用于制备氨基甲酰氧基甲基三唑环己酸化合物的方法与流程
用于制备氨基甲酰氧基甲基三唑环己酸化合物的方法
1.相关申请的交叉引用
2.本技术要求2019年4月16日提交的美国临时申请号62/834,538的优先权权益;将其内容通过引用以其整体并入本文。
技术领域
3.本发明涉及用于制备氨基甲酰氧基甲基三唑环己酸化合物的改进方法以及其新型中间体。
背景技术:
4.已经描述了可用于治疗纤维化的氨基甲酰氧基甲基三唑环己酸lpa(尤其是lpa1)拮抗剂。参见例如wo 2017/223016(us 2017/0360759)。需要制造氨基甲酰氧基甲基三唑环己酸化合物的改进方法,其提供实用的大规模合成以及改进的生产质量、效率和安全性。
技术实现要素:
5.本发明提供了用于制造氨基甲酰氧基甲基三唑环己酸化合物的新颖方法以及其新颖中间体。
6.还描述了制造中间体化合物、立体异构体和其盐的方法。
具体实施方式
7.在阅读下面详细描述时,本领域普通技术人员可以更容易地理解本发明的特征和优点。应当理解,出于清楚的原因,在单独的实施方案的上下文中所述的本发明的某些特征也可以组合以形成单个实施方案。相反,出于简洁的原因,在单个实施方案的上下文中所述的本发明的各种特征也可以被组合以形成其子组合。
8.技术人员将认识到本文所述的一些化学结构可以通过一种或多种其他共振形式在纸上呈现;或者可以以一种或多种其他互变异构形式存在,即使在动力学上,技术人员也认识到此类互变异构形式仅代表此类一种或多种化合物的非常小的一部分。此类化合物清楚地考虑在本公开文本的范围内,但此类共振形式或互变异构体在本文中未明确呈现。
9.在方面1a中,本发明提供了一种制造具有式(i)的结构的化合物的方法:
[0010][0011]
其中r1是c
1-6
烷基;并且r2和r
2a
是卤素;
[0012]
所述方法包括使式(iii)的化合物:
[0013][0014]
其中r1是c
1-6
烷基;r2是卤素;并且r3是卤素;
[0015]
与为有机锂的试剂2在为极性非质子或非极性非质子溶剂或其溶剂混合物的溶剂2中
[0016]
在一定温度下接触一定时间以足以进行锂卤素交换;接着与烷基化剂在为极性溶剂、极性非质子溶剂、或非极性非质子溶剂或其溶剂混合物的溶剂3中
[0017]
在一定温度下接触一定时间以足以进行烷基化;接着与强酸反应以产生所述式(i)的化合物。
[0018]
在方面1b中,本发明提供了一种制造具有式(i)的结构的化合物的方法:
[0019][0020]
其中r1是c
1-6
烷基;并且r2和r
2a
是卤素;
[0021]
所述方法包括(1)使式(ii)的化合物:
[0022][0023]
其中r2和r3独立地是卤素;
[0024]
与选自r
5-om(金属碱醇盐)、m-oh(金属碱氢氧化物)、m2co3(金属碱碳酸盐)、mhco3(金属碱碳酸氢盐)(r6)3n(叔胺)及其混合物的试剂1的混合物
[0025]
在为r
4-oh,极性质子、极性非质子或非极性非质子溶剂,或其溶剂混合物的溶剂1中接触;并且接着添加烷基化剂;
[0026]
其中m是选自li、na、k和cs的金属元素;并且r4、r5和r6[0027]
独立地是c
1-6
烷基;
[0028]
其时间和温度足以进行烷基化并且以产生式(iii)的化合物:
[0029][0030]
其中r1是c
1-6
烷基;r2是卤素;并且r3是卤素;
[0031]
以及(2)使所述式(iii)的化合物与为有机锂的试剂2在为极性非质子或非极性非质子溶剂或其溶剂混合物的溶剂2中
[0032]
在一定温度下接触一定时间以足以进行锂卤素交换;接着与烷基化剂在为极性溶剂、极性非质子溶剂、或非极性非质子溶剂或其溶剂混合物的溶剂3中
[0033]
在一定温度下接触一定时间以足以进行烷基化;接着与强酸反应以产生所述式(i)的化合物。
[0034]
在方面1a范围内的方面2a中,本发明提供了一种制造具有式(i)的结构的化合物的方法;
[0035]
所述方法包括使所述式(iii)的化合物与选自n-buli、n-hexli和phli的试剂2在选自己烷、thf、methf及其溶剂混合物的溶剂2中
[0036]
在-30℃至-10℃下接触一定时间以足以进行锂卤素交换;接着与选自苄基氯甲基醚的烷基化剂在-30℃至-10℃下接触一定时间以足以进行烷基化,并且与选自在乙酸中的hbr的强酸或乙酰溴和2-丙醇的混合物在选自thf、ch3cn、ipac、methf及其溶剂混合物的溶剂3中接触以产生所述式(i)的化合物。
[0037]
在方面1b范围内的方面2b方面中,本发明提供了一种制造具有式(i)的结构的化合物的方法;
[0038]
所述方法包括(1)使式(ii)的化合物,
[0039]
与选自liotbu、k2co3、khco3、et3n、naotbu、kotbu、lioh、lioh*h2o、liome及其混合物的金属碱醇盐的混合物在选自ch3ch2c(ch3)2oh、c(ch3)3oh、ch(ch3)2oh、ch3ch2oh、ch3oh、thf、etoac、ipac、methf、丙酮、mibk、ch3cn、nmp、dmf、dcm、h2o及其溶剂混合物的溶剂1中接触;并且接着与选自甲基碘、硫酸二甲酯、碳酸二甲酯、甲苯磺酸甲酯及其混合物的烷基化剂
[0040]
在一定温度下接触24至72小时以足以进行烷基化并且以产生式(iii)的化合物:
[0041]
并且(2)使所述式(iii)的化合物与选自n-buli、n-hexli和phli的试剂2在选自己烷、thf、methf及其溶剂混合物的溶剂2中
[0042]
在-30℃至-10℃下接触一定时间以足以进行锂卤素交换;并且接着与选自苄基氯甲基醚的烷基化剂在-30℃至-10℃下接触一定时间以足以进行烷基化,并且与选自在乙酸中的hbr的强酸或乙酰溴和2-丙醇的混合物在选自thf、ch3cn、ipac、methf及其溶剂混合物的溶剂3中接触以产生所述式(i)的化合物。
[0043]
在方面3a中,本发明提供了一种制造具有式(ia)的结构的化合物的方法:
[0044][0045]
所述方法包括使式(iiia)的化合物:
[0046][0047]
与在己烷中的n-buli
[0048]
在-30℃至-10℃下接触《1小时以足以进行锂卤素交换,接着与苄基氯甲基醚
[0049]
在-30℃至-10℃下接触5至24小时以足以进行烷基化并且与在乙酸中的33wt%hbr在ch3cn中接触以产生所述式(ia)的化合物。
[0050]
在方面3b中,本发明提供了一种制造具有式(ia)的结构的化合物的方法:
[0051][0052]
所述方法包括(1)使式(iia)的化合物:
[0053][0054]
与ch3ch2c(ch3)2oh和liotbu的混合物在thf中在《40c下接触1至2小时;接着添加ch3i并且继续老化24至48小时以产生式(iiia)的化合物:
[0055][0056]
以及(2)使式(iiia)的化合物与n-buli在己烷中
[0057]
在-30℃至-10℃下接触《1小时以足以进行锂卤素交换,接着与苄基氯甲基醚在-30℃至-10℃下接触5至24小时以足以进行烷基化并且与在乙酸中的33wt%hbr在ch3cn中接触以产生式(ia)的化合物。
[0058]
在方面4a中,本发明提供了一种制造具有式(iv)的结构的化合物或其盐的方法:
[0059][0060]
其中ra是-n(c
1-4
烷基)2;
[0061]
所述方法包括使式(v)的化合物:
[0062][0063]
其中r6是卤素;
[0064]
与式(vi)的化合物或其盐:
[0065][0066]
其中ra是-n(c
1-4
烷基)2;
[0067]
在无机碱和相转移催化剂的存在下在为极性非质子溶剂或其溶剂混合物的溶剂4中
[0068]
在一定温度下接触一定时间以足以完成反应以产生所述式(iv)的化合物或其盐。
[0069]
在方面4a范围内的方面5a中,本发明提供了一种制造具有式(iva)的结构的化合物或其盐的方法;
[0070][0071]
所述方法包括使式(v)的化合物与式(via)的化合物或其盐:
[0072][0073]
在khco3和(c
1-4
烷基)4nbr的存在下在选自dmf、ch3cn、nmp、dmac、hmpa、dmpu、dme、thf及其溶剂混合物的溶剂4中
[0074]
在25℃至35℃下接触一定时间以足以完成反应以产生所述式(iva)的化合物或其盐。
[0075]
在方面6a中,本发明提供了一种制造具有式(ivb)的结构的化合物的方法:
[0076][0077]
所述方法包括使式(va)的化合物:
[0078]
[0079]
与式(via)的化合物:
[0080][0081]
在khco3和(et)4nbr的存在下在dmf中
[0082]
在25℃至35℃下接触4至72小时以足以完成反应以产生所述式(ivb)的化合物。
[0083]
在方面7a中,本发明提供了一种制造式(vii)的化合物或其立体异构体或盐的方法:
[0084][0085]
其中ra是-n(c
1-4
烷基)2;
[0086]
所述方法包括使式(viii)的化合物或其盐:
[0087][0088]
在和选自和4-dmap的助催化剂的存在下与或不与碱的水溶液在为非极性溶剂或其溶剂混合物的溶剂5中
[0089]
在一定温度下接触一定时间以足以完成反应以产生所述式(vii)的化合物或其立体异构体或盐。
[0090]
在方面7a范围内的方面8a中,本发明提供了一种制造具有式(viia)的结构的化合物或其盐的方法;
[0091]
[0092]
所述方法包括使式(viii)的化合物或其盐;
[0093]
在和的存在下与或不与koh水溶液在选自甲苯、ch2cl2、三氟甲苯、1,2-二氯苯及其溶剂混合物的溶剂5中
[0094]
在25℃至35℃下接触一定时间以足以完成反应以产生所述式(viia)的化合物或其盐。
[0095]
在方面9a中,本发明提供了一种制造具有式(viib)的结构的化合物的方法:
[0096][0097]
所述方法包括使式(viii)的化合物或其盐;
[0098]
在和的存在下与或不与koh水溶液在甲苯中
[0099]
在25℃至35℃下接触24至48小时以足以完成反应以产生所述式(viib)的化合物。
[0100]
在方面10a中,本发明提供了一种制造式(ix)的化合物或其立体异构体或盐的方法:
[0101][0102]
其中ra是-n(c
1-4
烷基)2;
[0103]
所述方法包括使式(vii)的化合物或其盐:
[0104][0105]
其中ra是-n(c
1-4
烷基)2;
[0106]
与过渡金属催化剂在二质子酸的存在下在为质子或极性非质子溶剂或其溶剂混合物的溶剂6中
[0107]
在一定温度下接触一定时间以足以进行酮还原以产生所述式(ix)的化合物或其立体异构体或盐。
[0108]
在方面10a范围内的方面11a中,本发明提供了一种制造式(ixa)的化合物或其盐的方法;
[0109][0110]
所述方法包括使式(viia)的化合物或其盐:
[0111][0112]
与选自ircl4、ircl4*水合物或[ir(cod)cl]2的过渡金属催化剂在亚磷酸的存在下在选自ipa、meoh、etoh、t-amoh、h2o、nmp、dmf、dmac、环丁砜及其溶剂混合物的溶剂6中
[0113]
在65℃至100℃下接触一定时间以足以进行酮还原以产生所述式(ixa)的化合物或其盐。
[0114]
在方面12a中,本发明提供了一种制造式(ixb)的化合物的方法:
[0115][0116]
所述方法包括使式(viib)的化合物:
[0117][0118]
与ircl4*水合物或[ir(cod)cl]2在亚磷酸的存在下在ipa/h2o或其溶剂混合物中
[0119]
在80℃至85℃下接触24至96小时以足以进行酮还原以产生所述式(ixb)的化合物。
[0120]
在方面13a中,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0121][0122]
所述方法包括(1)使式(ixa)的化合物或其盐:
[0123][0124]
与选自naoh、koh、lioh、四烷基氢氧化铵及其混合物的试剂3在其中r7独立地是c
1-6
烷基的r
7-oh水溶液中
[0125]
在80℃至85℃下接触最高达48小时以足以水解所有三个酯部分以产生所述式(xi)的化合物
[0126][0127]
其中m选自金属元素和四烷基铵,所述金属元素选自li、na和k;
[0128]
(2)接触在质子溶剂中的酸;以及
[0129]
(3)接触在质子溶剂中的高碘酸;
[0130]
在20℃-25℃下持续最高达48小时以足以氧化以产生所述式(xa)的化合物或其盐。
[0131]
在方面13b中,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0132][0133]
所述方法包括(1)使式(xix)的化合物或其盐:
[0134][0135]
与烯还原酶生物催化剂在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机助溶剂的情况下
[0136]
在一定温度下接触一定时间以足以产生所述式(xx)的化合物;
[0137][0138]
接着(2)在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂的情况下添加酮还原酶生物催化剂
[0139]
在一定温度下持续另外的时间以足以产生所述式(xa)的化合物或其盐。
[0140]
在另一个方面,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0141][0142]
所述方法包括(1)使式(xix)的化合物或其盐:
[0143][0144]
与选自ered-302、ered-303和ered-211的烯还原酶生物催化剂在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机助溶剂的情况下
[0145]
在25℃至35℃下接触一定时间以足以产生所述式(xx)的化合物;
[0146][0147]
接着(2)在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂的情况下添加酮还原酶生物催化剂
[0148]
在25℃至35℃下持续另外的时间以足以产生式(xa)的化合物或其盐。
[0149]
在另一个方面,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0150][0151]
所述方法包括(1)使式(xix)的化合物或其盐:
[0152][0153]
与选自ered-302、ered-303和ered-211的烯还原酶生物催化剂在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机助溶剂的情况下
[0154]
在25℃至35℃下接触至少14小时以足以产生所述式(xx)的化合物;
[0155][0156]
接着(2)在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂的情况下添加kred-p2-g03
[0157]
在25℃至35℃下持续至少另外2小时以足以产生式(xa)的化合物或其盐。
[0158]
在方面13c中,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0159][0160]
所述方法包括使式(xix)的化合物或其盐:
[0161][0162]
