制备(3R,5R)-7-(2-(4-氟苯基)-5-异丙基-3-苯基-4-((4-羟甲基苯基氨基)羰基)-吡咯-1-基)-3,5-二羟基庚酸半钙盐的方法以及制备其所用中间体的方法与流程
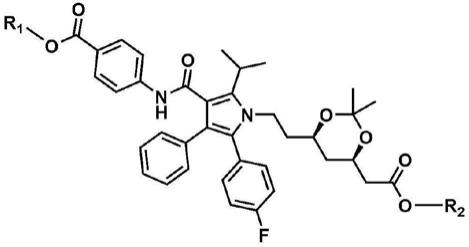
制备(3r,5r)-7-(2-(4-氟苯基)-5-异丙基-3-苯基-4-((4-羟甲基苯基氨基)羰基)-吡咯-1-基)-3,5-二羟基庚酸半钙盐的方法以及制备其所用中间体的方法
技术领域
1.本发明涉及一种制备(3r,5r)-7-(2-(4-氟苯基)-5-异丙基-3-苯基-4-((4-羟甲基苯基氨基)羰基)-吡咯-1-基)-3,5-二羟基庚酸半钙盐的方法,以及制备其所用中间体的方法。
背景技术:
2.(3r,5r)-7-(2-(4-氟苯基)-5-异丙基-3-苯基-4-((4-羟甲基苯基氨基)羰基)-吡咯-1-基)-3,5-二羟基庚酸半钙盐作为3-羟基-3-甲基戊二酰辅酶a还原酶的抑制剂并抑制胆固醇的细胞内合成,因此可用作降脂剂或降胆固醇剂。
3.韩国专利登记号10-1329112描述了一种制备(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐的方法,如以下反应式所示。
[0004][0005]
(在上述制备方法的反应式中,r为烷基、芳基或烷基芳基。)
[0006]
然而,根据该制备方法,由于其中使用的苯甲醛而产生相关物质,并且所得的相关物质难以去除。此外,当在一个接一个地顺序合成取代基的中间步骤中出现问题时,需要花费大量时间从头再次开始该过程。因此,常规制备方法的问题在于其不适于大规模合成。
[0007]
因此,本发明人研究出了一种有效合成(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐,将反应中间体合成为固体,并以汇聚合成方式容易地控制主要相关物质的方法,其中化合物的每个主要结构部分独立地合成,然后偶联,而不是通过如上所述的常规顺序合成方法,从而完成本发明。
技术实现要素:
[0008]
技术问题
[0009]
本发明的一个目的是提供一种制备(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐的新方法,其可有效地缓解常规顺序合成方法的弊端。
[0010]
本发明的另一个目的是提供一种制备用于制备(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐的中间体的方法。
[0011]
技术方案
[0012]
为了解决上述问题,本发明提供了一种制备本发明的(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐的方法,该方法包括:
[0013]
(s1)使下式a的化合物与下式b的化合物反应以制备下式c的化合物;
[0014]
(s2)使所述式c的化合物与下式d的化合物反应以制备下式e的化合物;
[0015]
(s3)由所述式e的化合物制备下式f的化合物;
[0016]
(s4)由所述式f的化合物制备下式g的化合物;
[0017]
(s5)由所述式g的化合物制备下式h的化合物;以及
[0018]
(s6)由所述式h的化合物制备下式1的化合物:
[0019]
[式a]
[0020][0021]
[式b]
[0022][0023]
[式c]
[0024][0025]
[式d]
[0026][0027]
[式e]
[0028][0029]
[式f]
[0030][0031]
[式g]
[0032][0033]
[式h]
[0034][0035]
[式1]
[0036][0037]
其中r1和r2各自独立地为c
1-c6烷基,且
[0038]
x为离去基团。
[0039]
所述离去基团可以是卤素诸如cl、br或i,甲苯磺酸酯基(-ots,-o-s(=o)
2-(c6h4)-ch3),三氟甲磺酸酯基(-otf,-o-s(=o)
