具有密度和/或泡孔形态梯度的膨胀珠粒以及由其获得的烧结泡沫的制作方法
1.本发明涉及制造由具有密度和/或泡孔(cell)形态梯度的膨胀珠粒获得的烧结的发泡的聚合物材料的方法。
2.特别地,该方法使用以时变(time-varying)条件为特征的使一种或更多种物理发泡剂在可膨胀聚合物材料的颗粒中溶解(solubilization)的步骤,随后使所述颗粒膨胀以及对由此获得的膨胀珠粒进行烧结的步骤。溶解步骤的时变条件产生在颗粒中物理发泡剂的浓度的非均匀分布,其在膨胀时在膨胀珠粒中产生相应的非均匀密度和/或形态,并且其在烧结时产生相对于由具有均匀泡孔密度和形态以及相等平均密度的膨胀珠粒获得的相同发泡且烧结的制品具有改善的结构和功能特性的发泡烧结的制品。
背景技术:
3.近来,人们对“梯度”发泡材料产生了兴趣,与以在密度和泡孔形态方面的均匀结构为特征的发泡材料相比,“梯度”发泡材料的结构和功能特性得到改善。
4.通过最近的科学研究(理论-数值和实验二者)都已证明这一点,例如liang cui等,"designing the energy absorption capacity of functionallygraded foam materials",materials science and engineering:a. 507(1-2):215-225,2009年5月。专利和科学文献描述了这样的层状泡沫结构或具有梯度形态和/或密度的用途和优点。
5.专利申请公开第us2015125663号描述了“梯度”组装的不同层的聚合物泡沫在吸收头盔中的撞击能量中的用途。
6.层状泡沫令人感兴趣的另一个领域是通过烧结的膨胀的领域(“蒸汽室模塑”或“珠粒发泡”的技术),这在烧结的发泡聚苯乙烯制品的生产中非常普遍,但最近,除了别的以外还使用聚丙烯、热塑性聚氨酯和聚乳酸。
7.在该领域中,使用预膨胀珠粒(具有基本上圆柱形、椭圆形或球形的形状和约数毫米的尺寸),在制品的生产中,将其插入模具中并通过水蒸气或热气包围,以进行珠粒的最终膨胀和烧结。最终膨胀通过仍然以溶液包含在预膨胀珠粒中的发泡剂(其随加热而放出)诸如例如戊烷进行,但也通过加热之后泡沫中包含的气体的热膨胀进行。
8.us2015252163描述了包含含有热塑性聚氨酯泡沫颗粒的聚氨酯基体的混杂材料、用于这样的混杂材料的制造方法、以及这样的混杂材料的用途(例如自行车鞍座、室内装饰品和鞋底)。
9.us9079360描述了使用包括圆柱形发泡中心层和覆盖其的非膨胀外层的预膨胀聚烯烃珠粒生产印刷制品的方法,其中预膨胀珠粒通过共挤出法来获得。
10.代表同一申请人的于2018年4月19日提交的专利申请it 102018000004727描述了用于获得层状聚合物发泡材料的方法,所述层状聚合物发泡材料包括不同密度和/或形态的至少两个层。
技术实现要素:
11.在进一步研究之后,申请人出乎意料地观察到,由将通过专利申请it 102018000004727中描述的方法获得的多层珠粒烧结而制成的最终制品具有与由具有均匀的形态和密度的珠粒开始可实现的那些不同的特别且独特的机械特性。
12.固定起始聚合物,就泡沫的数量和尺寸而言,均匀泡沫的机械特性在很大部分上取决于泡沫的密度,而少数上(通常可忽略不计)取决于形态。对于通过烧结产生的泡沫(例如,所谓的eps、可膨胀聚苯乙烯)以及对于例如通过挤出产生的整体式泡沫(所谓的xps)二者都是如此。
13.考虑到,例如,均匀泡沫的压缩弹性模量(或刚度),根据既定文献 [l.j.gibson,m.f.ashby.cellular solids.structure&properties.pergamonpress,oxford 1988]其随着泡沫与致密聚合物之间的密度比的平方而变化。诸如尺寸分布和平均孔径的泡沫的其他特性具有较小影响。然后,对于相同的密度,很难获得在刚度上的变化。类似的考虑适用于许多其他特性,例如屈服强度、疲劳强度、剪切模量、弯曲强度、平台应力、热导率等。
[0014]
另一方面,申请人出乎意料地观察到,对于相同的平均泡沫密度,由多层珠粒制成的最终产品的刚度根据所使用的珠粒的梯度形态和密度而变化。
[0015]
特别地,申请人观察到,对于相同的平均密度,当珠粒的外层比内层更致密时实现较大的刚度,而当珠粒的外层比内层不那么致密时实现较小的刚度。
[0016]
因此,本发明的第一目的是通过使用一种或更多种发泡剂制备包含烧结的膨胀珠粒的发泡聚合物材料的方法,其特征在于该方法包括以下步骤:
[0017]
提供呈颗粒形式的可膨胀聚合物材料,
[0018]
在时变压力分布(time-varying pressure profile)的情况下使所述一种或更多种发泡剂在可膨胀聚合物材料中溶解,
[0019]
通过将压力瞬时释放或者通过压力释放和随后的加热使所述颗粒膨胀以形成所述膨胀珠粒,以及
[0020]
将所述膨胀珠粒烧结在一起,优选在高于30℃的温度下将所述膨胀珠粒烧结在一起。
[0021]
本发明的第二目的由用根据本发明的第一目的的方法获得的包含烧结的膨胀珠粒的发泡聚合物材料表示,其中对于相同的平均密度,所述发泡聚合物材料显示出取决于所述时变压力分布的机械和功能特性。
[0022]
本发明的第三目的由特征在于所述烧结的膨胀珠粒之间的焊接层的密度大于或小于所述发泡聚合物材料的平均密度的包含烧结的膨胀珠粒的发泡聚合物材料表示。
[0023]