与烯还原酶生物催化剂与酮还原酶生物催化剂的组合在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂情况下
[0163]
在一定温度下接触一定时间以足以产生所述式(xa)的化合物或其盐。
[0164]
在另一个方面,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0165][0166]
所述方法包括使式(xix)的化合物或其盐:
[0167][0168]
与选自ered-302、ered-303和ered-211的烯还原酶生物催化剂与酮还原酶生物催化剂的组合在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂情况下
[0169]
在20℃至35℃下接触一定时间以足以产生式(xa)的化合物或其盐。
[0170]
在另一个方面,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0171][0172]
所述方法包括使式(xix)的化合物或其盐:
[0173][0174]
与选自ered-302、ered-303和ered-211的烯还原酶生物催化剂与kred-p2-g03的组合在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂情况下
[0175]
在20℃至35℃下接触至少14小时以足以产生式(xa)的化合物或其盐。
[0176]
在方面13d中,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0177][0178]
所述方法包括(1)使其式(xxi)的化合物:
[0179][0180]
与磺酰氯试剂在非质子极性溶剂和无机或有机碱中
[0181]
在一定温度下接触一定时间以足以进行o-磺酰化以产生所述式(xxii)的化合物;
[0182][0183]
(2)(a)使所述式(xxii)的化合物与过渡金属催化剂、膦配体和有机或无机碱在(c
1-4
烷基)-oh的存在下在用一氧化碳吹扫的情况下
[0184]
在一定温度和压力下接触一定时间以足以进行羰基化以产生所述式(xxiii)的化合物;
[0185][0186]
然后接着使式(xxiii)的化合物与碱的水溶液接触以产生式(xix)的化合物或其盐:
[0187][0188]
或(b)使所述式(xxii)的化合物与过渡金属催化剂、膦配体在无机或有机碱的存在下在水和另一种极性非质子溶剂中在用一氧化碳吹扫的情况下
[0189]
在一定温度和压力下接触一定时间以足以进行羰基化以产生所述式(xix)的化合物或其盐;
[0190]
(3)使所述式(xix)的化合物或其盐与烯还原酶生物催化剂在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机助溶剂的情况下
[0191]
在一定温度下接触一定时间以足以产生所述式(xx)的化合物;
[0192][0193]
接着(4)在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂的情况下添加酮还原酶生物催化剂
[0194]
在一定温度下持续另外的时间以足以产生所述式(xa)的化合物或其盐。
[0195]
在方面13e中,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0196][0197]
所述方法包括(1)使其式(xxi)的化合物:
[0198][0199]
与对甲苯磺酰氯在乙酸乙酯和三乙胺中
[0200]
在0℃至10℃下接触至少12小时以足以进行o-磺酰化以产生所述式(xxii)的化合物;
[0201][0202]
(2)(a)使式(xxii)的化合物与乙酸钯(ii)、1,3-双(二苯基膦基)丙烷和n,n-二异丙基乙胺在(c
1-4
烷基)-oh的存在下在用一氧化碳吹扫的情况下
[0203]
在20℃至60℃和一氧化碳压力下接触至少12小时以足以进行羰基化以产生式(xxiii)的化合物;
[0204][0205]
然后接着使式(xxiii)的化合物与碱的水溶液接触以产生式(xix)的化合物或其盐:
[0206][0207]
或(b)使式(xxii)的化合物与选自pd(oac)2、pdcl2(ch3cn)2和pd(dppp)cl2的过渡金属催化剂,选自1,3-双(二苯基膦基)丙烷、外消旋-binap、呫吨、josiphos sl-j001-1、josiphos sl-j009-1-g3钯环和biphep的膦配体,和选自k2co3、n,n-二异丙基乙胺、三乙胺、叔丁基四甲基胍、四甲基胍、khco3、dbu、na2co3和koac的无机或有机碱在水和选自etoac、2-methf、ch3cn、dmac、thf及其混合物的极性非质子溶剂中在用一氧化碳吹扫的情况下
[0208]
在20℃至60℃和一氧化碳压力下接触至少12小时以足以进行羰基化以产生式(xix)的化合物或其盐;
[0209]
(3)使式(xix)的化合物或其盐与选自ered-302、ered-303和ered-211的烯还原酶生物催化剂在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有诸如dmso、ipa、二噁烷或丙酮的有机助溶剂的情况下
[0210]
在20℃至35℃下接触一定时间以足以产生所述式(xx)的化合物;
[0211][0212]
接着(4)在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂的情况下添加酮还原酶生物催化剂
[0213]
在25℃至35℃下持续另外的时间以足以产生式(xa)的化合物或其盐。
[0214]
在方面13f中,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0215][0216]
所述方法包括(1)使其式(xxi)的化合物:
[0217][0218]
与磺酰氯试剂在非质子极性溶剂和无机或有机碱中
[0219]
在一定温度下接触一定时间以足以进行o-磺酰化以产生所述式(xxii)的化合物;
[0220][0221]
(2)(a)使所述式(xxii)的化合物与过渡金属催化剂、膦配体和有机胺碱在(c
1-4
烷基)-oh的存在下在用一氧化碳吹扫的情况下
[0222]
在一定温度和压力下接触一定时间以足以进行羰基化以产生所述式(xxiii)的化合物;
[0223][0224]
然后接着使式(xxiii)的化合物与碱的水溶液接触以产生式(xix)的化合物或其盐:
[0225][0226]
或(b)使所述式(xxii)的化合物与过渡金属催化剂、膦配体在无机或有机碱的存在下在水和另一种极性非质子溶剂中在用一氧化碳吹扫的情况下
[0227]
在一定温度和一氧化碳压力下接触一定时间以足以进行羰基化以产生所述式(xix)的化合物或其盐;
[0228]
(3)使式(xix)的化合物或其盐与烯还原酶生物催化剂与酮还原酶生物催化剂的组合在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂的情况下
[0229]
在一定温度下接触一定时间以足以产生所述式(xa)的化合物或其盐。
[0230]
在方面13g中,本发明提供了一种制造式(xa)的化合物或其盐的方法:
[0231][0232]
所述方法包括(1)使其式(xxi)的化合物:
[0233]
[0234]
与对甲苯磺酰氯在乙酸乙酯和三乙胺中
[0235]
在0℃至10℃下接触至少12小时以足以进行o-磺酰化以产生所述式(xxii)的化合物;
[0236][0237]
(2)(a)使式(xxii)的化合物与乙酸钯(ii)、1,3-双(二苯基膦基)丙烷和n,n-二异丙基乙胺在(c
1-4
烷基)-oh的存在下在用一氧化碳吹扫的情况下
[0238]
在20℃至60℃和一氧化碳压力下接触至少12小时以足以进行羰基化以产生式(xxiii)的化合物;
[0239][0240]
然后接着使式(xxiii)的化合物与碱的水溶液接触以产生式(xix)的化合物或其盐:
[0241][0242]
或(b)使式(xxii)的化合物与选自pd(oac)2、pdcl2(ch3cn)2和pd(dppp)cl2的过渡金属催化剂,选自1,3-双(二苯基膦基)丙烷、外消旋-binap、呫吨、josiphos sl-j001-1、josiphos sl-j009-1-g3钯环和biphep的膦配体,和选自k2co3、二异丙基乙胺、三乙胺、叔丁基四甲基胍、四甲基胍、khco3、dbu、na2co3和koac的无机或有机碱在水和选自etoac、2-methf、ch3cn、dmac、thf及其混合物的极性非质子溶剂中在用一氧化碳吹扫的情况下
[0243]
在20℃至60℃和一氧化碳压力下接触至少小时以足以进行羰基化以产生式(xix)的化合物或其盐;
[0244]
(3)使式(xix)的化合物或其盐与选自ered-302、ered-303和ered-211的烯还原酶生物催化剂与kred-p2-g03的组合在磷酸盐缓冲水溶液、gdh、nadph和葡萄糖的存在下在有或没有选自dmso、ipa、二噁烷、丙酮及其混合物的有机溶剂情况下
[0245]
在20℃至35℃下接触至少14小时以足以产生式(xa)的化合物或其盐。
[0246]
在方面14a中,本发明提供了一种制造式(xa)的化合物的方法:
[0247][0248]
所述方法包括(1)使式(ixb)的化合物或其盐:
[0249][0250]
与naoh在ipa水溶液中在80℃至85℃下接触至少12小时以足以产生所述式(xia)的化合物;
[0251][0252]
(2)接触hcl水溶液;以及
[0253]
(3)接触在ipa水溶液中的高碘酸;
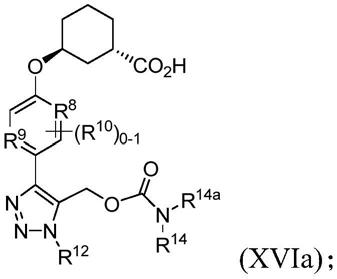
[0254]
在20℃-25℃下持续最高达48小时以足以氧化以产生所述式(xa)的化合物。
[0255]
在方面15a中,本发明提供了一种制造式(xii)的化合物或其立体异构体或盐的方法:
[0256][0257]
其中:r8和r9独立地是n、ch或c(c
1-6
烷基);
[0258]r10
独立地是c
1-4
烷基或卤素;并且
[0259]r11
独立地是br、cl或i;
[0260]
所述方法包括使式(xiii)的化合物或其盐:
[0261][0262]
其中:r8、r9、r
10
和r
11
与上式(xii)中的相同并且r
11a
是卤素;
[0263]
与所述式(x)的化合物或其立体异构体或盐:
[0264][0265]
在金属醇盐的存在下在为极性非质子或非极性溶剂或其溶剂混合物的溶剂7中;
[0266]
在一定温度下接触一定时间以足以完成反应以产生所述式(xii)的化合物或其立体异构体或盐。
[0267]
在方面15a范围内的方面16a中,本发明提供了一种制造式(xiia)的化合物或其盐的方法;
[0268][0269]
所述方法包括使式(xiiia)的化合物或其盐:
[0270][0271]
与所述式(xa)的化合物或其盐:
[0272][0273]
在选自kotbu、khmds、nahmds和戊醇钾的金属醇盐的存在下在选自dmf、mtbe、dmac、nmp、dmpu、thf、2-methf、cpme、二异丙醚、甲苯及其溶剂混合物的溶剂7中
[0274]
在20℃至35℃下接触一定时间以足以完成反应以产生所述式(xiia)的化合物或其盐。
[0275]
在方面17a中,本发明提供了一种制造式(xiib)的化合物或其盐的方法:
[0276][0277]
所述方法包括使式(xiiib)的化合物或其盐:
[0278][0279]
与所述式(xa)的化合物或其盐:
[0280][0281]
在kotbu的存在下在dmf/mtbe混合物中
[0282]
在20℃至35℃下接触18至》48小时以足以进行氟基置换以产生所述式(xiib)的化
合物或其盐。
[0283]
在方面18a中,本发明提供了一种制造式(xivc)的化合物或其立体异构体或盐的方法:
[0284][0285]
其中:r8和r9独立地是n、ch或c(c
1-6
烷基);
[0286]r10
独立地是c
1-4
烷基或卤素;并且
[0287]r12
是c
1-4
烷基;
[0288]
所述方法包括(1)使式(i)的化合物:
[0289][0290]
其中r1是c
1-6
烷基;并且r2和r
2a
是卤素;
[0291]
与有机金属试剂并且与或不与无机试剂在为极性非质子或非极性溶剂或其溶剂混合物的溶剂8中
[0292]
在一定温度下接触一定时间以足以进行金属-卤素交换;然后
[0293]
(2)使式(xii)的化合物或其立体异构体或盐:
[0294][0295]
其中:r8和r9独立地是n、ch或c(c
1-6
烷基);
[0296]r10
独立地是c
1-4
烷基或卤素;并且
[0297]r11
是br或cl;
[0298]
与金属-卤素交换产物和钯催化剂在溶剂8中
[0299]
在一定温度下接触一定时间以足以进行c-c偶联;以及
[0300]
(3)使金属结合剂在溶剂8中
[0301]
在一定温度下接触一定时间以足以淬灭反应以产生所述式(xivc)的化合物或其立体异构体或盐。
[0302]
在方面18a范围内的方面19a中,本发明提供了一种制造式(xivd)的化合物或其盐
的方法;
[0303][0304]
其中:r8和r9独立地是n、ch或c(c
1-4
烷基);
[0305]r10
独立地是c
1-4
烷基或卤素;并且
[0306]r12
是c
1-4
烷基;
[0307]
所述方法包括(1)使式(i)的化合物,
[0308]
与为选自i-prmgcl、i-prmgcl*licl和i-prmgbr的格氏试剂或选自甲基锂、正丁基锂、异丙基锂、仲丁基锂、叔丁基锂和苯基锂的有机锂试剂的所述有机金属试剂在有或没有选自zncl2、znbr2和zni2的无机试剂的情况下在选自thf、2-methf、dmf、dma、dmpu、nmp、1,4-二噁烷及其溶剂混合物的溶剂8中
[0309]
在-5℃至25℃下接触一定时间以足以进行金属-卤素交换和有机锌形成;
[0310]
(2)使式(xiia)的化合物或盐:
[0311][0312]
与为有机金属试剂的所述金属-卤素交换产物和选自pdcl2(呫吨)、pd(dppf)cl2或pd(oac)2+brettphos、binap、dppf、dpephos和呫吨的钯催化剂在溶剂8中
[0313]
在-5℃至40℃下接触一定时间以足以进行c-c偶联;以及
[0314]
(3)使选自乙二胺四乙酸三钠或乙二胺四乙酸二钠的金属结合剂在溶剂8中
[0315]
在-5℃至25℃下接触一定时间以足以淬灭反应以产生所述式(xivd)的化合物或其盐。
[0316]
在方面18a范围内的另一个方面,本发明提供了一种制造式(xivd)的化合物或其盐的方法,
[0317]
所述方法包括(1)使式(i)的化合物,
[0318]
与为选自i-prmgcl(在thf中2.15m)、i-prmgcl*licl(在thf中1.3m)和i-prmgbr(在2-methf中2.9m)的格氏试剂或选自甲基锂、正丁基锂、异丙基锂、仲丁基锂、叔丁基锂和苯基锂的有机锂试剂的所述有机金属试剂在有或没有选自zncl2、znbr2和zni2的无机试剂的情况下在选自thf、2-methf、dmf、dma、dmpu、nmp、1,4-二噁烷及其溶剂混合物的溶剂8中
[0319]
在-5℃至25℃下接触一定时间以足以进行金属-卤素交换和有机锌形成;
[0320]
(2)使式(xiia)的化合物或盐:
[0321][0322]