2-cf3)或乙酸酯基(-oac,-o(c=o)-ch3)。
[0040]
上述步骤(s1)可以是使活性亚甲基化合物烷基化的步骤,并在碱性或中性条件下进行。在这种情况下,用于碱性条件的碱的实例可包括碳酸钾、碳酸锂、碳酸钠、碳酸铯、氢氧化锂、氢氧化钠、氢氧化钾、1,8-二氮杂双环(5.4.0)十一碳-7-烯(dbu)、n,n-二异丙基乙胺(dipea)、三乙醇胺(tea)、吡啶、氢化钠、叔丁醇钾(k[oc(ch3)3])等,其可以单独使用或者以其两种或更多种的组合使用。
[0041]
另外,上述步骤(s1)可以在可用于活性亚甲基化合物的烷基化反应的溶剂中进行,且溶剂的实例可包括甲苯、二甲苯、苯、二氯甲烷、丁醇、四氢呋喃、乙酸乙酯、丙醇、环己酮、乙醚、二噁烷、乙醇、甲醇、吡啶、丙酮、乙腈、n,n-二甲基甲酰胺、二甲亚砜、其水溶液等,其可以单独使用或者以其两种或更多种的组合使用。
[0042]
上述步骤(s2)可以是通过将式c的化合物与式d的化合物偶联形成吡咯环的步骤,且能够用于吡咯环化反应的溶剂可包括例如甲醇、乙醚、乙酸乙酯、四氢呋喃、甲苯、庚烷、环己烷、己烷等,其可以单独使用或者以其两种或更多种的组合使用。在这种情况下,吡咯环化反应可以在酸性条件下进行,其中可以使用新戊酸、甲苯-4-磺酸等。
[0043]
上述步骤(s3)可以是从式e的化合物中除去烷基的步骤,并且可以在酸性条件下进行。在这种情况下,用于酸性条件的酸可包括盐酸、溴酸、碘酸、硫酸、乙酸、三氟乙酸等,其可单独使用或者以其两种或更多种的组合使用。
[0044]
上述步骤(s4)可以是选择性还原式f中的羧基的步骤,并且可以在作为还原剂的硼-二甲硫醚(boron-dimethylsulfide,dms)络合物或硼-四氢呋喃络合物的存在下进行,且其中所用的溶剂可以包括烃溶剂(例如,己烷、正庚烷或甲苯)、醚溶剂(例如,四氢呋喃、二噁烷或乙醚)等,其可以单独使用或者以其两种或更多种的组合使用。
[0045]
上述步骤(s5)可以是使式g的化合物中的缩酮基团被酸开环的步骤,且其中所用
的溶剂可以包括极性溶剂(例如,甲醇、乙醇或异丙醇)、醚溶剂(例如,四氢呋喃、二噁烷或乙醚)等,其可以单独使用或者以其两种或更多种的组合使用。
[0046]
上述步骤(s6)可以是制备由式1所示的(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐的步骤,所述(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐是通过将式h的化合物在碱性条件下水解,然后向其中加入钙离子源而获得的最终产物。在这种情况下,水解反应和盐制备反应可以在溶剂(诸如甲醇、四氢呋喃、水等,其可以单独使用或者以其两种或更多种的组合使用)中进行。
[0047]
在上述步骤(s6)中使用的钙离子源的实例可以包括乙酸钙、氯化钙、碳酸钙、磷酸三钙、柠檬酸钙、马来酸柠檬酸钙、乳酸钙、葡萄糖酸钙、氢氧化钙、草酸钙、氟化钙、硫酸钙、其水溶液等,其可以单独使用或者以其两种或更多种的组合使用。
[0048]
本发明的包括步骤(s1)至(s6)的制备式1化合物的方法可由以下反应式1表示。
[0049]
[反应式1]
[0050][0051]
式c的化合物可以用作用于制备本发明的(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐的中间体,并且可以通过使式a的化合物与式b的化合物反应来制备。
[0052]
制备式c的化合物的方法可由以下反应式2表示。
[0053]
[反应式2]
[0054][0055]
式a的化合物可通过使下式a-1的化合物与下式a-2的化合物反应来制备。
[0056]
[式a-1]
[0057]
(其中r1为c
1-c6烷基)
[0058]
[式a-2]
[0059]
(其中r3为-o-(c
1-c5烷基)、cl或br)
[0060]
式a-1的化合物可以通过使用下式a-3的化合物来制备。
[0061]
[式a-3]