本发明的第四目的由全部或部分由根据本发明的第二或第三目的的聚合物材料制成的制成制品表示。
附图说明
[0024]
图1a示出了本发明中使用的不连续发泡装置的照片。
[0025]
图1b示出了本发明中使用的不连续发泡装置的图。
[0026]
图1c示出了在其中安置颗粒的圆柱形模具的图。
[0027]
图2示出了由具有梯度形态的膨胀珠粒的烧结获得的产品,以及在每个烧结产品
下方对应的珠粒:a)实施例1;b实施例2;c)实施例3;d) 实施例4。
[0028]
图3a示出了在实施例1所示条件的情况下获得的tpu样品的截面的sem图像。
[0029]
图3b示出了图3a的显示膨胀珠粒之间的互连/焊接区域的放大。
[0030]
图4a示出了在实施例2所示条件的情况下获得的tpu样品的截面的 sem图像。
[0031]
图4b示出了图4a的显示膨胀珠粒之间的互连/焊接区域的放大。
[0032]
图5a示出了在实施例3所示条件的情况下获得的tpu样品的截面的 sem图像。
[0033]
图5b示出了图5a的显示膨胀珠粒之间的互连/焊接区域的放大图。
[0034]
图6a示出了在实施例4所示条件的情况下获得的tpu样品的截面的 sem图像。
[0035]
图6b示出了图6a的显示膨胀珠粒之间的互连/焊接区域的放大。
[0036]
图7a示出了在应力-应变图方面由实施例1至4中产生的tpu样品的单轴静态压缩测试获得的结果。
[0037]
图7b示出了在应力-应变图方面由实施例1至4中产生的tpu样品的静态单轴压缩测试获得的结果的小变形下的线性区域的放大。
[0038]
图8a示出了在实施例6所示条件下获得的单个ps颗粒的截面的光学显微镜图像。
[0039]
图8b和图8c示出了在实施例6所示条件下获得的烧结的ps样品的在不同放大倍数下的截面的光学显微镜图像。
[0040]
图9a示出了在实施例7所示条件下获得的单个ps颗粒的截面的光学显微镜图像。
[0041]
图9b和图9c示出了在实施例7所示条件下获得的烧结的ps样品的在不同放大倍数下的截面的光学显微图像。
[0042]
图10a和图10b示出了在实施例8所示条件下获得的烧结的ps样品的在不同放大倍数下的截面的光学显微图像。
[0043]
图11a、图11b和图11c示出了在实施例8所示条件下获得的烧结的 ps样品的在不同放大倍数下的截面的光学显微图像。
[0044]
图12a示出了在应力-应变图方面由实施例6和7中产生的样品的单轴静态压缩测试获得的的结果。
[0045]
图12b示出了在应力-应变图方面关于密度为230g/cm3的基于ps的泡沫由实施例6和7中产生的样品的静态单轴压缩测试获得的结果的小变形下的线性区域的放大。
[0046]
图12c示出了在应力-应变图方面由实施例8和9中产生的样品的单轴静态压缩测试获得的结果。
[0047]
图12d示出了在应力-应变图方面关于密度为110g/cm3的基于ps的泡沫由实施例8和9中产生的样品的静态单轴压缩测试获得的结果的小变形下的线性区域的放大。
[0048]
图13示出了在实施例11所示条件下获得的单个膨胀pp颗粒的截面的sem图像。
[0049]
图14a示出了在实施例12所示条件下获得的所得发泡pp珠粒的截面的sem图像。
[0050]
图14b示出了图14a的珠粒的焊接区的细节。
[0051]
图14c示出了图14a的珠粒的焊接区的细节。
[0052]
图15示出了对根据实施例12中描述的步骤发泡的所得珠粒进行的 dsc测试的图。
[0053]
图16示出了在实施例13所示条件下获得的单个膨胀pla颗粒的截面的sem图像。
[0054]
图17示出了在实施例14所示条件下获得的单个膨胀pla颗粒的截面的sem图像。
[0055]
图18示出了在实施例15所示条件下获得的单个膨胀pla颗粒的截面的sem图像。
[0056]
图19示出了在实施例16所示条件下获得的单个pla颗粒的截面的 sem图像。
[0057]
图20示出了在实施例17所示条件下获得的单个pla颗粒的截面的 sem图像。
[0058]
图21示出了在实施例18所示条件下获得的单个pla颗粒的截面的 sem图像。
[0059]
图22示出了对根据实施例12中描述的步骤发泡的所得珠粒进行的 dsc测试的图。
具体实施方式
[0060]
表述“聚合物材料”意指包括热塑性或热固性均聚物或共聚物或其混合物的聚合物材料。
[0061]
表述“发泡聚合物材料”是指其中例如通过发泡剂已经形成有泡沫的聚合物材料。
[0062]
表述“发泡剂”意指能够通过在聚合物材料内形成泡沫而引起聚合物材料膨胀的物质。
[0063]
表述“可膨胀聚合物材料”意指能够在一定温度下和在压力下吸收发泡剂,当释放压力时允许泡沫成核并在泡沫生长期间抵抗拉伸应力直至凝固的聚合物材料。
[0064]
术语“颗粒”表示基本上为球形、类球形、圆柱形或椭圆形形状的聚合物颗粒,优选最大直径与最小直径之间的平均变化低于20%,优选低于 15%。
[0065]
表述“多层结构”意指包括两个或更多个层,优选三个或更多个层的结构。
[0066]
表述“均匀组成”意指由在其所有点处均匀且恒定的组成的聚合物材料组成的组成。
[0067]
术语“不连续性”意指两个相邻层之间清晰且明显的边界,是单独制成的不同结构的两个层通过热接合或用粘合剂制成的复合材料的典型边界。