与为有机金属试剂的所述金属-卤素交换产物和选自pdcl2(呫吨)、pd(dppf)cl2或pd(oac)2+brettphos、binap、dppf、dpephos和呫吨的钯催化剂在溶剂8中
[0323]
在-5℃至40℃下接触一定时间以足以进行c-c偶联;以及
[0324]
(3)使选自乙二胺四乙酸三钠或乙二胺四乙酸二钠的金属结合剂在溶剂8中
[0325]
在-5℃至25℃下接触一定时间以足以淬灭反应以产生所述式(xivd)的化合物或其盐。
[0326]
在方面20a中,本发明提供了一种制造作为游离酸或选自钾盐、四甲基铵盐、叔丁胺盐、二环己胺盐和氨基丁三醇盐的其盐分离的式(xive)的化合物的方法:
[0327][0328]
所述方法包括(1)使式(ia)的化合物:
[0329][0330]
与i-prmgcl(在thf中2.15m)在有或没有zncl2的情况下在thf中
[0331]
在-5℃至25℃下接触一定时间以足以进行金属-卤素交换和有机锌形成(如果存在zncl2的话);
[0332]
(2)使式(xiib)的化合物或其盐:
[0333][0334]
与i-prmgcl(在thf中2.15m)、有或没有zncl2、有机镁(或有机锌)试剂和pdcl2(呫
吨)在thf中
[0335]
在-5℃至40℃下接触》12小时以足以进行c-c偶联;以及
[0336]
(3)顺序地接触在thf溶剂混合物中的乙二胺四乙酸三钠或乙二胺四乙酸二钠与或不与过碳酸钠和亚硫酸氢钠(或焦亚硫酸钠),
[0337]
在-5℃至25℃下持续》1小时以足以淬灭反应以产生所述式(xive)的化合物,所述式(xive)的化合物然后作为游离酸或选自钾盐、四甲基铵盐、叔丁胺盐、二环己胺盐和氨基丁三醇盐的其盐分离。
[0338]
在方面20b中,本发明提供了一种制造作为游离酸或选自钾盐、四甲基铵盐、叔丁胺盐、二环己胺盐和氨基丁三醇盐的其盐分离的式(xive)的化合物的方法:
[0339][0340]
所述方法包括(1)使式(ia)的化合物:
[0341][0342]
与i-prmgcl(在thf中2.15m)在thf中
[0343]
在-5℃至25℃下接触一定时间以足以进行金属-卤素交换以及;
[0344]
(2)使式(xiib)的化合物或其盐:
[0345][0346]
与i-prmgcl(在thf中2.15m)、zncl2、有机镁试剂和pdcl2(呫吨)在thf中
[0347]
在-5℃至40℃下接触》12小时以足以进行c-c偶联;以及
[0348]
(3)顺序地接触在thf溶剂混合物中的乙二胺四乙酸三钠或乙二胺四乙酸二钠、过碳酸钠和亚硫酸氢钠(或焦亚硫酸钠),
[0349]
在-5℃至25℃下持续》1小时以足以淬灭反应以产生所述式(xive)的化合物,所述式(xive)的化合物然后作为游离酸或选自钾盐、四甲基铵盐、叔丁胺盐、二环己胺盐和氨基丁三醇盐的其盐分离。
[0350]
在另一个方面,本发明提供了一种制造作为游离酸或选自钾盐、四甲基铵盐、叔丁胺盐、二环己胺盐和氨基丁三醇盐的其盐分离的式(xive)的化合物的方法:
[0351][0352]
所述方法包括(1)使式(ia)的化合物:
[0353][0354]
与i-prmgcl(在thf中2.15m)和zncl2在thf中
[0355]
在-5℃至25℃下接触一定时间以足以进行金属-卤素交换和有机锌形成;
[0356]
(2)使式(xiib)的化合物或其盐:
[0357][0358]
与i-prmgcl(在thf中2.15m)、有机锌试剂和pdcl2(呫吨)在thf中
[0359]
在-5℃至40℃下接触》12小时以足以进行c-c偶联;以及
[0360]
(3)接触在thf溶剂混合物中的乙二胺四乙酸三钠
[0361]
在-5℃至25℃下持续1小时以足以淬灭反应以产生所述式(xive)的化合物,所述式(xive)的化合物然后作为游离酸或选自钾盐、四甲基铵盐、叔丁胺盐、二环己胺盐和氨基丁三醇盐的其盐分离。
[0362]
在方面21a中,本发明提供了一种制造式(xv)的化合物或其立体异构体或盐的方法:
[0363][0364]
其中:r8和r9独立地是n、ch或c(c
1-6
烷基);
[0365]r10
独立地是c
1-4
烷基或卤素;并且
[0366]r12
是c
1-4
烷基;
[0367]
所述方法包括使式(xivc)的化合物或其立体异构体或盐:
[0368][0369]
与过渡金属催化剂并且与或不与无机或有机酸在为极性质子或极性非质子溶剂或其溶剂混合物的溶剂9中
[0370]
在一定温度下接触一定时间以足以进行氢解以产生所述式(xv)的化合物或其立体异构体或盐。
[0371]
在方面21a范围内的方面22a中,本发明提供了一种制造式(xva)的化合物或其盐的方法;
[0372][0373]
其中:r8和r9独立地是n、ch或c(c
1-4
烷基);
[0374]r10
独立地是c
1-4
烷基或卤素;并且
[0375]r12
是c
1-4
烷基;
[0376]
所述方法包括使式(xivd)的化合物或其盐:
[0377][0378]
与选自5-20wt%pd/c的过渡金属催化剂在有或没有选自柠檬酸、草酸、h2so4的无机或有机酸的情况下在选自etoh、meoh、水、thf、dmac、nmp、ipa、t-amoh、methf、dmf、ch3cn、etoac、ipoac及其溶剂混合物的溶剂9中
[0379]
在20℃至60℃下接触一定时间以足以进行氢解以产生所述式(xva)的化合物或其盐。
[0380]
在方面21a范围内的另一个方面,本发明提供了一种制造式(xva)的化合物或其盐的方法,
[0381][0382]
其中:r8和r9独立地是n、ch或c(c
1-4
烷基);
[0383]r10
独立地是c
1-4
烷基或卤素;并且
[0384]r12
是c
1-4
烷基;
[0385]
所述方法包括使式(xivd)的化合物或其盐:
[0386][0387]
与选自5-20wt%pd/c的过渡金属催化剂在有或没有选自柠檬酸、草酸、h2so4的无机或有机酸的情况下在选自etoh、meoh、thf、dmac、nmp、ipa、t-amoh、methf、methf、dmf、ch3cn、etoac、ipoac及其溶剂混合物的溶剂9中
[0388]
在20℃至60℃下接触一定时间以足以进行氢解以产生所述式(xva)的化合物或其盐。
[0389]
在方面23a中,本发明提供了一种制造式(xvb)的化合物的方法:
[0390][0391]
所述方法包括使式(xive)的化合物或其盐:
[0392][0393]
与10wt%pd/c并且与或不与柠檬酸在选自etoh、meoh、水、thf、dmac、nmp及其溶剂混合物的溶剂9中
[0394]
在20℃至60℃下接触》12小时以足以进行氢解以产生所述式(xvb)的化合物。
[0395]
在另一个方面,本发明提供了一种制造式(xvb)的化合物的方法:
[0396][0397]
所述方法包括使式(xivf)的化合物:
[0398]
[0399]
与10wt%pd/c和柠檬酸在选自etoh、meoh、thf、dmac、nmp及其溶剂混合物的溶剂9中
[0400]
在20℃至60℃下接触》12小时以足以进行氢解以产生所述式(xvb)的化合物。
[0401]
在方面24a中,本发明提供了一种制造式(xvi)的化合物或其立体异构体或盐的方法:
[0402][0403]
其中:r8和r9独立地是n、ch或c(c
1-6
烷基);
[0404]r10
独立地是c
1-4
烷基或卤素;
[0405]r12
是c
1-4
烷基;并且
[0406]r14
和r
14a
独立地是c
1-6
烷基;
[0407]
所述方法包括使式(xv)的化合物或其立体异构体或盐:
[0408][0409]
与式(xvii)的化合物或其盐:
[0410][0411]
其中r
14
和r
14a
独立地是c
1-6
烷基;
[0412]
在金属醇盐的存在下在为极性质子或极性非质子溶剂或其溶剂混合物的溶剂9中
[0413]
在一定温度下接触一定时间以足以形成氨基甲酸酯以产生所述式(xvi)的化合物或其立体异构体或盐。
[0414]
在方面24a范围内的方面25a中,本发明提供了一种制造式(xvia)的化合物或其盐的方法;
[0415][0416]
其中:r8和r9独立地是n、ch或c(c
1-4
烷基);
[0417]r10
独立地是c
1-4
烷基或卤素;
[0418]r12
是c
1-4
烷基;并且
[0419]r14
和r
14a
独立地是c
1-4
烷基;
[0420]
所述方法包括使式(xva)的化合物或其盐:
[0421][0422]
与式(xvii)的化合物或其盐:
[0423][0424]
在选自kotbu(在thf中20wt%)或kotbu(在thf中1m)的金属醇盐的存在下在选自t-amoh、dmf、thf、ch3cn、mek、nmp、dmac、丙酮、mibk、2-methf及其溶剂混合物的溶剂9中
[0425]
在20℃-75℃下接触一定时间以足以形成氨基甲酸酯以产生所述式(xvia)的化合物或其盐。
[0426]
在方面26a中,本发明提供了一种制造式(xvib)的化合物或其盐的方法:
[0427][0428]
所述方法包括使式(xvb)的化合物或其盐:
[0429][0430]
与式(xviia)的化合物或其盐:
[0431][0432]
在kotbu(在thf中20wt%)的存在下在选自t-amoh、dmf、thf、ch3cn、mek及其溶剂混合物的溶剂9中
[0433]
在20℃-75℃下接触一定时间以足以形成氨基甲酸酯以产生所述式(xvib)的化合物或其盐。
[0434]
在方面26b中,本发明提供了一种制造式(xvib)的化合物或其盐的方法:
[0435][0436]
所述方法包括使式(xvb)的化合物或其盐:
[0437][0438]
与式(xviia)的化合物:
[0439][0440]
在kotbu(在thf中20wt%)的存在下在选自t-amoh、dmf、thf、ch3cn、mek及其溶剂混合物的溶剂9中
[0441]
在20℃-75℃下接触一定时间以足以形成氨基甲酸酯以产生所述式(xvib)的化合物或其盐。
[0442]
在方面27a中,本发明提供了式(ib)的化合物或其盐:
[0443][0444]
其中r1是c
1-6
烷基;并且r2是卤素。
[0445]
在方面28a中,本发明提供了式(ic)的化合物或其盐:
[0446][0447]
在方面29a中,本发明提供了式(iv)的化合物或其盐:
[0448][0449]
其中ra是-n(c
1-4
烷基)2。
[0450]
在方面30a中,本发明提供了式(iva)的化合物或其盐:
[0451][0452]
在方面31a中,本发明提供了式(ivb)的化合物或其盐:
[0453][0454]
在方面32a中,本发明提供了式(vii)的化合物或其立体异构体或盐:
[0455][0456]
其中ra是-n(c
1-4
烷基)2。
[0457]
在方面33a中,本发明提供了式(viia)的化合物或其盐:
[0458][0459]
在方面34a中,本发明提供了式(viib)的化合物或其盐:
[0460][0461]
在方面35a中,本发明提供了式(ix)的化合物或其立体异构体或盐:
[0462][0463]
其中ra是-n(c
1-4
烷基)2。
[0464]
在方面36a中,本发明提供了式(ixa)的化合物或其盐:
[0465][0466]
在方面37a中,本发明提供了其式(ixb)的化合物:
[0467][0468]
在方面38a中,本发明提供了式(xii)的化合物或其立体异构体或盐:
[0469][0470]
其中:r8和r9独立地是n、ch或c(c
1-6
烷基);
[0471]r10
独立地是c
1-4
烷基或卤素;并且
[0472]r11
独立地是br、cl或i。
[0473]
在方面38a范围内的方面39a中,本发明提供了式(xiia)的化合物或其盐;
[0474][0475]
其中:r8和r9独立地是n、ch或c(c
1-4
烷基);
[0476]r10
独立地是c
1-4
烷基或卤素;并且
[0477]r11
独立地是br、cl或i。
[0478]
在方面40a中,本发明提供了式(xiib)的化合物或其盐:
[0479][0480]
在方面41a中,本发明提供了式(xiv)的化合物或其立体异构体或盐:
[0481][0482]
其中:r8和r9独立地是n、ch或c(c
1-6
烷基);
[0483]r10
独立地是c
1-4
烷基或卤素;
[0484]r12
是c
1-4
烷基;并且
[0485]r13
独立地是h、c
1-4
烷基或苄基。
[0486]
在方面41a范围内的方面42a中,本发明提供了式(xiva)的化合物或其盐;
[0487][0488]
其中:r8和r9独立地是n、ch或c(c
1-4
烷基);
[0489]r10
独立地是c
1-4
烷基或卤素;
[0490]r12
是c
1-4
烷基;并且
[0491]r13
独立地是h、c
1-4
烷基或苄基。
[0492]
在方面43a中,本发明提供了式(xivb)的化合物或其盐:
[0493][0494]
其中:r
13
独立地是h或苄基。
[0495]
在方面44a中,本发明提供了式(xviia)的化合物:
[0496][0497]
在方面45a中,本发明提供了一种制造式(xvii)的化合物或其盐的方法:
[0498][0499]
其中r
14
和r
14a
独立地是c
1-6
烷基;
[0500]
所述方法包括使式(xviii)的化合物:
[0501][0502]
与nhr
14r14a
在为极性质子、非质子或非极性溶剂或其溶剂混合物的溶剂10的存在下
[0503]
在-5℃至25℃下接触一定时间以足以形成甲酰胺以产生所述式(xvii)的化合物。
[0504]
在方面46a中,本发明提供了一种制造式(xviia)的化合物的方法:
[0505][0506]
所述方法包括使式(xviii)的化合物:
[0507]
[0508]
与n-甲基丙胺在选自dcm、t-amoh、水、mtbe、乙腈、thf、methf、丙酮、mek、mibk、meoac、etoac、ipac、dmf、nmp、dmac及其溶剂混合物的溶剂10的存在下
[0509]
在-5℃至25℃下接触至少1小时以足以形成甲酰胺;
[0510]
接着添加(ho2c)2以产生所述式(xviia)的化合物。
[0511]
在方面47a中,本发明提供了一种制造式(xviia)的化合物的方法;
[0512]
所述方法包括使式(xviii)的化合物;
[0513]
与n-甲基丙胺在选自dcm、t-amoh、水、mtbe、乙腈及其溶剂混合物的溶剂10的存在下
[0514]
在-5℃至25℃下接触至少1小时以足以形成甲酰胺;
[0515]
接着添加(ho2c)2以产生所述式(xviia)的化合物。
[0516]
在方面48a中,本发明提供了一种制造式(xvi)的化合物或其立体异构体或盐的方法,
[0517]
所述方法包括方面21a的步骤;
[0518]
然后是方面24a的步骤;
[0519]
其中所有式和变量如在方面21a和24a中所定义。
[0520]
在方面49a中,本发明提供了一种制造式(xvia)的化合物或其立体异构体或盐的方法,
[0521]
所述方法包括方面22a的步骤;
[0522]
然后是方面25a的步骤;
[0523]
其中所有式和变量如在方面22a和25a中所定义。
[0524]
在方面50a中,本发明提供了一种制造式(xvib)的化合物或其立体异构体或盐的方法,
[0525]
所述方法包括方面23a的步骤;
[0526]