[0062][0063]
式a-1的化合物可以通过酯化式a-3的化合物来制备。
[0064]
制备式a的化合物的方法可由以下反应式3表示。
[0065]
[反应式3]
[0066][0067]
式b的化合物可通过使下式b-1的化合物与下式b-2的化合物反应以制备式b-3的化合物,并卤化式b-3的化合物来制备。
[0068]
[式b-1]
[0069][0070]
[式b-2]
[0071][0072]
[式b-3]
[0073][0074]
制备式b的化合物的方法可由以下反应式4表示。
[0075]
[反应式4]
[0076][0077]
根据反应式4,以式b-1的化合物为起始原料,其可与式b-2的化合物进行friedel-crafts酰化反应以制备式b-3的化合物,然后可进行卤化反应以制备式b的化合物。
[0078]
有益效果
[0079]
根据本发明,用于制备(3r,5r)-7-[2-(4-氟苯基)-5-异丙基-3-苯基-4-[(4-羟甲基苯基氨基)羰基]-吡咯-1-基]-3,5-二羟基庚酸半钙盐的方法可以以汇聚合成方式进行,其中化合物的每个主要结构部分独立地合成然后偶联,由此与常规文献中公开的顺序合成方式相比,具有容易控制主要相关物质的优点。此外,本发明可以通过降低顺序合成路线中固有的风险因素(诸如在合成中途失败的情况下必须返回到第一路线并再次重复合成)来缩短制备时间。
具体实施方式
[0080]
在下文中,将通过具体实施例更详细地描述本发明。除非另外定义,否则本文使用的所有术语(包括技术或科学术语)具有与本发明所属领域的普通技术人员通常理解的相同的含义。除非在本技术中明确定义,否则在通常使用的字典中定义的这些术语应解释为具有与相关领域中的上下文含义相同的含义,并且不应被解释为具有理想的或过度正式的含义。
[0081]
实施例1:4-(2-(2-(4-氟苯基)-2-氧代-1-苯基乙基)-4-甲基-3-氧代戊酰胺基)苯甲酸甲酯的制备
[0082]
(步骤1-1):4-氨基苯甲酸甲酯的制备
[0083]
将4-氨基苯甲酸(120g,1.0eq)用甲醇(1.2l)稀释后,在0至5℃搅拌下溶解所得溶液。向其中缓慢加入socl2(125g,1.2eq)并升高温度以在回流下搅拌所得混合物2小时。在确认反应完成后,将所得溶液冷却至20至25℃,减压干燥,并在搅拌下加入水(1.2l)。加入
碳酸氢钠水溶液以在0至5℃达到ph 8以制备固体,之后加入水(1.2l)并搅拌30分钟。将所得固体用水充分洗涤,过滤,然后在50℃下真空干燥16小时,得到4-氨基苯甲酸甲酯。
[0084]
固体128g(收率96.8%),水分含量0.7%(《1.0%)
[0085]
(步骤1-2):4-(4-甲基-3-氧代戊酰胺基)苯甲酸甲酯的制备
[0086]
将上述步骤1-1中得到的4-氨基苯甲酸甲酯(80g,1.003eq)和4-甲基-3-氧代戊酸甲酯(76g,1.0eq)用甲苯(0.76l)稀释,并在室温下搅拌溶解。使用dean-stark分水器(dean-stark trap)将反应溶液回流一天以除去水分。在确认反应完成后,将所得产物冷却至20至25℃,减压干燥,并通过加入乙酸乙酯(0.76l)溶解,之后将有机层用20%盐酸水溶液洗涤两次。将萃取的有机层用水(0.15l)、10%碳酸氢钠水溶液(0.15l)和饱和氯化钠水溶液(0.06l)洗涤,用硫酸镁干燥,然后过滤。将滤液减压干燥后,加入庚烷(0.76l),并在室温下搅拌10小时,得到固体。将所得固体用庚烷彻底洗涤并过滤。将所得产物在50℃下真空干燥16小时,得到4-(4-甲基-3-氧代戊酰胺基)苯甲酸甲酯。
[0087]
固体118.3g(收率85%)
[0088]1h nmr(500mhz,dmso-d6)10.40(nh,br s,1h),7.92(d,j=8.5hz,2h),7.71(d,j=8.5hz,2h),3.82(s,3h),3.67(s,2h),2.75(sep,j=7.0hz,1h),1.05(d,j=7.0hz,6h),mh+264.