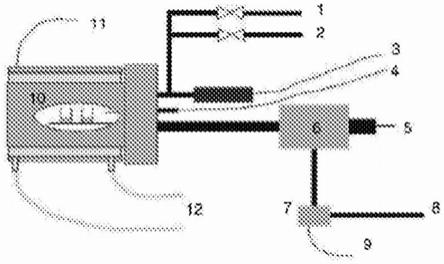
[0068]
术语“密度”意指给定要素的重量与该要素所占体积之间的比率,特别是本发明的发泡聚合物材料的层或区域的重量与其所占体积之间的比率。
[0069]
术语“平均密度”意指要素的表观密度,特别是本发明的包括具有不同密度和/或形态的区域和/或层的发泡聚合物材料的表观密度。
[0070]
术语“形态”表示发泡聚合物材料内形成的泡沫的形状、尺寸和每单位体积的数量。
[0071]
表述“焊接层”表示烧结的膨胀珠粒之间的珠粒间结合线。
[0072]
本发明的第一目的是通过使用一种或更多种发泡剂制备包含烧结的膨胀珠粒的发泡聚合物材料的方法,其特征在于该方法包括以下步骤:
[0073]
提供呈颗粒形式的可膨胀聚合物材料,
[0074]
在时变压力分布的情况下使所述一种或更多种发泡剂在可膨胀聚合物材料中溶解,
[0075]
通过将压力瞬时释放或者通过压力释放和随后的加热使所述颗粒膨胀以形成所述膨胀珠粒,以及
[0076]
将所述膨胀珠粒烧结在一起,优选在高于30℃的温度下将所述膨胀珠粒烧结在一起。
[0077]
根据本发明的第一目的,所述聚合物材料优选选自热塑性聚合物材料或热固性聚合物材料。
[0078]
有利地,所述热塑性聚合物材料选自包括聚烯烃、聚氨酯、聚酯和聚酰胺的组。
[0079]
优选地,所述热固性聚合物材料选自包括聚氨酯、环氧树脂、三聚氰胺树脂、聚酚(polyphenol)和聚酰亚胺的组。
[0080]
优选地,所述聚合物材料为苯乙烯、乙烯、丙烯和其他烯烃的聚合物和共聚物,例如聚苯乙烯、聚乙烯和聚丙烯。任选地,所述聚合物材料可以包含一种或更多种共聚单体。共聚单体可以包括例如烷基苯乙烯、二乙烯基苯、丙烯腈、二苯醚、α-甲基苯乙烯、或其组合。作为一个实例,聚合物材料可以包含约0重量%至约30重量%、优选按重量计约0.1%至按重量计约15%、并且更优选按重量计约1%至按重量计约10%的共聚单体。
[0081]
优选地,聚合物材料可以显示出约10,000道尔顿至约500,000道尔顿、更优选约150,000道尔顿至约400,000道尔顿、并且甚至更优选约200,000 道尔顿至约350,000道尔顿的分子量mw(通过gpc测量的)。
[0082]
有利地,聚合物材料显示出1.0g/10分钟至20g/10分钟的根据astmd 1238在200℃的温度和10kg的负载下测量的流动指数。
[0083]
根据本发明的第一目的,所述颗粒的最大直径为0.1mm至10mm,优选为0.5mm至5mm。
[0084]
根据本发明的第一目的,所述时变压力分布优选以周期性或非周期性方式随时间变化。
[0085]
根据本发明的第一目的,所述时变压力分布优选以具有选自正弦形、三角形、方形、锯齿型或其组合的波形的周期性方式随时间变化。
[0086]
根据本发明的第一目的,所述时变压力分布优选以按照线性、突变、曲线、抛物线、指数、脉冲形或其组合的非周期性方式随时间变化。
[0087]
根据本发明的第一目的,所述时变压力分布优选从等于大气压的最小压力变化至最大300巴,更优选从大气压变化至250巴,并且有利地从大气压变化至200巴。
[0088]
根据本发明的第一目的,所述时变压力分布优选包括具有压力分布随时间增加的至少一个步骤和具有压力分布随时间降低的至少一个步骤。
[0089]
根据本发明的第一目的,所述时变压力分布可以有利地包括具有压力分布随时间恒定的至少一个步骤。
[0090]
根据本发明的第一目的,溶解步骤用发泡剂、或者两种或更多种发泡剂的混合物进行,优选用两种发泡剂的混合物进行。溶解步骤可以有利地通过随时间改变发泡剂的浓度来进行。特别地,发泡剂混合物中发泡剂的浓度可以随时间变化。
[0091]
根据本发明的第一目的,溶解步骤优选在100℃至350℃、更优选 120℃至250℃、并且有利地130℃至200℃的温度下进行。在本发明的第一目的另一个实施方案中,例如当使用聚乳酸(pla)、聚(甲基丙烯酸甲酯)(pmma)、聚己内酯(pcl)和其他类似聚合物时,溶解步骤优选在
ꢀ‑
50℃至200℃、更优选0℃至150℃、并且有利地20℃至100℃的温度下进行。
[0092]
根据本发明的第一目的,一种或更多种发泡剂选自惰性气体、二氧化碳以及经取代或未经取代的具有3至8个碳原子的脂族烃(线性、支化或环状的)。
[0093]
发泡剂有利地选自氮气、二氧化碳、正丁烷、异丁烷、正戊烷和异戊烷。优选地,经取代的脂族烃包括卤代烃,特别是含氯烃、含氯氟烃和碳氟化合物,诸如,例如1,1,1,2-四氟乙烷(freon r-134a)、1,1-二氟乙烷 (freon r-152a)、二氟甲烷(freon r-32)、五氟乙烷(freon r-125)、六氟化硫。
[0094]
根据本发明的第一目的,膨胀步骤在第一实施方案中通过将压力瞬时释放或者在第二替代实施方案中通过压力释放和随后的加热来进行。
[0095]
如本领域中已知的,根据第一实施方案,当使用处于熔融、软化或溶胀状态的可膨胀聚合物材料时,膨胀珠粒的形成将在瞬时压力释放的同时发生。
[0096]
如在本领域中还已知的,根据第二实施例方案,当使用处于固态,诸如,例如玻璃态或半结晶态的可膨胀聚合物材料时,膨胀珠粒的形成将在加热时发生。
[0097]