然后是方面26a的步骤;
[0527]
其中所有式和变量如在方面23a和26a中所定义。
[0528]
在方面51a中,本发明提供了一种制造式(xvi)的化合物或其立体异构体或盐的方法,
[0529]
所述方法包括方面18a的步骤(1)、(2)和(3);
[0530]
方面21a的步骤;
[0531]
然后是方面24a的步骤;
[0532]
其中所有式和变量如在方面18a、21a和24a中所定义。
[0533]
在方面52a中,本发明提供了一种制造式(xvia)的化合物或其立体异构体或盐的方法,
[0534]
所述方法包括方面19a的步骤(1)、(2)和(3);
[0535]
方面22a的步骤;
[0536]
然后是方面25a的步骤;
[0537]
其中所有式和变量如在方面19a、22a和25a中所定义。
[0538]
在方面53a中,本发明提供了一种制造式(xvib)的化合物或其立体异构体或盐的方法,
[0539]
所述方法包括方面20a的步骤(1)、(2)和(3);
[0540]
方面23a的步骤;
[0541]
然后是方面26a的步骤;
[0542]
其中所有式和变量如在方面20a、23a和26a中所定义。
[0543]
在方面54a中,本发明提供了一种制造式(xvi)的化合物或其立体异构体或盐的方法,
[0544]
所述方法包括方面15a的步骤;
[0545]
方面18a的步骤(1)、(2)和(3);
[0546]
方面21a的步骤;
[0547]
然后是方面24a的步骤;
[0548]
其中所有式和变量如在方面15a、18a、21a和24a中所定义。
[0549]
在方面55a中,本发明提供了一种制造式(xvia)的化合物或其立体异构体或盐的方法,
[0550]
所述方法包括方面16a的步骤;
[0551]
方面19a的步骤(1)、(2)和(3);
[0552]
方面22a的步骤;
[0553]
然后是方面25a的步骤;
[0554]
其中所有式和变量如在方面16a、19a、22a和25a中所定义。
[0555]
在方面56a中,本发明提供了一种制造式(xvib)的化合物或其立体异构体或盐的方法,
[0556]
所述方法包括方面17a的步骤;
[0557]
方面20a的步骤(1)、(2)和(3);
[0558]
方面23a的步骤;
[0559]
然后是方面26a或26b的步骤;
[0560]
其中所有式和变量如在方面17a、20a、23a和26a或26b中所定义。
[0561]
在另一个方面,本发明提供了一种制造式(xvib)的化合物或其立体异构体或盐的方法,
[0562]
所述方法包括方面17a的步骤;
[0563]
方面20a的步骤(1)、(2)和(3);
[0564]
方面23a的步骤;
[0565]
然后是方面26a的步骤;
[0566]
其中所有式和变量如在方面17a、20a、23a和26a中所定义。
[0567]
在另一个方面,本发明提供了一种制造式(xvib)的化合物或其立体异构体或盐的方法,
[0568]
所述方法包括方面17a的步骤;
[0569]
方面20a的步骤(1)、(2)和(3);
[0570]
方面23a的步骤;
[0571]
然后是方面26b的步骤;
[0572]
其中所有式和变量如在方面17a、20a、23a和26b中所定义。
[0573]
定义
[0574]
反应杂质和/或加工杂质的存在可以通过本领域已知的分析技术来确定,所述分析技术例如色谱法、核磁共振波谱法、质谱法和/或红外光谱法。
[0575]
其他实施方案包括在具体实施方式和/或权利要求中所述的那些。
[0576]
为了便于理解本文阐述的公开内容,下面定义了许多附加术语。通常,本文使用的命名法和本文所述的有机化学、药物化学和药理学的实验室程序是本领域熟知和常用的那些。除非另外定义,否则本文使用的所有技术和科学术语通常具有与本公开文本所属领域的普通技术人员通常所理解的相同的含义。
[0577]
如本文所用的关于配制品、组合物或成分的术语“可接受的”意指对所治疗的受试者的总体健康没有持久的有害作用。
[0578]“api”是指活性药物成分。
[0579]
术语“卤基”是指氟(f)、氯(cl)、溴(br)或碘(i)。
[0580]
术语“烷基”是指可以是直链或支链的含有指示数量碳原子的烃链。例如,c
1-10
表示基团中可以在其中具有从1至10(包括)个的碳原子。非限制性例子包括甲基、乙基、异丙基、叔丁基、正己基。
[0581]
术语“卤代烷基”是指其中一个或多个氢原子被独立选择的卤基替代的烷基。
[0582]
术语“烷氧基”是指-o-烷基(例如,-och3)。
[0583]
术语“卤代烷氧基”是指-o-卤代烷基(例如,-ocf3)。
[0584]
术语“亚烷基”是指支化或未支化的二价烷基(例如,-ch
2-)。
[0585]
术语“芳基”是指6碳单环、10碳双环或14碳三环芳族环体系,其中每个环的0、1、2、3或4个原子可以被取代基取代,并且其中包含单环基团的环是芳族的,并且其中至少一个包含双环或三环基团的稠环是芳族的,例如四氢萘基。芳基的例子还包括苯基、萘基等。
[0586]
如本文所用,术语“环烷基”包括具有3至10个碳、优选3至8个碳、并且更优选3至6个碳的饱和环状烃基,其中环烷基可以任选地被取代。优选的环烷基包括但不限于环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环庚基和环辛基。如本文所用,术语“亚环烷基”是指二价环烷基。
[0587]
术语“杂芳基”是指具有1-3个杂原子(如果是单环的)、1-6个杂原子(如果是双环的)或1-9个杂原子(如果是三环的)的芳族5-8元单环、8-12元双环或11-14元三环体系,所述杂原子选自o、n或s(例如,碳原子和1-3、1-6或1-9个n、o或s杂原子(如果分别是单环、双环或三环的话)),其中每个环的0、1、2、3或4个原子可以被取代基取代,并且其中包含单环基团的环是芳族的,并且其中至少一个包含双环或三环基团的稠环是芳族的(但不必是含有杂原子的环,例如四氢异喹啉基。杂芳基的例子还包括吡啶基、呋喃基(furyl)或呋喃基(furanyl)、咪唑基、苯并咪唑基、嘧啶基、噻吩基(thiophenyl)或噻吩基(thienyl)、喹啉基、吲哚基、噻唑基等。
[0588]
术语“杂环基”是指具有1-3个杂原子(如果是单环的)、1-6个杂原子(如果是双环的)或1-9个杂原子(如果是三环的)的非芳族5-8元单环、8-12元双环或11-14元三环体系,所述杂原子选自o、n或s(例如,碳原子以及1-3、1-6或1-9个n、o或s的杂原子(如果分别是单环、双环或三环的话),其中每个环的0、1、2或3个原子可以被取代基取代。杂环基的例子包括哌嗪基、吡咯烷基、二噁烷基、吗啉基、四氢呋喃基等。术语“杂环亚烷基”是指二价杂环
基。
[0589]
术语“布朗斯台德酸”是指质子(h
+
)供体。
[0590]
术语“路易斯酸”是指可以接受来自电子供体化合物的电子对的化学物质。
[0591]
术语“布朗斯台德碱”是指质子(h
+
)受体。
[0592]
术语“路易斯碱”是指可以向电子受体化合物提供电子对的化学物质。
[0593]
术语“过渡金属催化剂”是指一种配位络合物,其具有在两个壳层而不是只有一个壳层中具有价电子的多种金属元素(诸如钯和镍)中的任一种,并且当添加到化学反应中时提高反应速率。
[0594]
质子溶剂是指具有与氧(诸如在羟基中)或氮(诸如在胺基中)结合的氢原子的溶剂。
[0595]
非质子溶剂是指不是氢键供体的溶剂。
[0596]
极性溶剂是指偶极矩大或带部分电荷的溶剂;它们含有在电负性非常不同的原子之间的键,诸如氧和氢。
[0597]
溶剂混合物是指两种或多种溶剂的组合。
[0598]
另外,构成本实施方案的化合物的原子旨在包括此类原子的所有同位素形式。如本文所用,同位素包括具有相同原子序数但不同质量数的那些原子。作为一般性例子而非限制,氢的同位素包括氚和氘,并且碳的同位素包括
13
c和
14
c。
[0599]
实施例
[0600]
提出以下实施例以向本领域普通技术人员提供如何制造和使用本发明的完整公开和描述,并且不旨在限制诸位发明人视为其发明的范围,它们也不旨在代表下面的实验被执行或它们都是可以执行的实验。应当理解,以现在时态书写的示例性描述不一定被执行,而是可以执行所述描述以生成其中描述的性质的数据等。已经努力确保关于所使用的数字(例如,量、温度等)的准确性,但是应当考虑一些实验误差和偏差。
[0601]
本发明合成序列中使用的起始材料是已知的、通过已知方法制造的、或可商购的。技术人员还将认识到,本文所述的条件和试剂可以与本领域公认的等效物互换。例如,在一个反应中,盐酸可以与诸如氢溴酸、硫酸等其他酸互换。
[0602]
技术人员将认识到可以用于表征本文所述化合物的多种分析方法,包括例如1h核磁共振波谱法(nmr)、异核nmr、质谱法(ms)、液相色谱法(lc)、和红外(ir)光谱法。前述列表是技术人员可用的表征方法的子集并且不旨在是限制的。
[0603]
为了进一步说明上述内容,包括以下非限制性示例性合成方案。在权利要求范围内的这些实施例的变化在本领域技术人员的能力范围内,并且被认为落入如本文所述和本文要求保护的发明的范围内。读者应认识到,被提供本公开文本的和本领域的技术人员能够在没有穷举实施例的情况下制备和使用本发明。
[0604]
以下缩写具有指示的含义:
[0605]
acoh=乙酸
[0606]
t-amoh=叔戊醇
[0607]
aq=水性
[0608]
binap=(2,2'-双(二苯基膦基)-1,1'-联萘)
[0609]
bomcl=苄基氯甲基醚
[0610]
brettphos=2-(二环己基膦基)3,6-二甲氧基-2',4',6'-三异丙基-1,1'-联苯
[0611]
n-bubr=丁基溴
[0612]
n-buli=丁基锂
[0613]
cdcl3=氘化氯仿
[0614]
ch3cn=乙腈
[0615]
ch3i=甲基碘
[0616]
cdi=羰基二咪唑
[0617]
co2=二氧化碳
[0618]
cpme=环戊基甲基醚
[0619]
d=双峰
[0620]
dcm=二氯甲烷
[0621]
dmac=二甲基乙酰胺
[0622]
4-dmap=4-二甲氨基吡啶
[0623]
dme=1,2-二甲氧基乙烷
[0624]
dmf=二甲基甲酰胺
[0625]
dmpu=n,n'-二甲基丙烯脲
[0626]
dmso=二甲基亚砜
[0627]
dmso-d6=氘化二甲基亚砜
[0628]
dpephos=双[(2-二苯基膦基)苯基]醚
[0629]
dppf=1,1'-二茂铁二基-双(二苯基膦)
[0630]
dppp=1,3-双(二苯基膦基)丙烷
[0631]
equiv=当量
[0632]
ered=烯还原酶
[0633]
esi=电喷雾电离
[0634]
et3n=三甲胺
[0635]
(et)4nbr或et4nbr=四乙基溴化铵
[0636]
etoac=乙酸乙酯
[0637]
etoh=乙醇
[0638]
g=克
[0639]
gdh=葡萄糖脱氢酶
[0640]
h=小时
[0641]
h2=二氢
[0642]
hcl=氯化氢(通常作为溶液)
[0643]
hbr=溴化氢(通常作为溶液)
[0644]
n-hexli=己基锂
[0645]
hio3=碘酸
[0646]
hio4=高碘酸
[0647]
hmpa=六甲基磷酰胺
[0648]
h2o=水
[0649]
h3po4=磷酸
[0650]
h2so4=硫酸
[0651]
i2=碘
[0652]
ipa=异丙醇
[0653]
ipac=乙酸异丙酯
[0654]
ircl4=四氯化铱
[0655]
ircl4*水合物=四氯化铱水合物
[0656]
[ir(cod)cl]2=双(1,5-环辛二烯)二铱(i)二氯化物
[0657]
iprmgcl=异丙基氯化镁
[0658]
iprmgcl*licl=异丙基氯化镁氯化锂络合物
[0659]
iprmgbr=异丙基溴化镁
[0660]
kcl=氯化钾
[0661]
k2co3=碳酸钾
[0662]
khco3=碳酸氢钾
[0663]
khmds=双(三甲基甲硅烷基)氨基钾
[0664]
kotbu=叔丁醇钾
[0665]
koh=氢氧化钾
[0666]
kg=千克
[0667]
kred=酮还原酶
[0668]
l=升
[0669]
lcap=液相色谱面积百分比
[0670]
lc/ms=液相色谱质谱仪
[0671]
licl=氯化锂
[0672]
liotbu=叔丁醇锂
[0673]
lioh=氢氧化锂
[0674]
lioh*h2o=氢氧化锂水合物
[0675]
liome=甲醇锂
[0676]
lrms=低分辨率质谱法
[0677]
m=多重峰
[0678]
m=摩尔
[0679]
mg=毫克
[0680]
mek=甲基乙基酮
[0681]
meoh或ch3oh=甲醇
[0682]
methf=2-甲基四氢呋喃
[0683]
2-methf或ch3thf=2-甲基四氢呋喃
[0684]
mgcl2=氯化镁
[0685]
mgclbr=氯化溴化镁
[0686]
mhz=兆赫兹
[0687]
mibk=甲基异丁基酮
[0688]
min=分钟
[0689]
ml=毫升
[0690]
mmol=毫摩尔
[0691]
mtbe=甲基叔丁基醚
[0692]
nacl=氯化钠
[0693]
na2co3=碳酸钠
[0694]
nadph=烟酰胺腺嘌呤二核苷酸磷酸氢
[0695]
na3edta*xh2o=乙二胺四乙酸三钠盐水合物
[0696]
na2edta*2h2o=乙二胺四乙酸二钠盐二水合物
[0697]
nahmds=双(三甲基甲硅烷基)氨基钾
[0698]
naotbu=叔丁醇钠
[0699]
naoh=氢氧化钠
[0700]
naome=甲醇钠
[0701]
nh2c(ch3)3=叔丁胺
[0702]
nh4oh或nh3h2o=氢氧化铵
[0703]
nh4oac=乙酸铵
[0704]
nmp=n-甲基-2-吡咯烷酮
[0705]
pd/c=钯碳
[0706]
pdcl2(呫吨)=二氯[9,9-二甲基-4,5-双(二苯基膦基)-呫吨]钯(ii)
[0707]
pd(dppf)cl2=[1,1'-双(二苯基膦基)二茂铁]二氯钯(ii)
[0708]
pd(oac)2=乙酸钯(ii)
[0709]
phli=苯基锂
[0710]
p(oh)3=亚磷酸
[0711]
ppm=百万分率
[0712]
ptfe=聚四氟乙烯
[0713]
rt=保留时间
[0714]
s=单峰
[0715]
t=三重峰
[0716]
t-buoh=叔丁醇
[0717]
thf=四氢呋喃
[0718]
tfa=三氟乙酸
[0719]
℃=摄氏度
[0720]
uplc/ms=超高效液相色谱质谱仪
[0721]
vol=体积
[0722]
wt=重量
[0723]
呫吨=[5-(二苯基膦基)-9,9-二甲基-9h-呫吨-4-基](二苯基)-膦
[0724]
znbr2=溴化锌
[0725]
znclbr=氯化溴化锌
[0726]
zncl2=氯化锌
[0727]
zni2=碘化锌
[0728]
已经描述了本发明的许多实施方案。然而,应该理解,在不脱离本发明的精神和范围的情况下,可以进行各种修改。因此,其他实施方案也在以下权利要求书的范围内。
[0729]
实验方法
[0730]
实施例1:4,5-二溴-1-甲基-1h-1,2,3-三唑的制备
[0731][0732]