[0089]
(步骤1-3):2-溴-1-(4-氟苯基)-2-苯基乙-1-酮的制备
[0090]
步骤a:1-(4-氟苯基)-2-苯基乙-1-酮的制备
[0091]
在0℃下在氮气下将氟苯(100g)加入反应器中,然后加入氯化铵(103g)。冷却至-10℃后,滴加苯乙酰氯(110.75g)并搅拌2小时。向反应溶液中加入冰水(600g)和盐酸(0.03l)以终止反应。加入二氯甲烷(1l)以分离有机层,并将水层用二氯甲烷(0.5l)再洗涤一次。将萃取的有机层用5%碳酸氢钠水溶液(0.6l)和5%饱和氯化钠水溶液(0.4l)洗涤,用硫酸镁干燥,然后通过硅藻土过滤。将滤液加入到反应器中并干燥,得到1-(4-氟苯基)-2-苯基乙-1-酮。
[0092]
固体150g(收率98%)
[0093]1h nmr(500mhz,dmso-d6)8.15-8.11(m,2h),7.39-7.34(m,2h),7.33-7.30(m,2h),7.27-7.21(m,3h),4.39(s,2h),mh+215
[0094]
步骤b:2-溴-1-(4-氟苯基)-2-苯基乙-1-酮的制备
[0095]
将溴(109g)用二氯甲烷(0.2l)稀释,滴加到上述步骤a中得到的1-(4-氟苯基)-2-苯基乙-1-酮中,搅拌10分钟。冷却至15℃后,加入5%硫酸钠(0.4l)和二氯甲烷以分离有机层。将萃取的有机层用5%碳酸氢钠水溶液(0.4l)和5%饱和氯化钠水溶液洗涤,并减压浓缩。将浓缩物用己烷(2l)稀释以制备浆液,然后静置一天。将所得固体过滤并在50℃下真空干燥4小时,得到2-溴-1-(4-氟苯基)-2-苯基乙-1-酮。
[0096]
固体171g(收率81.4%)
[0097]1h nmr(500mhz,dmso-d6):8.18-8.14(m,2h),7.55-7.53(m,2h),7.40-7.31(m,5h),7.18(s,1h).
[0098]
(步骤1-4):4-(2-(2-(4-氟苯基)-2-氧代-1-苯基乙基)-4-甲基-3-氧代戊酰胺基)苯甲酸甲酯的制备
[0099]
将在上述步骤1-2中得到的4-(4-甲基-3-氧代戊酰胺基)苯甲酸甲酯(167g)用丙
酮(600g)稀释,然后加入碳酸钾(131.5g)并在20℃下搅拌。冷却至18℃后,滴加上述步骤1-3中得到的2-溴-1-(4-氟苯基)-2-苯基乙-1-酮(bfpe,186g)(其用丙酮(0.2l)稀释),并将反应溶液搅拌6小时,过滤并干燥。将浓缩物用乙酸乙酯(1.6l)和蒸馏水(0.8l)洗涤,并将水层用乙酸乙酯(0.3l)再洗涤一次。将萃取的有机层减压浓缩,用甲醇:蒸馏水=9:1(v:v,1.25l)溶液稀释,并回流直至完全溶解。将反应溶液冷却至0至5℃,搅拌2小时,用甲醇:蒸馏水=9:1(0.167l)冷溶液和冰水(0.167l)洗涤,并过滤。将滤液用己烷(0.835l)稀释以制备浆液,并在50℃下真空干燥一天,得到4-(2-(2-(4-氟苯基)-2-氧代-1-苯基乙基)-4-甲基-3-氧代戊酰胺基)苯甲酸甲酯。
[0100]
固体224g(收率74.3%)
[0101]1h nmr(500mhz,dmso-d6)10.52(nh,br s,1h),8.15-8.11(m,2h),7.84(d,j=9.0hz,2h),7.47(d,j=9.0hz,2h),7.35(d,j=7.5hz,2h),7.33-7.30(m,2h),7.24-7.21(m,3h),5.43(d,j=11.0hz,1h),4.91(d,j=11.0hz,1h),3.80(s,3h),2.90(sep,j=7.0hz,1h),1.17(d,j=7.0hz,3h),0.93(d,j=7.0hz,3h),mh+476.