根据本发明的第一目的,烧结步骤有利地在高于所述可膨胀聚合物材料的玻璃化转变温度的温度下进行。优选地,烧结步骤在20℃至250℃、更优选50℃至150℃的温度下进行,诸如例如对于聚乳酸在40℃至230℃、优选60℃至200℃的温度下进行,对于聚(甲基丙烯酸甲酯)在50℃至 180℃、优选70℃至160℃的温度下进行,对于聚己内酯在25℃至100℃、优选35℃至90℃的温度下进行,对于聚苯乙烯在90℃至130℃、优选100℃至110℃的温度下进行,对于热塑性聚氨酯在90℃至130℃、优选100℃至110℃的温度下进行,以及对于聚丙烯在110℃至160℃、优选120℃至 140℃的温度下进行。
[0098]
本发明的第二目的由用根据本发明的第一目的的方法获得的包含烧结的膨胀珠粒的发泡聚合物材料表示,其中对于相同的平均密度,所述发泡聚合物材料显示出取决于所述时变压力分布的机械和功能特性。
[0099]
换言之,通过本发明的方法实现的压力分布随时间的变化产生发泡剂的浓度分布,这导致发泡聚合物材料的机械和功能特性所依赖的密度和/ 或形态分布。
[0100]
特别地,根据本发明的第二目的,对于相同的平均密度,所述发泡聚合物材料显示出大于或小于在均匀压力分布的情况下获得的机械特性值的机械特性值。
[0101]
有利地,根据本发明的第二目的,当时变压力分布包括具有大于随后的第二饱和步骤的压力的压力的第一饱和步骤时,对于相同的平均密度,所述发泡聚合物材料显示出大于在均匀压力分布的情况下获得的机械特性值的机械特性值。
[0102]
或者,根据本发明的第二目的,当时变压力分布包括具有小于第二饱和步骤的压力的压力的第一饱和步骤时,对于相同的平均密度,所述发泡聚合物材料显示出小于在均匀压力分布的情况下获得的机械特性值的机械特性值。
[0103]
类似地,申请人发现通过随时间变化以不同的扩散率和溶解度为特征的两种或更多种发泡剂(例如氮气和二氧化碳)的分压,可以适当地调节发泡气体的组成的变化。
[0104]
以这种方式,根据本发明的第二目的,通过随时间变化以不同的扩散率和溶解度为特征的两种或更多种发泡剂(例如氮气和二氧化碳)的分压,可以获得这样的发泡聚合物材料:对于相同的平均密度,其显示出大于或小于在均匀压力分布的情况下获得的机械特性值的机械特性值。
[0105]
特别地,当时变压力分布包括具有大于具有较大溶解度的一种或更多种发泡剂的分压的第一饱和步骤和具有大于具有较小溶解度的一种或更多种发泡剂的分压的随后第二饱和步骤时,对于相同的平均密度,所述发泡聚合物材料显示出大于在均匀压力分布的情况下获得的机械特性值的机械特性值。
[0106]
或者,当时变压力分布包括具有大于具有较小溶解度的一种或更多种发泡剂的分压的第一饱和步骤和具有大于具有较大溶解度的一种或更多种发泡剂的分压的随后第二饱和步骤时,对于相同的平均密度,所述发泡聚合物材料显示出小于在均匀压力分布的情
况下获得的机械特性值的机械特性值。
[0107]
本发明的第三目的由以所述烧结的膨胀珠粒之间的焊接层为特征的包含烧结的膨胀珠粒的发泡聚合物材料表示,所述焊接层的密度大于或小于所述发泡聚合物材料的平均密度。
[0108]
优选地,所述焊接层显示出0.01μm至1000μm、更优选0.1μm至500 μm、并且甚至更优选1μm至100μm的厚度。
[0109]
有利地,根据本发明的第二方面和第三方面,所述烧结的膨胀珠粒包括焊接层和所述焊接层的内部,所述焊接层的内部包括至少一个膨胀层,其中所述焊接层的密度大于所述内部的密度。
[0110]
或者,根据本发明的第二方面和第三方面,所述烧结的膨胀珠粒包括焊接层和所述焊接层的内部,所述焊接层的内部包括至少一个膨胀层,其中所述焊接层的密度小于所述内部的密度。
[0111]
根据本发明的第二方面和第三方面,所述烧结的膨胀珠粒可以有利地包括包括有具有不同密度和/或形态的至少两个层并且显示出逐渐变化的密度和/或形态的所述焊接层的内部。
[0112]
所述烧结的膨胀珠粒有利地包括所述焊接层的内部,所述焊接层的内部包括至少一个具有较低密度和较精细形态的层以及至少一个具有较高密度和较粗糙形态的层。
[0113]
所述烧结的膨胀珠粒有利地包括所述焊接层的内部,所述焊接层的内部包括至少一个具有较低密度和较粗糙形态的层以及至少一个具有较高密度和较精细形态的层。
[0114]
有利地,所述烧结的膨胀珠粒包括所述焊接层的内部,所述焊接层的内部包括具有均匀形态的至少一个具有较低密度的层和至少一个具有较高密度的层。
[0115]
有利地,所述烧结的膨胀珠粒包括所述焊接层的内部,所述焊接层的内部包括具有均匀密度的至少一个具有较粗糙形态的层和至少一个具有较精细形态的层。
[0116]
有利地,所述具有不同密度和/或形态的至少两个层之间的界面未显示出形态和/或密度的不连续性。
[0117]
此外,申请人注意到由于不同的发泡程度和用不同发泡剂的处理,根据本发明的第二和第三目的的烧结的膨胀珠粒具有包括具有不同结晶结构和/或结晶度的层的内部。
[0118]
更特别地,所述烧结的膨胀珠粒包括内部,所述内部包括具有不同结晶度的至少两个层,诸如例如其中结晶度高于外层的内层,或者反之亦然。
[0119]
因此,根据本发明的第二和第三目的的发泡聚合物材料包括以包括具有不同结晶度的至少两个层的内部为特征的烧结的膨胀珠粒。
[0120]
此外,申请人还注意到,烧结的膨胀珠粒的焊接层的结晶度与发泡聚合物材料的平均结晶度不同。
[0121]