向在氮气保护下的10l反应器中加入t-amoh(2.0l,4.0ml/g)并且将温度调节至20℃。向反应混合物中顺序地加入4,5-二溴-1h-1,2,3-三唑(500g,1.0当量,限制试剂)和thf(175g,1.1当量),然后向反应器中分批加入liotbu(195g,1.1当量),维持内部温度《40℃。将反应混合物温热至40℃并且搅拌1小时;并且然后加入ch3i(470g,1.5当量),并且在40℃下继续老化24h。将反应混合物温热至50℃并且在真空(80托)下浓缩至3.0ml/g,同时维持内部温度《60℃;并且然后加入ipa(1.0l,2.0ml/g)和水(4.0l,8.0ml/g)。将反应混合物温热至60℃并且老化2h。然后将所得均质溶液经5h冷却至0℃并且老化过夜。将所得浆料过滤,顺序地用预冷至0℃-5℃的ipa:h2o(35:65)(1.0l,2ml/g)、然后用水(1.5l,3ml/g)在环境温度下洗涤,并且在真空下在50℃下干燥以得到430g(82%-81%产率)呈白色固体的4,5-二溴-1-甲基-1h-1,2,3-三唑。
[0733]1h nmr(400mhz,cdcl3)δ4.10(s,3h)。
13
c nmr:(100mhz,cdcl3)δ122.70,112.98,36.89。
[0734]
lc/ms(esi)计算值[c3h3br2n3+h]
+
=239.88,实测值239.88。
[0735]
uhplc方法条件:柱:phenomenex kinetex c8,1.7μm,2.1x50mm;流动相a:在乙腈中的0.05%tfa:水(5:95);流动相b:在乙腈中的0.05%tfa:水(95:5);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;220nm;uplc rt 1.02min。
[0736]
实施例2:5-((苄氧基)甲基)-4-溴-1-甲基-1h-1,2,3-三唑溴化物盐的制备
[0737]
[0738]
在20℃下向在氮气保护下的10l反应器中加入thf(4.0l,4.0ml/g)和4,5-二溴-1-甲基-1h-1,2,3-三唑(1.00kg,1.0当量,限制试剂)。将溶液冷却至-25℃并且逐滴加入n-buli(在正己烷溶液中2.5m,1.26kg,1.1当量),维持内部温度《-20℃。另外20min后,向浓稠悬浮液中加入bomci(0.75kg,1.15当量),维持内部温度《-20℃。2h后,在-20℃下向反应混合物中加入在acoh溶液中的33%hbr(0.10kg,0.10当量)并且搅拌30min。然后将反应混合物温热至15℃并且在真空(50托)下浓缩至2.0-2.5ml/g,维持内部温度《40℃;并且然后将ch3cn(5.0l,5ml/g)加入浓缩溶液并且继续将批料蒸馏至2.0-2.5ml/g,维持内部温度《40℃。向反应混合物中加入ch3cn(3.0l,3.0ml/g),将悬浮液温热至20℃并且老化1h。将反应浆料过滤并且将废固体用ipac(2x2.0l,2x2.0ml/g)洗涤。然后将合并的滤液调节至20℃并且经至少1h逐滴加入在acoh溶液中的33%hbr(1.07kg,1.05当量)。另外1h后,将所得浆料过滤,顺序地用ipac:ch3cn(1:1)(3.0l,3ml/g)、然后用ipac(3.0l,3ml/g)洗涤,并且在真空下在40℃下干燥以得到1.27g(83%产率)呈白色固体的5-((苄氧基)甲基)-4-溴-1-甲基-1h-1,2,3-三唑溴化物盐。
[0739]1h nmr:(500mhz,dmso-d6)δ7.29-7.37(m,5h),6.35(br s,1h),4.61(s,2h),4.51(s,2h),4.04(s,3h)。
13
c nmr:(100mhz,dmso-d6)δ137.48,132.61,128.42,127.86(2c’s),121.01,71.86,58.62,36.02.lc/ms(esi)计算值[c
11h12
brn3o+h]
+
=282.02,实测值282.02。
[0740]
uhplc方法条件:柱:phenomenex kinetex c8,1.7μm,2.1x50mm;流动相a:在乙腈中的0.05%tfa:水(5:95);流动相b:在乙腈中的0.05%tfa:水(95:5);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;220nm;uplc rt 1.32min。
[0741]
实施例3:2-((4-(二甲基氨基)苯甲酰基)氧基)丙二酸二甲酯的制备
[0742][0743]
向在氮气保护下的5l反应器中加入dmf(900ml,3.0ml/g)并且将温度调节至20℃。顺序地加入4-二甲基氨基苯甲酸(299.4g,1.05当量,限制)、碳酸氢钾(203.5g,1.20当量)和et4nbr(35.6g,0.10当量),然后另外加入作为冲洗液的dmf(900ml,3.0l/kg)。然后将反应混合物温热至30℃并且加入氯丙二酸二甲酯(300.0g,94.0wt%,1.69摩尔,限制试剂),并且继续在30℃下老化过夜。将反应混合物冷却至15℃-20℃并且经2h缓慢加入水(2.7l,9.0ml/g)。另外4h后,将所得浆料过滤,顺序地用dmf:h2o(1:1.5)(750ml,2.5ml/g)、然后用2-丙醇(2x750ml,2x2.5ml/g)洗涤,并且在真空下在45℃-50℃下干燥以得到473g(95%产率)呈白色固体的2-((4-(二甲基氨基)苯甲酰基)氧基)丙二酸二甲酯。1h nmr(400mhz,dmso):δ7.81(d,j=9.1hz,2h),6.76(d,j=9.1hz,2h),5.80(s,1h),3.78(s,6h),3.02(s,6h)。
13
c nmr(100mhz,dmso):δ165.17,164.36,153.82,131.29,113.48,110.88,71.50,53.04,39.53。实测值:295.11g/mol。lc/ms(esi)计算值[c
14h17
no6+h]
+
=296.11,实测值
296.11。
[0744]
uhplc方法条件:柱:supelco ascentis express c18,2.7μm,2.1x50mm;流动相a:在meoh中的0.05%tfa:水(20:80);流动相b:在meoh中的0.05%tfa:乙腈(20:80);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;229nm;uplc rt 1.25min。
[0745]
实施例4:(s)-2-((4-(二甲基氨基)苯甲酰基)氧基)-2-(3-氧代环己基)丙二酸二甲酯的制备
[0746][0747]
在20℃下向在氮气保护下的2l反应器中加入甲苯(100ml,1.0ml/g)和1-((1s,2s)-2-氨基环己基)-3-(3,5-双(三氟甲基)苯基)硫脲盐酸盐(7.07g,0.05当量)。加入1n koh水溶液(50ml,0.5ml/g)并且搅动1h。然后去除下部水层,并且向剩余的上部甲苯层中顺序地加入2-((4-(二甲基氨基)苯甲酰基)氧基)丙二酸二甲酯(100.0g,335.3mmol,1.0当量,限制试剂)、4-吡咯烷基吡啶(2.54g,0.05当量)和另外的甲苯(300ml,3.0ml/g)。
[0748]
将反应混合物温热至33℃以产生均质溶液并且然后一次性加入2-环己烯-1-酮(39.42g,1.2当量)。48h后,向所得浆料中一次性加入2-丙醇(63ml,0.63ml/g),然后经3h缓慢加入庚烷(800ml,8.0ml/g)。将浆料冷却至20℃,并且在过夜老化后,过滤,顺序地用在2:1庚烷:甲苯中的5%ipa(300ml,3.0ml/g)、然后用庚烷(300ml,3.0ml/g)洗涤,并且在真空下在45℃-50℃下干燥以得到114.7g(87%-88%产率)呈白色固体的(s)-2-((4-(二甲基氨基)苯甲酰基)氧基)-2-(3-氧代环己基)丙二酸二甲酯。
[0749]1h nmr(400mhz,dmso-d6)δ:7.81(d,j=8.8hz,2h),6.75(d,j=9.1hz,2h),3.72(s,3h),3.72(s,3h)3.02(s,6h),2.72-2.58(m,1h),2.54-2.34(m,3h),2.28-2.16(m,2h),2.08-1.95(m,1h),1.93-1.80(m,1h),1.69-1.52(m,1h)。注:在3.72处的单峰对应于甲酯峰。虽然理论上它们不应该是非对映异位的并且因此应该是磁性等效的并且产生6h单峰,但由于缓慢的旋转,它们实际上表现为两个重叠的3h单峰,并且因此3.72峰的重复不是排
字错误。
13
c nmr:(100mhz,dmso-d6)δ:208.29,165.92,164.47,153.74,131.29,114.11,110.85,82.94,52.77,43.19,41.95,40.18,25.37,23.41。(未观察到一个碳信号。)lc/ms(esi)计算值[c
20h25
no7+h]
+
=392.17,实测值392.17。
[0750]
uhplc方法条件:柱:supelco ascentis express c18,2.7μm,2.1x50mm;流动相a:在meoh中的0.05%tfa:水(20:80);流动相b:在meoh中的0.05%tfa:乙腈(20:80);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;229nm;uplc rt 1.28min
[0751]
手性hplc方法条件:柱:phenomenex lux cellulose-2,3μm,4.6x150mm;流动相a:在meoh中的0.05%tfa:水(20:80);流动相b:在meoh中的0.05%tfa:乙腈(20:80);温度:30℃;梯度:0min(20%b),2.0min(20%b),5.0min(55%b),12.0min(63%b),18.0min(100%b),21.0min(100%b);流量:1.2ml/min;229nm;hplc rt希望的对映异构体11.89min;hplc rt不希望的对映异构体9.94min。
[0752]
实施例5:2-((4-(二甲基氨基)苯甲酰基)氧基)-2-((1s,3s)-3羟基环己基)丙二酸二甲酯的制备
[0753][0754]
在20℃下向在氮气保护下的5l反应器中按以下顺序加入(s)-2-((4-(二甲基氨基)苯甲酰基)氧基)-2-(3-氧代环己基)丙二酸二甲酯(364g,930mmol,1.0当量,限制试剂)、亚磷酸(114.4g,1.50当量)、2-丙醇(730ml,2.0ml/g)、h2o(1100ml,3ml/g)和[ir(cod)cl]2(3.27g,0.005当量)。然后将反应混合物温热至温和回流(约80℃内部温度)并且观察到混合物在温热后变成均质的。24h后,将反应混合物冷却至20℃以产生悬浮液。然后向反应混合物中顺序地一次性加入h2o(182ml,0.5ml/g),老化5h,并且经2h加入h2o(1638ml,4.5ml/g)。然后将所得浆料老化过夜,过滤,并且顺序地用1:4 2-丙醇:h2o(1100ml,3.0ml/g)并且然后用h2o(1100ml,3.0ml)洗涤。将湿饼在真空下在50℃下在n2吹扫下干燥以得到333g(91%校正分离产率)呈白色固体的2-((4-(二甲基氨基)苯甲酰基)氧基)-2-((1s,3s)-3羟基环己基)丙二酸二甲酯。
[0755]
可替代地,在20℃下向在氮气保护下的4l反应器中按以下顺序加入2-丙醇(200ml,2.0ml/g)、(s)-2-((4-(二甲基氨基)苯甲酰基)氧基)-2-(3-氧代环己基)丙二酸二甲酯(100g,255.5mmol,1.0当量,限制试剂)、亚磷酸(33g,1.50当量)、ircl4·
xh2o(0.89g,0.01当量)、2-丙醇(200ml,2.0ml/g)和h2o(50ml,0.5ml/g)。反应混合物在20℃下不是均质的。然后将反应混合物温热至温和回流(80℃-82℃内部温度)并且观察到混合物在温热期间变成均质的。48h后,将反应混合物冷却至20℃。向反应器中经2h加入h2o(350ml,3.5ml/g)。在此添加后,在20℃下加入2-((4-(二甲基氨基)苯甲酰基)氧基)-2-((1s,3s)-3羟基环己基)丙二酸二甲酯晶种(100mg,0.01当量)并且将混合物老化过夜。第二天,经6h加入h2o
(1200ml,12.0ml/g)。将所得浆料老化过夜。第二天,将浆料过滤并且顺序地用1:4 2-丙醇:h2o(300ml,3.0ml/g)并且然后用h2o(300ml,3.0ml)洗涤。将湿饼在真空下在50℃下在n2吹扫下干燥以得到90g(82%校正分离产率)呈固体的2-((4-(二甲基氨基)苯甲酰基)氧基)-2-((1s,3s)-3羟基环己基)丙二酸二甲酯。
[0756]1h nmr(400mhz,dmso-d6)δ7.79(d,j=9.1hz,2h),6.75(d,j=9.1hz,2h),4.03-3.96(m,1h),3.67(s,6h),3.01(s,6h),2.66-2.54(m,1h),1.74-1.56(m,4h),1.54-1.43(m,1h),1.43-1.11(m,3h)。
13
c nmr(100mhz,dmso-d6):166.32,164.64,153.63,131.14,114.61,110.82,84.06,63.79,52.40,52.39,38.28,33.62,31.75,26.78,19.52.lc/ms(esi)计算值[c
20h27
no7+h]
+
=394.19,实测值394.19。
[0757]
uhplc方法条件:柱:supelco ascentis express c18,2.7μm,2.1x50mm;流动相a:在meoh中的0.05%tfa:水(20:80);流动相b:在meoh中的0.05%tfa:乙腈(20:80);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;229nm;uplc rt 1.24min
[0758]
手性hplc方法条件:柱:phenomenex lux cellulose-2,3μm,4.6x150mm;流动相a:在meoh中的0.05%tfa:水(20:80);流动相b:在meoh中的0.05%tfa:乙腈(20:80);温度:30℃;梯度:0min(50%b),2.0min(50%b),11.0min(80%b),12.0min(100%b),15.0min(100%b);流量:1.2ml/min;229nm;hplc rt希望的反式对映异构体10.8min;hplc rt不希望的反式对映异构体5.9min;hplc rt不希望的主要顺式对映异构体5.0min;hplc rt不希望的次要顺式对映异构体9.8min。
[0759]
实施例6:(1s,3s)-3-羟基环己烷-1-甲酸的制备
[0760][0761]
向在氮气保护下在20℃下的5l夹套反应器中加入2-丙醇(300ml,1ml/g)、水(450ml,1.5l/kg、2-((4-(二甲基氨基)苯甲酰基)氧基)-2-((1s,3s)-3羟基环己基)丙二酸二甲酯(300g,1.00当量,限制试剂)和作为冲洗液的2-丙醇(60ml,0.2l/kg)。
[0762]
然后向所得悬浮液中加入10n naoh水溶液(460ml,6.0当量)和作为冲洗液的水