[0102]
实施例2:4-(2-(2-(4-氟苯基)-2-氧代-1-苯基乙基)-4-甲基-3-氧代戊酰胺基)苯甲酸甲酯的制备
[0103]
(步骤2-1):2-氯-1-(4-氟苯基)-2-苯基乙-1-酮的制备
[0104]
在氯化钙保护管下加入硫酰氯(100g)。将通过与步骤1-3中的步骤a基本相同的方法得到的1-(4-氟苯基)-2-苯基乙-1-酮(150g)用二氯甲烷(0.23l)稀释并在10至15℃下滴加4分钟。在室温下搅拌2.5小时后,向反应溶液中加入冰水(900g)以终止反应。用二氯甲烷(0.6l)稀释后,将有机层用5%碳酸氢钠水溶液(0.3l)和5%饱和氯化钠水溶液(0.3l)洗涤,用硫酸镁干燥,并通过硅藻土过滤。将滤液减压干燥,然后真空干燥,得到2-氯-1-(4-氟苯基)-2-苯基乙-1-酮。
[0105]
固体156g(收率90%)
[0106]1h nmr(500mhz,dmso-d6)8.15-8.11(m,2h),7.50-7.47(m,2h),7.40-7.31(m,5h),7.13(s,1h).
[0107]
(步骤2-2):4-(2-(2-(4-氟苯基)-2-氧代-1-苯基乙基)4-甲基-3-氧代戊酰胺基)苯甲酸甲酯的制备
[0108]
将在上述步骤1-2中得到的4-(4-甲基-3-氧代戊酰氨基)苯甲酸甲酯(110g)用丙酮(0.66l)稀释,然后加入碳酸钾(86.6g),并在20至25℃下搅拌。在保持相同温度的同时,滴加用丙酮(0.22l)稀释的2-氯-1-(4-氟苯基)-2-苯基乙-1-酮(cfpe,104g)。将反应溶液回流8小时,冷却至20至25℃并过滤。将滤液浓缩,用乙酸乙酯(1.0l)和蒸馏水(0.5l)稀释,搅拌10分钟,并洗涤。将水层用乙酸乙酯(0.18l)再洗涤一次。将萃取的有机层减压浓缩,用甲醇:蒸馏水=4:1(v:v,0.55l)溶液稀释,并回流1小时。将反应溶液缓慢冷却至0至5℃,搅拌2小时,用甲醇:蒸馏水=4:1(0.11l)溶液和冰水(0.11l)洗涤,并过滤。将滤液用己烷(0.55l)稀释以制备浆液,并在50℃下真空干燥8小时,得到4-(2-(2-(4-氟苯基)-2-氧代-1-苯基乙基)-4-甲基-3-氧代戊酰胺基)苯甲酸甲酯。
[0109]
固体157g(收率79%)
[0110]1h nmr(500mhz,dmso-d6)10.52(nh,br s,1h),8.15-8.11(m,2h),7.84(d,j=9.0hz,2h),7.47(d,j=9.0hz,2h),7.35(d,j=7.5hz,2h),7.33-7.30(m,2h),7.24-7.21
(m,3h),5.43(d,j=11.0hz,1h),4.91(d,j=11.0hz,1h),3.80(s,3h),2.90(sep,j=7.0hz,1h),1.17(d,j=7.0hz,3h),0.93(d,j=7.0hz,3h),mh+476.