因此,根据本发明的第二和第三目的的发泡聚合物材料包括烧结的膨胀珠粒,其特征在于所述烧结的膨胀珠粒之间的焊接层的结晶度高于或低于所述发泡聚合物材料的平均结晶度。
[0122]
根据本发明的第二和第三目的的聚合物材料适用于生产具有改善的机械特性的复杂形状的制成制品,特别是对于相同的密度具有更高的弹性模量(或刚度)或者对于相同的弹性模量(或刚度)具有更高的明度 (lightness)的复杂形状的制成制品。
[0123]
本发明的第四目的由全部或部分由根据本发明的第二或第三目的的聚合物材料制成的制成制品表示。
[0124]
特别地,根据本发明的第四目的,所述制成制品由例如以下表示:防护系统(护胫、护背、护肩和肘部、护膝垫、壳体和护垫、防弹背心)、头盔(自行车、摩托车、工作和战斗)、矫形假体、义齿、表皮修补物、组织工程支架、吸声和隔音板和系统、隔热板和系统、运动鞋的鞋底和元素、汽车面板、运动设备、家具、包装、膜和过滤系统、用于陶瓷材料和多孔金属的牺牲泡沫、用于扩散器和曝气器的泡沫、生物医学系统、用于受控药物递送的垫和贴剂、渐进式机械响应系统、渐进式功能响应系统、电磁屏蔽系统、催化系统、航天和航空泡沫、用于光电子学的泡沫、浮选系统、框架和底盘、以及眼镜框架。
[0125]
现将参照在以下实验部分中通过说明而非限制的方式描述的材料和方法来对本发明进行说明。
[0126]
实验部分
[0127]
使用图1所示的分批膨胀系统来制备泡沫样品,其详细信息在以下示出。图1示出了本发明中使用的不连续发泡设备的照片(图1a)和图(图 1b)。
[0128]
反应器是圆柱形、温度受控且加压的,容积为0.3l(hip,型号bc-1)。对反应器进行改进以允许测量和控制关注的工艺参数。
[0129]
对于温度控制,使用电加热器(11)作为加热元件,并使用具有油浴的热交换器(12)作为冷却元件。
[0130]
加热器(11)和热交换器(12)由pid温度控制器(ascon,型号x1) 控制,该pid温度控制器使用pt100探针(4)读取反应器内部的温度。
[0131]
使用schaevitz压力传感器(型号p943)(3)测量饱和步骤期间的压力并记录发泡剂释放期间的压力趋势。阀(1)连接至发泡气体供应,而阀(2)连接至真空泵。
[0132]
泄压系统由hip球阀(型号15-71nfb)(5)、hip机电致动器(型号15-72nfb tsr8)(6)、以及连接至压缩空气管线(8)和用于电磁阀致动信号(7)的电缆(9)的电磁阀(7)组成。该系统允许阀开启的重现性。使用national instruments,austin,tx,美国的daq数据采集系统 pci6036e记录压力释放期间随时间的压力趋势p(t)。
[0133]
压力工序由teledyne isco容积泵型号500d(lincoln ne,美国)控制。通过泵控制器的串行接口,可以经由计算机控制泵并执行任何压力工序。此外,控制器可以控制多达四个用于不同流体的泵。
[0134]
溶解步骤的可变条件的实现可以通过以周期性趋势(例如,三角形波或正弦波)或者以非周期性趋势(例如,线性或曲线分布)改变发泡剂或数种发泡剂的溶解压力而发生,如通过以下实施例所述的。
[0135]
实施例1-比较
[0136]
在该实施例中使用的聚合物为由great eastern resins industrial co., ltd.(greco)(台中市,中国台湾)供应的平均分子量为500kda且密度为 1.14g/cm3的热塑性聚氨酯(tpu)(代码3080au)。
[0137]
该材料以特征尺寸为约3mm的椭圆形tpu颗粒供应。
[0138]
在室温下,将总固定重量为0.95g的一些颗粒安置在直径为25mm 且厚度为9mm的圆柱形钢模具(图1c)内部,然后用两块平坦钢板封闭并插入图1所示且以上描述的分批膨
胀设备的反应器内部。然后关闭反应器并使其达到140℃的温度。
[0139]
然后使用表1中描述的均匀压力分布使系统经历发泡气体溶解步骤 (n2/co2混合物,80/20v/v)。
[0140]
表1
[0141][0142]
如表1所示,在释放步骤3之前,均匀压力分布包括两个步骤:
[0143]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体 n2/co2的压力从大气压达到110巴;
[0144]
·
在步骤2中,将n2/co2发泡气体混合物的压力保持在110巴持续 90分钟,以允许发泡剂的完全溶解。
[0145]
在步骤3中,以1000巴/秒的速度释放压力以进行发泡。
[0146]
临压力释放之前,单个tpu颗粒内的发泡剂的浓度分布是平坦的(空间中恒定的浓度)。
[0147]
在140℃的温度下释放压力后膨胀时,单个颗粒以取决于发泡剂的浓度的泡沫的形态膨胀(形成膨胀珠粒)。在这种情况下,由于均匀的发泡剂浓度,因此形态是均匀的。通过膨胀,珠粒与相邻的珠粒烧结在一起(图 3a),形成焊接线,其细节在图3b中示出。
[0148]
然后,珠粒组填充圆柱形模具中的可用容积,形成与模具相同尺寸的圆柱形膨胀烧结的样品。
[0149]
图2a)示出了通过相同步骤获得的圆柱形膨胀烧结的样品和膨胀珠粒。
[0150]
实施例2-发明
[0151]
在室温下将在几何形状和在模具中的定位上与实施例1中描述的样品类似的tpu样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其达到140℃的温度。