(300ml,1.0l/kg)。然后将反应混合物加热至80℃-85℃并且老化至少16h。然后将反应混合物冷却至5℃-15℃并且经至少2h加入6m盐酸水溶液(620ml,4.88当量),维持内部温度《15℃。另外4h后,将反应混合物过滤并且用水(2x300ml,2x1ml/g)洗涤。然后将合并的滤液转移到新的反应器中,并且用mtbe(1500ml,5ml/g)洗涤。然后将下部富含产物的水层转移到新反应器中,用水(150ml,0.5ml/g)冲洗,然后加入2-丙醇(300ml,1ml/g)。然后经至少2h向反应流中加入h5io6(435g,2.50当量)在水(300ml,1ml/g)中的溶液,维持内部温度《22℃。另外17h后,加入氯化钾(600g,2g/g),并且另外1.5h后,将混合物过滤并且将固体用20wt%氯化钾水溶液(2x300ml,2x1ml/g)洗涤。然后将合并的滤液依次用2-甲基四氢呋喃(2
×
1200ml,2x4ml/g)洗涤两次。然后向合并的有机层中加入20wt%氯化钠水溶液(1200ml,4ml/g)和半胱氨酸盐酸盐(120g,0.40g/g)。混合至少0.5h后,将各层分开,并且将上部有机层用20wt%氯化钠水溶液(1200ml,4ml/g)洗涤。
[0763]
然后将富含产物的有机层在真空中浓缩(内部温度《40℃),加入2-甲基四氢呋喃(900ml,3ml/g),再浓缩并且然后加入2-甲基四氢呋喃(900ml,3ml/g)。然后将溶液浓缩至1g/ml,接着用另外的2-甲基四氢呋喃(3x300ml,3x1ml/g)进行放取(put-take)蒸馏。然后将所得约1ml/g溶液精过滤并且加入庚烷:甲苯混合物(3:1,300ml,1g/ml)。然后将所得浆料在15℃-25℃下老化至少2h,接着经至少1h添加庚烷:甲苯(3:1,900ml,3g/ml)。老化过夜后,将浆料冷却至0℃,老化至少3h,过滤,用甲苯(300ml,1g/ml)洗涤并且在真空烘箱中在50℃下在n2吹扫下干燥过夜以得到72.2g呈灰白色固体的(1s,3s)-3-羟基环己烷-1-甲酸(66%“原样”产率)。
[0764]
可替代地,向在氮气保护下在20℃下的1l夹套反应器中加入10m氢氧化钠(200ml,2ml/g,约8当量),接着加入水(300ml,3ml/g)。然后加入2-((4-(二甲基氨基)苯甲酰基)氧基)-2-((1s,3s)-3羟基环己基)丙二酸二甲酯(91.12g,95.13%qnmr效力,1.00当量,限制试剂),接着加入2-丙醇(100ml,1ml/g)。将悬浮液加热至回流(内部温度约82℃)并且老化14.5h。然后经4h将反应混合物冷却至7℃-8℃并且加入3m盐酸水溶液(569ml,约5.7ml/g,约7.75当量)。在7℃-8℃下另外26h后,将反应混合物温热至20℃,过滤并且用水(200ml,2ml/g)洗涤。然后将合并的滤液转移到新反应器中,并且加入水(50ml,0.5ml/g)和2-丙醇(200ml,2ml/g)。然后经2.5h向反应流中加入高碘酸(232.09g,4.00当量)在水(200ml,2ml/g)中的溶液以控制排气,接着老化另外27h。然后加入氯化钾(199.9g,2g/g),并且另外18h后,将混合物过滤并且将固体用20wt%氯化钾水溶液(200ml,2ml/g)洗涤。然后将合并的滤液依次用2-甲基四氢呋喃(2
×
400ml,总共800ml,8ml/g)洗涤两次。然后将下部水层用另外的2-甲基四氢呋喃(800ml,8ml/g)洗涤。然后向合并的有机层中加入20wt%氯化钾水溶液(500ml,5ml/g)和半胱氨酸盐酸盐(25g,0.25g/g)。将各层分开,并且将上部有机层用20wt%氯化钾水溶液(500ml,5ml/g)和半胱氨酸盐酸盐(25g,0.25g/g)的溶液洗涤。然后将上部有机层用20wt%氯化钾水溶液(500ml,5ml/g)洗涤。
[0765]
然后将浑浊的富含产物的有机层在真空中浓缩(旋转蒸发器;浴温度:50℃)至2ml/g,接着添加2-甲基四氢呋喃(200ml,2ml/g)。然后向混合物中加入活性碳(darco g-60;2g;0.02g/g),并且在搅动过夜(约18h)后,将混合物过滤并且用2-甲基四氢呋喃(2
×
50ml,2
×
0.5ml/g)洗涤。然后将合并的滤液在真空中浓缩(内部压力:200托,稳态蒸馏时的内部溶液温度:39℃-41℃)至约100ml(约1ml/g),冷却至20℃,并且顺序地经25min加入庚
烷(45ml,0.45ml/g),接着加入(1s,3s)-3-羟基环己烷-1-甲酸晶种(369mg,0.0037g/g)。老化过夜(约16h)后,经4-6h向浆料中加入另外的庚烷(355ml,3.55ml/g),过滤并且用1:4 2-甲基四氢呋喃:庚烷(100ml,1ml/g)、接着用庚烷(100ml,1ml/g)洗涤。然后将湿饼用甲苯(100ml,1ml/g)重新浆化(在搅动下),并且在真空下去除溶剂后,在真空烘箱中在50℃下在n2吹扫下进一步干燥过夜以得到20.76g呈灰白色固体的(1s,3s)-3-羟基环己烷-1-甲酸(95.01%效力,通过1h qnmr,62.1%校正分离产率)。
[0766]1h nmr(400mhz,d
4-meoh)δ3.93-4.00(m,1h),2.67-2.75(m,1h),1.65-1.85(m,4h),1.45-1.63(m,4h)。
13
c nmr:(100mhz,d4-meoh)δ179.64,66.88,39.11,36.51,33.75,29.27,21.05.lc/ms(dci)计算值[c7h
12
o3+h]
+
=145.086,实测值145.0865。
[0767]
hplc方法条件:柱:supelco ascentis express c18,2.7μm,4.6x150mm;流动相a:在水中的0.05%msa:ch3cn(98:2);流动相b:在水中的0.05%msa:ch3cn(10:90);温度:25℃;梯度:0min(0%b),7.0min(100%b),9.0min(100%b);流量:0.8ml/min;210nm;hplc rt 4.62min。
[0768]
手性hplc方法条件:柱:chiralpak ad-3,3μm,4.6x150mm;流动相a:在庚烷中的0.03%msa:etoh(85:15),等度15min;温度:25℃;流量:1.0ml/min;210nm;hplc rt希望的反式对映异构体7.67min;hplc rt不希望的反式对映异构体9.29min;hplc rt不希望的主要/次要顺式非对映异构体6.89min。
[0769]
实施例7:1-((1s,2s)-2-氨基环己基)-3-(3,5-双(三氟甲基)苯基)硫脲盐酸盐的制备
[0770][0771]
向在氮气保护下的4l反应器中顺序地加入2-methf(280ml,1.0ml/g)、(1s,2s)-(+)-1,2-二氨基环己烷(235.8g,2.0当量)在2-methf(840ml,3ml/g)中的溶液和作为冲洗液的2-methf(280ml,1.0ml/g)。将所得溶液冷却至-10℃并且然后经4.5h加入3,5-双(三氟甲基)苯基异氰酸酯(280g,1032mmol,1.0当量,限制试剂)在2-methf(840ml,3ml/g)中的溶液。然后经30min向反应混合物中缓慢加入6n hcl水溶液(560ml,2.0ml/g),维持内部温度《20℃。另外10min后,将各层分开并且弃去下部水层。然后将上部有机层在真空(320毫巴)下浓缩至4.0ml/g,维持内部温度在40℃-45℃之间。加入新鲜的2-methf(560ml,2.0ml/g)并且继续浓缩至3.0ml/g。然后将反应混合物置于1atm n2下并且调节至40℃-45℃的内部温
度。经3.5h缓慢地加入庚烷(2520ml,9.0ml/g),并且经2h将所得浆料从40℃-45℃冷却至20℃。老化过夜后,将浆料过滤,顺序地用庚烷:2-methf(3:1)(840ml,3.0ml/g)、接着用庚烷(840ml,3.0ml/g)洗涤并且在真空下在45℃-50℃下干燥以得到298g(74%-75%产率)呈白色固体的1-((1s,2s)-2-氨基环己基)-3-(3,5-双(三氟甲基)苯基)硫脲盐酸盐。
[0772]1h nmr(400mhz,dmso-d6)δ10.75-11.24(m,1h),8.77-8.99(m,1h),8.26-8.45(m,2h),7.96-8.26(m,3h),7.60-7.76(m,1h),5.70-5.73(m,1h),4.23-4.43(m,1h),2.95-3.17(m,1h),1.94-2.18(m,2h),1.59-1.80(m,2h),1.39-1.57(m,1h),1.11-1.39(m,3h)。
13
c nmr:(100mhz,dmso-d6)δ181.3,142.7,130.5(q,j=33.0hz),122.0,123.7(q,j=272.9hz),116.3,55.0,53.5,30.9,29.6,24.3,23.6。lc/ms(esi)计算值[c
15h17
f6n3s+h]
+
=386.11,实测值386.11。
[0773]
hplc方法条件:柱:supelco ascentis express c18,2.7μm,4.6x50mm;流动相a:在乙腈中的0.05%tfa:水(5:95);流动相b:在乙腈中的0.05%tfa:水(95:5);温度:50℃;梯度:0min(0%b),15.0min(100%b),18.0min(100%b);流量:1.2ml/min;229nm;hplc rt 6.53min。
[0774]
实施例8:(1s,3s)-3-((6-溴-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸的制备
[0775][0776]
在20℃下向在氮气保护下的100ml反应器中加入mtbe(45ml,7.5ml/g)、接着加入kotbu(11.2g,2.4当量)。在单独的加料漏斗中加入dmf(45ml,7.5ml/g)、接着加入(1s,3s)-3-羟基环己烷-1-甲酸(6.0g,1.0当量,限制试剂),其易于溶解并且产生澄清的溶液。然后经90-120min将加料漏斗的内容物逐滴添加到mtbe/kotbu混合物中,维持内部温度低于30℃。另外30min后,向所得分散良好的悬浮液中加入6-溴-3-氟-2-甲基吡啶(7.84g,1.00当量)并且将反应器的内容物加热至33℃。6h后,添加呈固体的kotbu(2.4g,1.00当量)、接着加入6-溴-3-氟-2-甲基吡啶(1.96g,0.25当量)老化另外24h后,将反应器的内容物冷却至25℃并且逐滴加入水(48ml,8.0ml/g),维持内部温度低于30℃。另外15min后,加入mtbe(24ml,4.0ml/g)并且在25℃下搅拌所得双相溶液15min。然后将各层分开,并且弃去上部有机层。然后将下部富含产物的水层用另外的mtbe(24ml,4.0ml/g)洗涤,并且弃去上部有机层。然后向富含产物的水层中加入methf(60ml,10.0ml/g)并且使用6n hcl(18ml,3.0ml/g)酸化至ph 5-7。然后将各层分开,并且将所得上部富含产物的有机层用水(2x18ml,2x3ml/g)洗涤两次,在真空下在45℃下浓缩至6.0ml/g。在恒定体积蒸馏条件下将所得溶液溶剂交换为甲苯(48ml,8.0ml/g),并且将所得溶液用碳zeta plus 55sp(0.9g,0.15当量)处理2小时并且过滤。然后将所得溶液在减压下浓缩,使用1wt%所希望的产物接种以进行结晶,接着添加庚烷(108ml,18.0ml/g)经4-6h。老化过夜后,将所得浆料过滤,顺序地用甲苯/庚烷(1:3)混合物(18ml,3.0ml/g)、庚烷(18ml,3.0ml/g)洗涤,并且在真空下在45℃下干燥以得到10.1g(78%产率)呈棕色固体的所希望的产物。
[0777]
可替代地,
[0778][0779]
在20℃下向在氮气保护下的100ml反应器中加入mtbe(45ml,7.5ml/g)、接着加入kotbu(14.0g,3.0当量)。在单独的加料漏斗中加入dmf(45ml,7.5ml/g)、接着加入(1s,3s)-3-羟基环己烷-1-甲酸(6.0g,1.0当量,限制试剂)。将(1s,3s)-3-羟基环己烷-1-甲酸容易地溶解并且产生澄清的溶液。经15-30min然后将加料漏斗的内容物逐滴添加到mtbe/kotbu混合物,维持内部温度低于30℃。另外30min后,向所得分散良好的悬浮液中加入6-溴-3-氟-2-甲基吡啶(9.8g,1.25当量)并且将反应器的内容物加热至33℃。24h后,将反应器的内容物冷却至25℃并且逐滴加入水(48ml,8.0ml/g)以维持内部温度低于30℃。另外15min后,加入mtbe(18ml,3.0ml/g)并且在25℃下搅拌所得双相溶液15min。然后将各层分开,并且弃去上部有机层。然后向富含产物的水层中加入甲苯(48ml,8.0ml/g)并且使用6n hcl(18ml,3.0ml/g)酸化至ph 5-7。然后将各层分开,并且将所得上部富含产物的有机层在45℃下在真空下浓缩至6.0ml/g。然后经2-4h向所得溶液中逐滴加入庚烷(108ml,18.0ml/g)并且老化过夜。然后将所得浆料过滤并且顺序地用甲苯/庚烷(1:3)混合物(18ml,3.0ml/g)和庚烷(18ml,3.0ml/g)洗涤,并且在真空下在45℃下干燥以得到7.9g(61%产率)呈棕色固体的(1s,3s)-3-((6-溴-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸。
[0780]1h nmr(400mhz,cdcl3)δ11.61-10.99(m,1h),7.27-7.14(m,1h),7.07-6.95(m,1h),4.68-4.50(m,1h),2.91-2.77(m,1h),2.56-2.39(m,3h),2.15-2.04(m,1h),2.04-1.82(m,3h),1.81-1.57(m,4h)。
13
c nmr(100mhz,cdcl3)δ181.3,151.2,150.9,130.1,125.5,121.9,72.3,38.2,31.8,29.4,27.9,20.2,19.1.lc/ms(esi)计算值[c
13h16
brno3+h]
+
=314.04,实测值314.04。
[0781]
分析型:柱:phenomex kinetex c8,150x4.6mm,2.6μm;流动相a:在水中的0.05%tfa;流动相b:在甲醇中的0.05%tfa:乙腈(80:20);温度:27℃;梯度:0min(44%b),1.0min(44%b),17.0min(60%b),18.0min(60%b),25.0min(90%b),28.0min(90%b),30.0min(100%b),30.1min(44%b),35min(44%b);流量:0.8ml/min;220nm;hplc rt 14.88min。
[0782]
实施例9:(1s,3s)-3-((6-(5-((苄氧基)甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸叔丁胺盐的制备
[0783][0784]
[0785]
有机镁制备-向配备有顶置式搅拌桨的带夹套的250ml chemglass反应器中加入5-((苄氧基)甲基)-4-溴-1-甲基-1h-1,2,3-三唑溴化物盐(36.05g,1.3当量)和四氢呋喃(209ml,8.5ml/g)。将反应器密封,配备氮气入口,并且用氮气的表面下喷射使浆料脱气30分钟。将浆料冷却至5℃的内部温度,并且允许老化10分钟。经30分钟加入异丙基氯化镁(103ml,2.55当量,在thf中1.90m)(警告:气体逸出!)。添加完成后,将反应器温热至20℃。3小时后,将均质的有机镁溶液立即用于随后的根岸(negishi)偶联(参见下文)。
