理性质的效果也变差。
[0289]
实验例2
[0290]
通过以下方法测量实施例和比较例中所用的混杂负载型催化剂以及使用所述催化剂制备的聚乙烯的物理性质,结果在下表3中示出。
[0291]
(1)催化活性(活性,kg pe/g cat
·
hr):在实施例和比较例的各种聚乙烯聚合期间评价催化活性,并且基于单位时间(h),通过所产生的聚合物的重量(kg pe)与所用负载型催化剂的重量(g)之比来确定。
[0292]
(2)重均分子量(mw,g/mol)和分子量分布(pdi,多分散性指数):分别通过凝胶渗透色谱法(gpc)测量实施例和比较例中制备的各种聚乙烯的重均分子量(mw)和数均分子量(mn),并测定mw/mn之比作为分子量分布(pdi)。
[0293]
具体而言,通过使用来自polymer laboratories的长度300mm的plgel mix
‑
b柱的waters pl
‑
gpc220仪器进行测量。此时,评价温度为160℃,使用1,2,4
‑
三氯苯作为溶剂,流速为1ml/min。样品以10mg/10ml的浓度制备,并以200μl的量供应。使用通过聚苯乙烯标样建立的校准曲线来导出mw和mn值。聚苯乙烯标样的分子量(g/mol)为2,000/10,000/30,000/70,000/200,000/700,000/2,000,000/4,000,000/10,000,000九种类型。
[0294]
(3)mi
5.0
和mfrr(21.6/5):根据astm d1238(条件e,190℃,在5.0kg的负荷下)标准测量实施例和比较例中制备的各种聚乙烯的熔融指数(mi
5.0
)。此外,通过用mfr
21.6
除以mfr5计算聚乙烯的熔体流动速率比(mfrr,21.6/5)。根据astm d 1238在21.6kg的负荷下于190℃的温度测量mfr
21.6
,并且根据astm d 1238在5kg的负荷下于190℃的温度测量mfr5。
[0295]
(4)复数粘度(η*(ω500)):必须调节门尼粘度,以在电线和电缆中使用cpe。因此,在500rad/s的频率下测得的复数粘度(η*(ω500))为800pas时,门尼粘度预计会一致,因此将其设为同等水平。
[0296]
具体而言,利用ta仪器的ares(advanced rheometric expansion system,ares g2),在0.05rad/s的频率(ω)下测量聚乙烯的复数粘度。使用直径为25.0mm的平行板在190℃下设置样品,使板之间的间隙变为2.0mm。在动态应变频率扫描模式下以5%的应变进行测量,频率为0.05rad/s至500rad/s,每个decade 10个点,总共41个点。
[0297]
(5)密度:根据astm d
‑
1505的方法测量各种聚乙烯的密度(g/cm3)。
[0298]
(6)分数(%):进行gpc分析,分数计算为所得分子量分布曲线中的log mw区间相对于总面积所占的面积(%)。分数的总和为100
±
1,其可能不会恰好为100。
[0299]
gpc分析具体在以下条件下进行。
[0300]
使用waters pl
‑
gpc220作为凝胶渗透色谱法(gpc)仪器,并使用长度为300mm的polymer laboratories plgel mix
‑
b柱。评价温度为160℃,使用1,2,4
‑
三氯苯作为溶剂,流速为1ml/min。通过使用gpc分析仪(pl
‑
gp220)将各聚乙烯样品溶解在含有0.0125%bht的1,2,4
‑
三氯苯中10小时来进行预处理,并且以200μl的量供应浓度为10mg/10ml的样品。通过采用聚苯乙烯标样建立的校准曲线获得mw和mn。使用9种聚苯乙烯标样,分子量为2000g/mol、10000g/mol、30000g/mol、70000g/mol、200000g/mol、700000g/mol、2000000g/mol、4000000g/mol和10000000g/mol。
[0301]
(7)mdr扭矩(m
h
‑
m
l
):
[0302]
使用αlpha technologies production mdr(动模流变仪)测量各聚乙烯样品的mdr扭矩,以便评价聚乙烯的交联度。
[0303]
特别是,在将100g实施例和比较例中制备的各种聚乙烯、0.4g酚类抗氧化剂(ao)
和1.2g交联剂(dcp,过氧化二异丙苯)在80℃混合后,在140℃下制备样品片10分钟。然后,利用mdr(动模流变仪)在180℃下测量样品片的m
h
值和m
l
值10分钟。通过从m
h
值中减去m
l
值来计算mdr扭矩(m
h
‑
m
l
)。在此,m
h
表示在完全固化时测得的最大硫化扭矩,而m
l
表示最小硫化扭矩。
[0304]
表3
[0305][0306]
作为实验结果,实施例1至3中所用的混杂负载型催化剂显示出相当于或高于比较例中所用的催化剂的优异催化活性,特别是,实施例1和2的混杂负载型催化剂显示出高于比较例的催化剂的催化活性。
[0307]
此外,与比较例相比,利用混杂负载型催化剂制备的实施例1至3的聚乙烯显示出显著减少的低分子含量,其中log mw为3.5以下的分数为1.2%以下,log mw超过3.5至4.0以下的分数为4.65%,同时其显示出了6.1以上的宽分子量分布。另外,与比较例相比,它们的mdr扭矩为7.7以上,表明交联度高。
[0308]
实施例3优于比较例,但与实施例1和2相比显示出减少低分子含量和提高交联度的效果略微降低。这些结果归因于聚乙烯的制备中所用的催化剂的差异。实施例3包含与实施例2中相同组合的过渡金属化合物,但它们在混合比上彼此不同。另外,实施例3包含与实施例1中混合比相同的过渡金属化合物,但第二过渡金属化合物的结构彼此不同。因此,与现有催化剂相比,本发明的包含第一和第二过渡金属化合物的组合的混杂负载型催化剂在催化活性和改善聚乙烯物理性质方面可以展现出更优异的效果。可见,当根据第一和第二过渡金属化合物的组合优化混合比时,可进一步改善催化活性和制备的聚乙烯的物理性质。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1