[0152]
然后使用两种发泡气体(第一种为n2/co2以80/20v/v组成的混合物;第二种为he)使用表2中描述的时变压力分布使系统经历溶解步骤。
[0153]
表2
[0154][0155]
如表2所示,在释放步骤4之前,时变压力分布包括三个步骤:
[0156]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体 n2/co2的压力从大气压达到140巴;
[0157]
·
在步骤2中,将n2/co2发泡气体混合物的压力保持在140巴持续 90分钟,
[0158]
·
在步骤3中,将第一发泡气体(n2/co2混合物)的压力在2分钟的时间内从140巴降低至0巴;同时,在同一步骤中,首先使用第二发泡气体(he)使压力平衡,然后将最终压力增加至150巴。
[0159]
在步骤3结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0160]
如图4a所示,与实施例1类似,在膨胀期间各珠粒与相邻的珠粒烧结在一起,形成焊接线,其细节在图4b中示出。然后,珠粒组填充圆柱形模具中的可用容积,形成与模具相同尺寸的圆柱形膨胀烧结的样品。
[0161]
图2b)示出了通过相同步骤获得的圆柱形膨胀烧结的样品和膨胀珠粒。
[0162]
如专利申请it 102018000004727、实施例9(图10a和10b)中所述,这种类型的溶解步骤在样品的外围处形成致密层(具有低膨胀度或零膨胀度)。该致密层在图4a和图4b中可见,当与其中珠粒之间的焊接层较小的图3a和图3b的均匀情况相比时明显。
[0163]
如在it 102018000004727中描述以及如在以下实施例中详细说明的,使用特定的溶解工序可以设计不同层的数量、厚度、形态和密度。
[0164]
实施例3-发明
[0165]
在室温下将在几何形状和在模具中的定位上与实施例1中描述的样品类似的tpu样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其达到140℃的温度。
[0166]
然后使用两种发泡气体(第一种为n2/co2以80/20v/v组成的混合物;第二种为he)使用表3中描述的时变压力分布使系统经历溶解步骤。
[0167]
表3
[0168][0169]
如表3所示,在释放步骤4之前,时变压力分布包括三个步骤:
[0170]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体 n2/co2的压力从大气压达到100巴;
[0171]
·
在步骤2中,将n2/co2发泡气体混合物的压力保持在100巴持续 90分钟,
[0172]
·
在步骤3中,将第一发泡气体(混合物n2/co2)的压力在2分钟的时间内从100巴降低至0巴;同时,在同一步骤中,使用第二发泡气体 he使压力平衡,将总压力始终保持等于100巴。
[0173]
在步骤3结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0174]
如图5a所示,与实施例1类似,在膨胀期间各珠粒与相邻的珠粒烧结在一起,形成焊接线,其细节在图5b中示出。
[0175]
然后,珠粒组填充圆柱形模具中的可用容积,形成与模具相同尺寸的圆柱形烧结膨胀的样品。图2c)示出了使用相同步骤的圆柱形烧结膨胀的样品和膨胀珠粒。
[0176]
与实施例2相比,在这种情况下,使用he的溶解步骤延长(5分钟而不是2分钟),这导致非膨胀层增厚,如图5a和图5b中的图像所示 (与具有较薄外层的梯度烧结的产品的对应图像4a和4b以及与均匀烧结的产品的对应图像3a和3b相比),。
[0177]
实施例4-发明
[0178]
在室温下将在几何形状和在模具中的定位上与实施例1中描述的样品类似的tpu样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其达到140℃的温度。
[0179]
然后使用表4中描述的时变压力分布使系统经历发泡气体溶解步骤 (n2/co2混合物,80/20v/v)。
[0180]
表4
[0181][0182]
如表4所示,在释放步骤5之前,时变压力分布包括四个步骤:
[0183]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内将发泡气体压力(n2/co2)从大气压达到150巴;
[0184]
·
在步骤2中,将发泡气体压力(n2/co2)保持在150巴持续90分钟;
[0185]
·
在步骤3中,将发泡气体(n2/co2)的压力从150巴突然(0.1秒) 持续抵达至220巴;
[0186]
·
在步骤4中,将发泡气体压力(n2/co2)保持在220巴持续2分钟。
[0187]
在步骤4结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0188]
如图6a所示,与实施例1类似,在膨胀期间各珠粒与相邻的珠粒烧结在一起,形成焊接线,其细节在图6b中示出。
[0189]
然后,珠粒组填充圆柱形模具中的可用容积,形成与模具相同尺寸的圆柱形烧结膨胀的样品。图2d)示出了使用相同步骤的圆柱形烧结膨胀的样品和膨胀珠粒。