[0786]
根岸偶联-向配备有顶置式搅拌桨的单独的带夹套的1l chemglass反应器中加入(1s,3s)-3-((6-溴-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸(25.00g,1.0当量,限制试剂)、无水氯化锌(3.65g,0.35当量)和四氢呋喃(85ml,3.4ml/g)。将反应器密封并且配备氮气入口,并且将顶部空间用氮气冲扫5分钟。将所得均质溶液冷却至0℃并且在搅动下老化10分钟。经15分钟加入异丙基氯化镁(37.8ml,0.95当量,在thf中1.90m)(警告:气体逸出!)。另外15分钟后,将反应器温热至20℃并且一次性加入pdcl2(呫吨)(1.44g,0.025当量)。将所得非均质溶液用氮气的表面下喷射脱气30分钟。脱气后,将溶液温热至40℃并且在搅动下老化10分钟。经10-15分钟以缓慢稳定的流加入有机镁溶液(参见上文)。将所得溶液在40℃下搅动16小时并且随后冷却至23℃。将反应经1小时用在水(300ml)中的乙二胺四乙酸三钠(11.67g,0.45当量)淬灭。用6n hcl(1.1当量)将溶液的ph调节至4.0-5.0,并且将双相混合物搅动30分钟。允许各相静置30分钟,并且随后分开。将有机层用过碳酸钠(2.43g,0.20当量)在水(80ml,3.2ml/g)中的新鲜制备的溶液处理[警告:气体逸出!(co2)]。将所得均质混合物在20℃下搅动4小时。用焦亚硫酸钠(2.87g,0.20当量)在15wt%nacl
水溶液
(80ml,3.2ml/g)中的溶液淬灭氧化。加入甲苯(75ml,3.0ml/g),并且将所得双相混合物搅动30分钟。允许各相静置30分钟,并且随后分开。将有机流在减压(100-150托)下浓缩至4ml/g的溶液体积。将富含产物的有机层用新鲜甲苯(150ml,6.0ml/g)稀释并且随后在减压下浓缩(100-150托)至6ml/g的溶液体积。将富含产物的有机层用甲苯(25ml,1.0ml/g)和四氢呋喃(25ml,1.0ml/g)稀释。将所得溶液精滤到干净的250ml带夹套的chemglass反应器中并且温热至55℃。一次性添加叔丁胺(1.90ml,1.2当量)在甲苯(83ml,3.3ml/g)和四氢呋喃(17ml,0.7ml/g)中的溶液的三分之一,接着接种所希望的产物(0.125g,0.5wt%)。经60分钟以逐滴方式加入剩余的三分之二的胺溶液。30分钟后,将浆料经2小时缓慢冷却至0℃。将浆料在0℃下老化14小时。过滤并且收集固体。将固体顺序地用甲苯:四氢呋喃(5:1)(75ml,3.0ml/g)、接着用甲苯(75ml,3.0ml/g)洗涤,并且在烘箱真空中在65℃下在氮气吹扫下干燥以得到35.96g(93.4%)呈白色固体的所希望的产物。
[0787]
可替代地,
[0788]
[0789][0790]
向20ml小瓶中加入5-((苄氧基)甲基)-4-溴-1-甲基-1h-1,2,3-三唑溴化物盐(1.7059g,1.5当量)。将小瓶用配备有ptfe隔膜的帽密封。为小瓶配备氮气入口,并且将顶部空间用氮气冲扫5分钟。将四氢呋喃(8.90ml,8.95ml/g)加入小瓶中。将所得浆料冷却至0℃并且允许老化10分钟。经15分钟加入异丙基氯化镁(4.11ml,3.0当量,在thf中2.15m)(警告:气体逸出!)。2小时后,将小瓶温热至23℃并且放入氮气气氛手套箱中。一次性加入无水氯化锌(0.601g,1.5当量),并且在氮气气氛手套箱中在23℃下搅动溶液。1小时后,从氮气气氛手套箱中取出均质的有机锌溶液并且立即用于随后的根岸偶联(参见下文)。
[0791]
根岸偶联-向单独的40ml小瓶中加入(1s,3s)-3-((6-溴-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸(1.00g,1.0当量,限制试剂)。将小瓶用配备有ptfe隔膜的帽密封。为小瓶配备氮气入口,并且将顶部空间用氮气冲扫5分钟。将四氢呋喃(4.03ml,4.0ml/g)加入小瓶中。将所得均质溶液冷却至0℃并且允许老化10分钟。经15分钟加入异丙基氯化镁(1.30ml,0.95当量,在thf中2.15m)(警告:气体逸出!)。15分钟后,将小瓶温热至23℃并且一次性加入pdcl2(呫吨)(0.0665g,0.03当量)。将所得非均质溶液通过氮气的表面下喷射脱气5分钟。在将溶液脱气后,经10分钟添加有机锌溶液(参见上文)。将所得溶液在23℃下剧烈搅动。21小时后,经1小时添加乙二胺四乙酸三钠(2.16g,2.0当量)在水(12.0ml)中的溶液。将各相分开,并且将有机层在减压(100托)下浓缩至4ml/g的溶液体积。将富含产物的有机层用新鲜四氢呋喃(8.0ml,8ml/g)稀释。在减压(100托)下去除溶剂至4ml/g的溶液体积。将富含产物的有机层用新鲜四氢呋喃(8.0ml,8ml/g)稀释。在减压(100托)下去除溶剂至4ml/g的溶液体积。将富含产物的有机层用甲苯(8.0ml,8.0ml/g)稀释。一次性添加叔丁胺(0.33ml,1.0当量)在甲苯(1.0ml,1.0ml/g)中的溶液的三分之一,接着接种(1s,3s)-3-((6-(5-((苄氧基)甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸叔丁胺盐(0.010g,1.0wt%)。经20分钟以逐滴方式加入剩余的三分之二的胺溶液。
15小时后,将浆料过滤并且收集固体。将固体顺序地用甲苯:四氢呋喃(2:1)(6.0ml,6.0ml/g)、接着用甲苯(6.0ml,6.0ml/g)洗涤,并且在烘箱真空中在50℃下在氮气吹扫下干燥以得到1.22g(76%)呈白色固体的(1s,3s)-3-((6-(5-((苄氧基)甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸叔丁胺盐。
[0792]1h nmr(400mhz,dmso-d6)δ8.54-7.37(br s,3h),7.81(d,j=8.6hz,1h),7.54(d,j=8.6hz,1h),7.33-7.19(m,5h),5.18(s,2h),4.69(br s,1h),4.53(s,2h),4.06(s,3h),2.33-2.35(m,1h),2.32(s,3h),2.00-1.42(m,8h),1.19(s,9h)。
13
c nmr:(100mhz,dmso-d6)δ178.3,151.2,148.1,144.5,141.8,138.4,131.4,128.7,128.1,128.0,120.4,119.5,73.2,72.0,59.7,49.8,40.4(与dmso-d6重叠)35.4,33.4,30.6,29.1,29.0,21.3,19.8。lc/ms(esi)计算值[c
24h28
n4o4+h]
+
=437.22,实测值437.22。
[0793]
分析型:柱:phenomenex kinetex c8 2.6um 4.6x150mm;流动相a:在h2o中的0.05%tfa;流动相b:在meoh中的0.05%tfa:ch3cn(80:20);温度:27℃;梯度:0min(44%b),1.0min(44%b),17.0min(60%b),18.0min(60%b),25.0min(90%b),28.0min(90%b),30.0min(100%b),30.1min(44%b),35.0min(44%b);流量:0.8ml/min;220nm;hplc rt 17.849min。
[0794]
实施例10:(1s,3s)-3-((6-(5-(羟甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸的制备
[0795][0796]
向不锈钢高压反应器中加入(1s,3s)-3-((6-(5-((苄氧基)甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸叔丁胺盐(1.00g,1.0当量,限制试剂)、柠檬酸一水合物(0.411g,1.00当量)、pd/c(10wt%,湿)(0.100g,0.10g/g)、乙醇(6.0ml,6.0ml/g)和水(2.0ml,2.0ml/g)。将反应器密封并且开始顶置式搅动。首先用氮气置换反应器中的气氛。然后将反应器中的气氛加压至30psi氢气,并且将反应器温热至40℃。25h后,将反应器冷却至20℃-25℃并且过滤。然后将反应器和滤饼用乙醇:水(3:1)(2x2.0ml,2x2.0ml/g)洗涤。然后将合并的滤液在减压(《10托)下浓缩至5.0ml/g的溶液体积。然后经2小时添加水(7.0ml,9.0ml/g)。将所得浆料过滤,将固体顺序地用水:乙醇(3:1)(3.0ml,3.0ml/g)、接着用水(3.0ml,3.0ml/g)洗涤,并且然后在真空烘箱中在50℃下在氮气吹扫下干燥以得到0.622g(90%)呈白色固体的所希望的产物。
[0797]
可替代地,
[0798][0799]
向不锈钢高压反应器中加入(1s,3s)-3-((6-(5-((苄氧基)甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸叔丁胺盐(1.00g,1.0当量,限制试剂)、无水柠檬酸(0.508g,1.34当量)、pd/c(10wt%,湿)(0.100g,0.10g/g)和乙醇(200proof)(10.0ml,10.0ml/g)。将反应器密封并且开始顶置式搅动。用氮气置换反应器中的气氛。将反应器中的气氛加压至30psi氢气,并且将反应器温热至40℃。25小时后,将反应器冷却至23℃并且过滤。将反应器和滤饼用乙醇(2x2.0ml,2x2.0ml/g)洗涤两次。将合并的有机层在减压(《10托)下浓缩至3.0ml/g的溶液体积。经2小时添加水(9.0ml,9.0ml/g)。将固体过滤并且顺序地用水:乙醇(3:1)(3.0ml,3.0ml/g)、接着用水(3.0ml,3.0ml/g)洗涤,并且在烘箱真空中在50℃下在氮气吹扫下干燥以得到0.491g(71%)呈白色固体的(1s,3s)-3-((6-(5-(羟甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸。
[0800]1h nmr(500mhz,dmso-d6)δ12.09(br s,1h),7.73(d,j=8.4hz,1h),7.35(d,j=8.5hz,1h),5.69(br s,1h),4.89(br s,2h),4.63(br s,1h),3.94(s,3h),3.28(br s,1h),2.37(br s,2h),2.31(s,3h),1.90-1.80(m,1h),1.75-1.55(m,3h),1.55-1.31(m,4h)。
13
c nmr:(125mhz,cdcl3)δ176.4,150.2,147.7,143.1,142.0,134.6,120.6,119.2,71.5,51.8,37.7,35.0,31.7,28.8,27.8,19.9,19.4。lc/ms(esi)计算值[c
17h22
n4o4+h]
+
=347.17,实测值347.17。
[0801]
分析型:柱:phenomenex kinetex c8 2.6um 4.6x150mm;流动相a:在h2o中的0.05%tfa;流动相b:在meoh中的0.05%tfa:ch3cn(80:20);温度:27℃;梯度:0min(44%b),1.0min(44%b),17.0min(60%b),18.0min(60%b),25.0min(90%b),28.0min(90%b),30.0min(100%b),30.1min(44%b),35.0min(44%b);流量:0.8ml/min;220nm;hplc rt 5.003min。
[0802]
实施例11:n-甲基-n-丙基-1h-咪唑-1-甲酰胺草酸盐的制备
[0803][0804]
向4l反应器中加入t-amoh(900ml,6.0ml/g)和1,1
’‑
羰基二咪唑(353g,1.05当量)。将加料口用另外的t-amoh(150ml,1.0ml/g)冲洗。将所得非均相浆料冷却至0℃并且经1小时添加n-甲基丙胺(150g,1.0当量,限制试剂)。17小时后,将溶液温热至20℃。用水(525ml,3.5ml/g)淬灭反应(警告:气体逸出!)。一旦气体逸出停止,就向反应器中加入6n hcl
水溶液
(228ml,0.67当量)和氯化钠(92g,0.76当量)。分离各相并且将有机层保留在反应器中。向反应器中加入15wt%nacl
水溶液
(525ml,3.5ml/g)和6n hcl
水溶液
(51ml,0.15当量)。分离各相并且将有机层保留在反应器中。向反应器中加入15wt%nacl
水溶液
(525ml,3.5ml/g)和6n hcl
水溶液
(41ml,0.12当量)。分离各相并且将有机层保留在反应器中。向反应器中加入15wt%nacl
水溶液
(525ml,3.5ml/g)和6n hcl
水溶液
(10ml,0.03当量)。分离各相。将溶液温热至45℃并且在减压(75托)下浓缩至4ml/g的溶液体积。向反应器中加入新鲜的甲苯(750ml,5.0ml/g)并且在减压(75托)下进一步浓缩至4ml/g的溶液体积。然后向反应器中加入新鲜的甲苯(900ml,6.0ml/g)并且允许冷却至23℃。将所得浆料精过滤并且将所得富含产物的有机层加入干净的4l反应器中。将溶液温热至65℃并且经3小时添加无水草酸(190g,1.0当量)在2-丙醇(750ml,5.0ml/g)中的溶液。将所得浆料在65℃下老化8小时并且随后经4小时冷却至0℃。将固体过滤并且顺序地用甲苯:2-丙醇(7:1)(450ml,3.0ml/g)、接着用甲苯(450ml,3.0ml/g)洗涤,并且在烘箱真空中在50℃下在氮气吹扫下干燥以得到406g(77%)呈白色固体的n-甲基-n-丙基-1h-咪唑-1-甲酰胺草酸盐。
[0805]1h nmr(400mhz,dmso-d6)δ14.41-13.65(br s,2h),8.31-8.04(m,1h),7.62-7.50(m,1h),7.19-7.06(m,1h),3.32(t,j=7.3hz,2h),3.00(s,3h),1.60(六重峰,j=7.3hz,2h),0.84(t,j=7.5hz,3h)。
13
c nmr:(100mhz,cdcl3)δ162.02,151.28,137.40,128.01,119.27,51.79,36.48,20.29,11.31.lc/ms(esi)计算值[c8h
13
n3o+h]
+
=168.11,实测值168.11。
[0806]
分析型:柱:supelco ascentis express c18 2.7um 4.6x100mm;流动相a:在ch3cn中的0.01m nh4oac:h2o(5:95);流动相b:在ch3cn中的0.01m nh4oac:h2o(95:5);温度:30℃;梯度:0min(0%b),2.0min(0%b),15.0min(25%b),20.0min(100%b),24.0min(100%b);流量:1.2ml/min;210nm;hplc rt 8.934min。
[0807]
实施例12:(1s,3s)-3-((2-甲基-6-(1-甲基-5-(((甲基(丙基)氨基甲酰基)氧基)甲基)-1h-1,2,3-三唑-4-基)吡啶-3-基)氧基)环己烷-1-甲酸的制备
[0808][0809]