[0111]
实施例3:(3r,5r)-7-(2-(4-氟苯基)-5-异丙基-3-苯基-4-((4-羟甲基苯基氨基)羰基)-吡咯-1-基)-3,5-二羟基庚酸半钙盐的制备
[0112]
(步骤3-1):4-(1-(2-((4r,6r)-6-(2-(叔丁氧基)-2-氧代乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-4-苯基-1h-吡咯-3-甲酰胺基)苯甲酸甲酯的制备
[0113]
将根据实施例1得到的4-(2-(2-(4-氟苯基)-2-氧代-1-苯基乙基)-4-甲基-3-氧代戊酰胺基)苯甲酸甲酯(800g)、2-((4r,6r)-6-(2-氨基乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙酸叔丁酯(552.5g)和特戊酸(177.2g)用庚烷(6.72l)、甲苯(2.4l)和四氢呋喃(2.4l)稀释。使用dean-stark分水器将反应溶液回流一天以除去水分。将反应溶液冷却至室温并减压浓缩。将浓缩物溶于乙酸乙酯(8.0l)中,搅拌,然后用蒸馏水(4l)洗涤两次。将萃取的有机层减压浓缩以得到固体,然后将其用异丙醇(4l)稀释并在50℃下浆化1小时。向反应混合物中加入己烷(4l),并在相同温度下进一步浆化1小时。将反应物冷却至室温并进一步搅拌1小时。将所得固体过滤,用己烷(4l)洗涤两次,并在50℃下干燥一天,得到4-(1-(2-((4r,6r)-6-(2-(叔丁氧基)-2-氧代乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-4-苯基-1h-吡咯-3-甲酰胺基)苯甲酸甲酯。
[0114]
固体845.5g(收率70.5%)
[0115]1h nmr(500mhz,dmso-d6)10.17(nh,br s,1h),7.83(d,j=9.0hz,2h),7.64(d,j=8.0hz,2h),7.28-7.25(m,2h),7.20(t,j=9.0hz,2h),7.08-7.03(m,4h),7.01-6.97(m,1h),4.13-4.08(m,1h),3.95-3.89(m,1h),3.80(s,3h),3.80-3.74(m,2h),3.25-3.19(m,1h),2.30(dd,j=15.0hz,5.0hz,1h),2.17(dd,j=15.0hz,8.0hz,1h),1.64-1.50(m,2h),1.38(s,9h),1.36(d,j=7.0hz,6h),1.35-1.30(m,1h),1.31(s,3h),1.16(s,3h),0.92(q,j=12.0hz,1h),mh-711.
[0116]
(步骤3-2):4-(1-(2-((4r,6r)-6-(2-(叔丁氧基)-2-氧代乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-4-苯基-1h-吡咯-3-甲酰胺基)苯甲酸的制备
[0117]
将上述步骤3-1中得到的4-(1-(2-((4r,6r)-6-(2-(叔丁氧基)-2-氧代乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-4-苯基-1h-吡咯-3-甲酰胺基)苯甲酸甲酯(700g)用乙腈(7l)稀释并搅拌。将氢氧化钠(98.3g)用蒸馏水(2.1l)稀释,滴加到反应溶液中,并在50℃下搅拌5小时。将反应溶液冷却至室温,并加入蒸馏水(7l)以终止反应。将20%乙酸水溶液(0.263l乙酸,1.05l蒸馏水)滴加到反应溶液中以调节ph至5.5-6.0,并在室温下搅拌3小时。将所得固体过滤,用蒸馏水(2.8l)洗涤四次,再用乙腈(2.1l)洗涤两次。将所得固体用95%乙醇(3.5l)稀释,并回流1小时,将反应溶液冷却至室温并搅拌3小时。将所得固体过滤,并在50℃下真空干燥一天,得到4-(1-(2-((4r,6r)-6-(2-(叔丁氧基)-2-氧代乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-4-苯基-1h-吡咯-3-甲酰胺基)苯甲酸。
[0118]
固体790g(收率71.5%)
[0119]1h nmr(500mhz,dmso-d6)10.13(nh,br s,1h),7.80(d,j=8.5hz,2h),7.62(d,j=
9.0hz,2h),7.28-7.25(m,2h),7.22-7.18(m,2h),7.08-7.04(m,4h),7.01-6.98(m,1h),4.13-4.08(m,1h),3.96-3.89(m,1h),3.81-3.75(m,2h),3.22(sep,j=7.0hz,1h),2.30(dd,j=15.0hz,5.0hz,1h),2.18(dd,j=15.0hz,8.0hz,1h),1.64-1.50(m,2h),1.38(s,9h),1.36(d,j=7.0hz,6h),1.36-1.30(m,1h),1.31(s,3h),1.16(s,3h),0.93(q,j=12.0hz,1h),mh-698.