为40ml小瓶配备磁力搅拌棒,并且向n2入口顺序地加入(1s,3s)-3-((6-(5-(羟甲基)-1-甲基-1h-1,2,3-三唑-4-基)-2-甲基吡啶-3-基)氧基)环己烷-1-甲酸(0.446g,1.29mmol,1.,0当量,限制试剂)、n-甲基-n-丙基-1h-咪唑-1-甲酰胺草酸盐(0.596g,2.32mmol,1.8当量)和2-甲基-2-丁醇(5.4ml,12l/kg)。然后将所得浓稠的反应浆料温热至55℃-60℃并且通过注射泵经45min逐滴加入kotbu(在thf中的20wt%,4.7ml,7.8mmol,6.0当量)。另外2h后,将所得的较稀反应混合物冷却至环境温度并且加入水(6.7ml,15l/kg)。然后逐滴添加6n hcl水溶液(1ml,1.2l.kg)以将水层的ph调节至4.0(目标ph 3.0-4.5)。将各层分离并且将下部水层用2-甲基-2-丁醇(4.0ml,9l/kg)反萃。然后将合并的富含产物的水层用nacl水溶液(2g/l,4.0ml,9.0l/kg)洗涤,并且将所得有机层在旋转蒸发器上浓缩至接近干燥。然后将残余物溶解在dcm(5.5ml,12l/kg)中并且用水(4.0ml,9l/kg)洗涤。然后分离各层,并且将下部富含产物的dcm层精过滤。然后将过滤器用dcm(2.0ml,4.5l/kg)洗涤以得到(1s,3s)-3-((2-甲基-6-(1-甲基-5-(((甲基(丙基)氨基甲酰基)氧基)甲基)-1h-1,2,3-三唑-4-基)吡啶-3-基)氧基)环己烷-1-甲酸在dcm中的溶液。
[0810]1h nmr(500mhz,cdcl3)δ7.96(d,j=8.4hz,1h),7.22(d,j=8.5hz,1h),5.76(d,j=9.9hz,2h),4.71(br s,1h),4.14(s,3h),3.28-3.22(m,1h),3.15-3.07(m,1h),2.99-2.85(m,3h),2.81(br s,1h),2.50(s,3h),2.19-2.11(m,1h),2.09-1.86(m,3h),1.82-1.72(m,1h),1.71-1.62(m,3h),1.60-1.54(m,1h),1.48-1.38(m,1h),0.90-0.71(m,3h)*。注:0.90-0.71的峰实际上观察到是两个宽单峰,每个都整合为1.5h,支持api以旋转异构体在cdcl3中的混合物的形式存在。
13
c nmr:(125mhz,cdcl3)δ179.7,155.8,155.7,150.7,148.8,145.3,141.2,129.9,119.4,118.9,71.4,55.1,50.7,38.0,35.2,35.1,34.5,33.8,31.8,29.2,27.9,20.8,20.4,20.1,19.5,10.84,10.76。由于观察到的旋转异构体混合物,27个碳(相对于理论的22个)。lc/ms(esi)计算值[c
22h31
n5o5+h]
+
=446.24,实测值446.24。
[0811]
hplc方法条件:柱:waters xbridge c8,3.5μm,4.6x150mm;流动相a:在乙腈中的0.01m nh4oac:水(20:80);流动相b:在乙腈中的0.01m nh4oac:水:meoh(75:5:20);温度:40℃;梯度:0min(0%b),9.6min(25%b),19.2min(100%b),27.0min(100%b);流量:1.0ml/min;233nm;hplc rt 14.20min。
[0812]
实施例13.(1s,3s)-3-((2-甲基-6-(1-甲基-5-(((甲基(丙基)氨基甲酰基)氧基)甲基)-1h-1,2,3-三唑-4-基)吡啶-3-基)氧基)环己烷-1-甲酸的制备
[0813][0814]
为40ml小瓶配备磁力搅拌棒,并且向n2入口顺序地加入cdi(0.842g,1.8当量)和thf(4.0ml,4.0ml/g)。将反应混合物冷却至0℃并且经20-45分钟逐滴添加n-甲基丙胺
(0.40g,1.89当量)。在0℃-5℃下老化另外30min后,将反应混合物温热至室温并且加入13-c(1.0g,1.0当量,限制试剂)和2-甲基-2-丁醇(8.0ml,8l/kg)。然后将所得溶液温热至45-50℃并且通过注射泵经45min逐滴加入kotbu(在thf中20wt%,3.8ml,2.4当量)。另外2h后,将所得的较稀反应混合物冷却至环境温度并且加入水(3.0ml,3l/kg)。然后逐滴添加6n hcl水溶液(2.4ml,2.4l.kg)以将水层的ph调节至4.0(目标ph 3.0-4.5)。弃去下部水层,并且然后将富含产物的有机层用nacl水溶液(3g/l,3.0ml,3.0l/kg)洗涤以获得最终产物在t-amoh中的溶液。
[0815]
(1s,3s)-3-羟基环己烷-1-甲酸的生物催化合成:
[0816][0817]
实施例14.4-甲基苯磺酸3-氧代环己-1-烯-1-基酯的制备
[0818]
向在氮气下的5l chemglass反应器中顺序地加入1,3-环己二酮(140.0g,1249mmol,1.0当量,限制试剂)、对甲苯磺酰氯(250.0g,1311mmol,1.05当量)和乙酸乙酯(1400ml,10l/kg)。然后将所得浆料冷却至0℃-5℃并且通过加料漏斗经约35分钟逐滴加入三甲胺(151.6g,1498mmol,1.2当量),保持内部温度《10℃。添加完成后,将加料漏斗用乙酸乙酯(10ml,1l/kg)冲洗。在0℃-5℃下老化另外1h后,将反应混合物温热至环境温度。老化过夜后,加入水(700ml,5l/kg)和乙酸乙酯(700ml,5l/kg)并且将所得层分开。然后将上部富含产物的有机层用在水(140ml,2l/kg)中的氯化钠(14质量%)洗涤,并且将所得富含产物的有机层经mgso4(140g,1kg/kg)干燥,过滤并且用乙酸乙酯(78ml,0.6l/kg)洗涤以得到所希望的产物在etoac中的粗溶液。基于所希望的产物与乙酸乙酯的峰比较,确定溶液含有约15.6wt%4-甲基苯磺酸3-氧代环己-1-烯-1-基酯,基于2114.36g的总溶液质量=329g所希望的产物(99%溶液产率)。
[0819]1h nmr(500mhz,cdcl3)δ7.75(d,j=8.3hz,2h),7.33(d,j=8.3hz,2h),5.70(s,1h),2.41-2.38(m,5h),2.25-2.22(m,2h),1.93-1.87(m,2h)。
[0820]
hplc方法条件:柱:supelco ascentis express c18,50x2.1mm,2.7μm;流动相a:在乙腈中的0.05%tfa:水(5:95);流动相b:在乙腈中的0.05%tfa:水(95:5);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;232nm;uhplc rt 1.35min。
[0821]
实施例15.3-氧代环己-1-烯-1-甲酸甲酯的制备
[0822]
制备乙酸钯(ii)(935.0mg,4.165mmol,0.05当量)、1,3-双(二苯基膦基)丙烷(2.06g,4.99mmol,0.06当量)和甲醇(40ml,1.8l/kg)的混合物。向300ml高压釜反应器中加入在etoac中的4-甲基苯磺酸3-氧代环己-1-烯-1-基酯(141.7g溶液,约15.6wt%,22.1g 4-甲基苯磺酸3-氧代环己-1-烯-1-基酯,83.03mmol,1.0当量,限制试剂)。向高压釜中加入pd(oac)2/dppp/meoh混合物,接着加入n,n-二异丙基乙胺。将高压釜密封并且用30psi氮气吹扫3次,接着用一氧化碳吹扫3次。然后将高压釜加压至30psi并且在搅动=700rpm下加热至60℃。15h后,将高压釜用氮气吹扫并且将内容物转移到琥珀色玻璃瓶中。然后将以上过程重复另外7次,并且将所有8个反应流合并用于纯化(理论产物产率=102.35g)。将合并的反应混合物在真空中浓缩,并且向所得残余物中首先加入正庚烷(160ml)、etoac(80ml)和水(160ml)。残余物没有完全溶解,并且将液体倾析并且保存。然后通过添加etoac(240ml)、水(160ml)并且温热至40℃将剩余的残余物溶解。然后将保存的正庚烷/etoac/水混合物添加到etoac/水的双相溶液中,并且分离各层。将下部水层用正庚烷(160ml)和etoac(160ml)的混合物反萃。然后将合并的有机层在旋转蒸发器上浓缩以得到110.39g橙色残余物。然后将残余物溶解在etoac(200ml)中,加入硅胶60(150g),过滤并且用etoac(160ml)洗涤。将合并的滤液在旋转蒸发器上浓缩以得到98.16g橙色液体,然后通过isco色谱法使用dcm/etoac作为流动相将其纯化以得到79.16g(77%产率)呈黄色油状物的3-氧代环己-1-烯-1-甲酸甲酯。
[0823]1h nmr(400mhz,cdcl3)δ6.74(s,1h),3.84(s,3h),2.61-2.58(m,2h),2.47-2.44(m,2h),2.10-2.03(m,2h)。
[0824]
hplc方法条件:柱:acquity uhplc hss c18,50x2.1mm,1.8μm;流动相a:在乙腈中的0.05%tfa:水(5:95);流动相b:在乙腈中的0.05%tfa:水(95:5);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;240nm;uhplc rt 1.16min。
[0825]
实施例16.3-氧代环己-1-烯-1-甲酸的制备
[0826][0827]
向干净的500ml反应器中加入碳酸钠(54.6g,515mmol,1.2当量)和水(400ml,约6l/kg)。完全溶解后,加入3-氧代环己-1-烯-1-甲酸甲酯(69.58g,429mmol,1.0当量,限制试剂)并且温热至35℃。约24h后,添加另外的水(70ml,约1l/kg),并且将从少量上部橙色油状物分离下部水层。缓慢加入3n hcl水溶液直至达到ph约1,并且然后将产物用dcm(5x100ml)反萃。然后将合并的有机层经na2so4干燥,过滤并且在旋转蒸发器上浓缩以得到46.72g黄色固体。然后向固体中加入dcm(125ml,约1.8l/kg),加热至30℃-35℃。并且然后经》1h逐滴加入正庚烷(650ml,约9.3l/kg)以诱导结晶。然后将所得浆料过滤,用正庚烷(2x50ml,2x0.7l/kg)洗涤并且在真空烘箱中在50℃下干燥以得到42.60g(67%产率)呈淡黄色固体的3-氧代环己-1-烯-1-甲酸。
[0828]
可替代地,
[0829][0830]
向在手套箱中的额定压力的4ml小瓶中加入pd(dppp)cl2在dce中的储备溶液(337.5mg溶液,用1.67g dce和7.96mg pd(dppp)cl2制备,其对应于1.60mg pd(dppp)cl2,0.0027mmol,0.04当量)和在etoac中的4-甲基苯磺酸3-氧代环己-1-烯-1-基酯溶液(150mg溶液,约13.4wt%,20.1mg 4-甲基苯磺酸3-氧代环己-1-烯-1-基酯,0.075mmol,1.0当量,限制试剂)。将混合物浓缩至干(genevac,全真空,约1h),并且然后顺序地加入koac(18.4mg,0.187mmol,2.5当量)和湿ch3cn(0.75ml,用3.7ml ch3cn和67.5mg水制备储备溶液,其对应于约10当量水)。然后将小瓶密封并且吹扫/排气至30psi氮气3次。然后将小瓶用一氧化碳加压至55psi并且在设定为500rpm的定轨振荡器上温热至60℃。》12h后,将小瓶放空/再置于氮气下,并且通过uplc分析判断已得到99ap 3-氧代环己-1-烯-1-甲酸。
[0831]1h nmr(500mhz,cdcl3)δ6.86(t,j=1.9hz,1h),2.61(td,j=6.1,1.9hz,2h),2.52-2.47(m,2h),2.14-2.06(m,2h)。
[0832]
hplc方法条件:柱:acquity uhplc hss c18,50x2.1mm,1.8μm;流动相a:在乙腈中的0.05%tfa:水(5:95);流动相b:在乙腈中的0.05%tfa:水(95:5);温度:40℃;梯度:0min(0%b),2.0min(100%b),2.5min(100%b);流量:1.0ml/min;240nm;uhplc rt 0.85min。
[0833]
实施例17.(1s,3s)-3-羟基环己烷-1-甲酸
[0834][0835]
逐步反应(途径a):
[0836]
向50ml反应器中添加ered-302(450mg,45wt%)、gdh-105(100mg,10wt%)和nadph(50mg,0.05wt%)。然后添加0.5m磷酸钠缓冲液ph 8.0(17ml,17l/kg)并且将搅动打开至500rpm。允许反应老化30分钟以允许生物催化剂的溶解。然后检查ph并且用2.5m naoh调节至ph 8.0,接着加入d-葡萄糖(3.75g,3当量)。此时,将温度设定为32.5℃并且使用ph稳定器维持反应ph=8.0。在第二容器中,加入3-氧代环己-1-烯-1-甲酸(1.0g,限制试剂)、0.5m磷酸钠缓冲液ph 8.0(3ml,3l/kg),并且搅动10分钟。然后通过添加2.5m naoh将此溶液的ph调节至ph 8.0。然后通过注射泵经6小时将此溶液加入生物催化剂溶液中。16小时后,判断反应完成(《1lcap 3-氧代环己-1-烯-1-甲酸)并且加入4m hcl水溶液以将ph调节至6.0。然后添加kred-p2-g03(200mg,20wt%)在0.4m磷酸钠缓冲液(1ml,1l/kg)的溶液,使用ph稳定器维持ph=6.0。16h后,判断反应完成(《1lcap 3-氧代环己-1-烯-1-甲酸)。
[0837]
级联反应(途径b):
[0838]
向100ml反应器中添加ered-302(750mg,45wt%)、gdh-105(100mg,10wt%)、nadph(50mg,0.05wt%)和kred-p2-g03(100mg,10wt%)。然后添加0.5m磷酸钠缓冲液ph 7.0(57ml,57l/kg)并且将搅动设定为500rpm。允许反应老化30分钟以允许生物催化剂的溶解。然后用2.5m naoh水溶液将ph调节至ph 7.0,接着添加d-葡萄糖(3.75g,2当量)。此时,将温度设定为32.5℃并且使用ph稳定器维持反应ph=7.0。在第二容器中,加入bmt-203387-01(1.0g,限制试剂)、0.5m磷酸钠缓冲液ph 8.0(3ml,3l/kg),并且搅动10分钟。然后通过添加2.5m naoh将此溶液的ph调节至ph 7.0。然后通过注射泵经6小时将此溶液加入生物催化剂溶液中。16小时后,判断反应完成(》99lcap(1s,3s)-3-羟基环己烷-1-甲酸)。
[0839]
分析型:工作浓度1mg/ml,注入体积:1μl。柱:db-ffap 15m x 0.32mm x 0.50μm,载气:氦气,流量:1.4ml/min(恒定流量),前入口温度:250℃,注入方式:分流,分流比:10,分流量:14。烘箱程序:50℃,保持1分钟,然后以10℃/min至230℃,保持3分钟,然后以20℃/min至250℃,保持2分钟(25分钟运行时间)。检测器类型:fid,检测器温度:250℃,燃料流量:30ml/min,氧化剂流量:300ml/min,补充气:氦气,补充气流量:30ml/min。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1