[0120]
(步骤3-3):2-((4r,6r)-6-(2-(2-(4-氟苯基)-4-((4-(羟基甲基)苯基)氨基甲酰基)-5-异丙基-3-苯基-1h-吡咯-1-基)乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙酸叔丁酯的制备
[0121]
将在上述步骤3-2中得到的4-(1-(2-((4r,6r)-6-(2-(叔丁氧基)-2-氧代乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙基)-5-(4-氟苯基)-2-异丙基-4-苯基-1h-吡咯-3-甲酰胺基)苯甲酸(1000g)用四氢呋喃(10l)稀释并冷却至0℃。加热至40-45℃后,滴加硼烷二甲硫醚(2m四氢呋喃溶液,1.79l),然后搅拌一天。将反应溶液冷却至0℃后,缓慢滴加甲醇(2l)以终止反应。将反应溶液减压浓缩,并用异丙醇:蒸馏水=4:1(v:v,12l)溶液稀释,然后滴加乙酸(0.087l)。向反应溶液中加入氢氧化钙(213g),回流1小时,然后在高温下过滤。将过滤的固体用异丙醇:蒸馏水=1:1(5l)溶液再洗涤一次。将滤液用蒸馏水(4l)稀释,并在室温下搅拌3小时,然后将所得固体过滤并用冷异丙醇:蒸馏水=1:1(2l)洗涤。将所得固体在50℃下真空干燥一天,得到2-((4r,6r)-6-(2-(2-(4-氟苯基)-4-((4-(羟基甲基)苯基)氨基甲酰基)-5-异丙基-3-苯基-1h-吡咯-1-基)乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙酸叔丁酯。
[0122]
固体884g(收率90%)
[0123]1h nmr(500mhz,dmso-d6)9.78(nh,br s,1h),7.46(d,j=8.5hz,2h),7.27-7.24(m,2h),7.21-7.15(m,4h),7.09-7.05(m,4h),7.02-6.99(m,1h),5.05(oh,t,j=5.5hz,1h),4.39(d,j=5.5hz,2h),4.13-4.08(m,1h),3.95-3.89(m,1h),3.80-3.74(m,2h),3.21(sep,j=7.0hz,1h),2.30(dd,j=15.0hz,5.0hz,1h),2.19-2.15(m,1h),1.63-1.50(m,2h),1.38(s,9h),1.36(d,j=7.0hz,6h),1.35-1.30(m,1h),1.31(s,3h),1.16(s,3h),0.96-0.89(m,1h).
[0124]
(步骤3-4):(3r,5r)-7-(2-(4-氟苯基)-4-((4-(羟基甲基)苯基)氨基甲酰基)-5-异丙基-3-苯基-1h-吡咯-1-基)-3,5-二羟基庚酸叔丁酯的制备
[0125]
将上述步骤3-3中得到的2-((4r,6r)-6-(2-(2-(4-氟苯基)-4-((4-(羟基甲基)苯基)氨基甲酰基)-5-异丙基-3-苯基-1h-吡咯-1-基)乙基)-2,2-二甲基-1,3-二噁烷-4-基)乙酸叔丁酯(605g)用四氢呋喃(3l)、甲醇(3l)和蒸馏水(3l)稀释,然后滴加1n稀盐酸(0.442l)并在45℃下搅拌2小时。通过向反应溶液中加入碳酸氢钠(48.3g)以终止反应,冷却至室温,并搅拌。将所得混合物减压浓缩直至溶剂的体积减少约1/2,然后加入10%乙酸乙酯/己烷(6l)。将反应溶液在50℃下搅拌30分钟,冷却至室温,并再搅拌1小时。将所得固体过滤并用蒸馏水(0.6l)洗涤,然后将所得固体用己烷(1.2l)稀释以制备浆液,然后将其在40℃下搅拌2小时并再次过滤。将所得固体在50℃下真空干燥一天,得到(3r,5r)-7-(2-(4-氟苯基)-4-((4-(羟基甲基)苯基)氨基甲酰基)-5-异丙基-3-苯基-1h-吡咯-1-基)-3,5-二羟基庚酸叔丁酯。
[0126]
固体516.5g(收率90.6%)
[0127]1h nmr(500mhz,dmso-d6)9.76(nh,br s,1h),7.45(d,j=8.5hz,2h),7.26-7.23(m,2h),7.20-7.15(m,4h),7.09-7.05(m,4h),7.01-6.98(m,1h),5.05(oh,t,j=6.0hz,1h),4.68(d,j=5.0hz,1h),4.62(d,j=5.0hz,1h),4.39(d,j=6.0hz,2h),3.96-3.90(m,1h),3.83-3.73(m,2h),3.54-3.48(m,1h),3.22(sep,j=7.0hz,1h),2.26-2.17(m,2h),1.65-1.58(m,1h),1.56-1.48(m,1h),1.45-1.39(m,1h),1.39(s,9h),1.37(d,j=7.0hz,6h),1.31-1.25(m,1h).
[0128]
(步骤3-5):(3r,5r)-7-(2-(4-氟苯基)-5-异丙基-3-苯基-4-((4-羟甲基苯基氨基)羰基)-吡咯-1-基)-3,5-二羟基庚酸半钙盐的制备
[0129]
将以上步骤3-4中得到的(3r,5r)-7-(2-(4-氟苯基)-4-((4-(羟基甲基)苯基)氨基甲酰基)-5-异丙基-3-苯基-1h-吡咯-1-基)-3,5-二羟基庚酸叔丁酯(1040g)用四氢呋喃(5l)和蒸馏水(5l)稀释并搅拌。在25至30℃下将氢氧化钠(77.5g)用蒸馏水(1.632l)稀释,然后将其滴加到反应溶液中并搅拌3小时。将反应溶液减压浓缩,然后加入蒸馏水(5l)和乙酸乙酯(5l)并搅拌10分钟以终止反应。分离有机层,并将水层用乙酸乙酯(5l)洗涤两次。将乙酸钙(153g)溶解于蒸馏水(5l)中,然后缓慢滴加到水层中。将反应溶液在室温下制成浆液一天,然后将所得固体过滤并用少量蒸馏水洗涤。将所得固体用乙酸乙酯(7l)、甲醇(0.7l)和蒸馏水(7l)稀释,然后向其中加入丁羟茴醚(1.5g)并回流。将反应溶液冷却至室温并搅拌3小时。将所得固体过滤,用少量水洗涤,并在50℃下真空干燥一天,得到(3r,5r)-7-(2-(4-氟苯基)-5-异丙基-3-苯基-4-((4-羟甲基苯基氨基)羰基)-吡咯-1-基)-3,5-二羟基庚酸半钙盐。
[0130]
固体852g(收率81.5%)
[0131]1h nmr(500mhz,dmso-d6)9.82(nh,br s,1h),7.46(d,j=8.5hz,2h),7.26-7.23(m,2h),7.21-7.14(m,4h),7.09-7.05(m,4h),7.01-6.98(m,1h),5.05(t,j=5.5hz,1h),4.39(d,j=5.5hz,2h),3.97-3.91(m,1h),3.79-3.73(m,1h),3.72-3.67(m,1h),3.54-3.50(m,1h),3.25-3.19(m,1h),2.02(dd,j=15.0hz,4.0hz,1h),1.87(dd,j=15.0hz,8.0hz,1h),1.63-1.56(m,1h),1.53-1.46(m,1h),1.40-1.35(m,1h),1.36(d,j=7.0hz,6h),1.22-1.16(m,1h),mh-587(acid mass).
[0132]
本文已经参考优选的示例性实施方案描述了本发明,但是本领域技术人员将理解,在不脱离本发明的精神和领域的情况下,可以对本发明进行各种改变和修改,如在所附专利权利要求的范围中所描述的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1