[0190]
与实施例2和3中的先前情况相比,珠粒具有反向密度梯度,在外层中比内层更膨胀。此外,如图6a和图6b所示,焊接线的特征在于不存在致密层。
[0191]
实施例5-机械特性
[0192]
使用metravib dma+1000(acoem)圆柱形板配置作为测量仪器使由以上实验产生的膨胀圆柱形样品(其代表性图像在图2中示出)经历静态单轴压缩测试。以0.3mm/分钟的位移速度使各样品经历高达50%变形的压缩。
[0193]
图7a和图7b表示由获取的数据获得的曲线。特别地,我们突出了不同烧结泡沫的压缩弹性模量的变化,全部均具有230kg/m3的平均密度。以下表5示出了所描述的不同实施例的弹性模量值。
[0194]
表5
[0195]
样品弹性模量(mpa)实施例11.01实施例21.12实施例31.32实施例40.98
[0196]
发现对于相同的平均密度,由最外层中膨胀较少的梯度珠粒产生的实施例2和3中的圆柱形样品比实施例1中的样品具有更大的刚度。
[0197]
相反,发现对于相同的平均密度,由最外层中膨胀较多的梯度珠粒产生的实施例4中的圆柱形样品比实施例1中的样品具有更小的刚度。
[0198]
实施例6-比较
[0199]
在该实施例中使用的聚合物为由versalis spa(mantua,意大利)供应的聚苯乙烯(ps)(代码n2380),其平均分子量、密度和熔体流动指数分别为300kda、1.05g/cm3以及在200℃和10kg下的2.0g/10分钟。
[0200]
该材料以特征尺寸为约3mm的椭圆形ps颗粒供应。在室温下,将总固定重量为0.95g的一些颗粒安置在直径为25mm且厚度为9mm的圆柱形钢模具(图1c)内部,然后用平坦钢板封闭并插入图1所示且以上描述的分批膨胀设备的反应器内部。然后关闭反应器并使其达到110℃的温度。
[0201]
然后使用表6中描述的均匀压力分布使系统经历发泡气体(co2)溶解步骤。
[0202]
表6
[0203][0204]
如表6所示,在释放步骤3之前,均匀压力分布包括两个步骤:
[0205]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体 (co2)的压力从大气压达到130巴;
[0206]
·
在步骤2中,将发泡气体压力(co2)保持在130巴持续90分钟;
[0207]
在步骤2结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0208]
在图8a中,示出了根据表6中描述的相同步骤制成的并且其中可以看出均匀形态的单个膨胀颗粒。与实施例1类似,使用模具并对于每个模具使用0.95g聚合物,在膨胀期间,各珠粒与相邻的珠粒烧结在一起,形成焊接线,其细节在图8b中示出并且在图8c中甚至更详细地示出。如在图3a和图3b的情况下,也示出不同珠粒之间的薄烧结区域和均匀的形态。
[0209]
实施例7-发明
[0210]
在室温下将在几何形状和在模具中的定位上与实施例6中描述的样品类似的ps样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其达到110℃的温度。
[0211]
然后使用表7中描述的时变压力分布使系统经历发泡气体(co2)溶解步骤。
[0212]
表7
[0213][0214]
如表7所示,在释放步骤5之前,时变压力分布包括四个步骤:
[0215]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体压力从大气压达到130巴;
[0216]
·
在步骤2中,将发泡气体压力保持在130巴持续90分钟;
[0217]
·
在步骤3中,将发泡气体压力在5分钟内从130巴增加至100巴;
[0218]
·
在步骤4中,将发泡气体压力保持在100巴持续2分钟。
[0219]
在步骤4结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0220]
图9a示出了根据表7中描述的相同步骤制成的并且其中可以看出梯度形态的单个膨胀颗粒,其由于较低的气体浓度而具有较致密的外层和较粗糙的形态。与实施例1类似,使用模具并对于每个模具使用0.95g聚合物,在膨胀期间,各珠粒与相邻的珠粒烧结在一起,形成焊接线,其细节在图9b中示出并且在图9c中甚至更详细地示出。与实施例6的情况相比,显示出具有从珠粒的中心向边缘延伸的更粗糙形态和更高密度的界面处的梯度形态。
[0221]
实施例8-比较
[0222]
在室温下将关于几何形状和在模具中的定位与实施例6中描述的样品类似但以较少的数量(0.42g)的ps样品安置在图1所示且以上描述的分批膨胀系统的反应器中。该材料以特征尺寸为约3mm的椭圆形ps颗粒供应。与其中使用0.95g来评估泡沫密度对通过对梯度泡沫珠粒进行烧结而获得的泡沫特性的影响的两个先前的实施例不同,在室温下,将总固定重量为0.42g的一些颗粒安置在25mm直径、9mm厚度的圆柱形钢模具(图1c)中,然后用平坦钢板封闭并插入图1所示且以上描述的分批膨胀设备的反应器内。然后关闭反应器并使其达到120℃的温度。
[0223]
然后使用表8中描述的均匀压力分布使系统经历发泡气体(co2)溶解步骤。
[0224]
表8
[0225][0226]
如表8所示,在释放步骤3之前,均匀压力分布包括两个步骤:
[0227]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体 (co2)的压力从大气压达到130巴;
[0228]
·
在步骤2中,将发泡气体压力(co2)保持在130巴持续90分钟;
[0229]
在步骤2结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0230]
在图10a中,示出所得烧结泡沫的照片,以及在图10b中示出焊接区域的细节。如在图3a和图3b的情况下,也示出了不同珠粒之间的薄烧结区域和均匀的形态。
[0231]
实施例9-发明
[0232]
在室温下将在几何形状和在模具中的定位上与实施例8中描述的样品类似的ps样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其达到120℃的温度。
[0233]
然后使用表9中描述的时变压力分布使系统经历发泡气体(co2)溶解步骤。
[0234]
表9
[0235][0236]
如表9所示,在释放步骤5之前,时变压力分布包括四个步骤:
[0237]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体压力从大气压达到130巴;
[0238]
·
在步骤2中,将发泡气体压力保持在130巴持续90分钟;
[0239]
·
在步骤3中,将发泡气体压力在5分钟内从130巴降低至100巴;
[0240]
·
在步骤4中,将发泡气体压力保持在100巴持续2分钟。
[0241]
在步骤4结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0242]
图11a示出了所得烧结泡沫的照片,以及在图11b和图11c中示出了焊接区域的两种细节。与实施例8的情况相比,其显示出具有从珠粒的中心向边缘延伸的更粗糙形态和更
高密度的界面处的梯度形态。
[0243]
实施例10-机械特性
[0244]
使由实验6、7、8和9产生的膨胀圆柱形样品经历如实施例5中所述的静态单轴压缩测试。
[0245]
图12a和图12b表示由对平均密度为230g/cm3的实施例6和7的烧结样品的压缩测试所获取的数据获得的曲线。特别地,图12b示出了用于计算压缩弹性模量的区域。图12c和12d表示由对平均密度等于110g/cm3的实施例8和9的烧结样品的压缩测试所采集的数据获得的曲线。特别地,图12d示出了用于计算压缩弹性模量的区域。以下表10示出了所描述的不同实施例的弹性模量值。
[0246]
表10
[0247]
样品弹性模量(gpa)实施例60.95实施例71.06实施例80.126实施例90.143
[0248]
对于230g/cm3的相同平均密度,由具有更致密外层的梯度珠粒产生的实施例7中的圆柱形样品比实施例6中的样品更加刚硬约12%。对于 110g/cm3的相同平均密度,由具有更致密外层的梯度珠粒产生的实施例9 中的圆柱形样品比实施例8中的样品更加刚硬约13%。
[0249]
实施例11-比较
[0250]
在该实施例中,使用的聚合物为由borealis供应的密度为0.95g/cm3的聚丙烯(pp)rd734mo。在室温下,将总固定重量为2g的一些颗粒安置在直径为25mm且厚度为9mm的圆柱形钢模具(图1c)内部,然后用平坦钢板封闭并插入图1所示且以上描述的分批膨胀设备的反应器内部。然后关闭反应器并使其达到140℃的温度。
[0251]
然后使用表11中描述的压力分布使系统经历发泡气体(co2)溶解步骤。
[0252]
表11
[0253][0254]
在压力步骤3之前,使反应器的温度在15分钟内达到125℃。如表 11所示,在释放步骤3之前,压力分布包括两个步骤:
[0255]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内将发泡气体压力从大气压达到150巴;
[0256]
·
在步骤2中,将发泡气体压力保持在150巴持续90分钟。
[0257]
在步骤2结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0258]
在图13中,示出了根据表11中描述的步骤制成的并且其中可以看出均匀形态的单个膨胀颗粒。
[0259]
实施例12-发明
[0260]
在室温下将在几何形状和在模具中的定位上与实施例11中描述的样品类似的pp样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其达到140℃的温度。然后使用两种发泡气体(第一种为co2;第二种为he)使用表12中描述的压力分布使系统经历溶解步骤。
[0261]
表12
[0262][0263]
在压力步骤3之前,使反应器的温度在15分钟内达到125℃。如表 12所示,在释放步骤5之前,压力分布包括四个步骤:
[0264]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使co2气体压力从大气压达到150巴;
[0265]
·
在步骤2中,将发泡气体压力保持在150巴持续90分钟;
[0266]
·
在步骤3中,将第一发泡气体(co2)的压力在0.5分钟的时间内从150巴降低至0巴;同时,在同一步骤中,使用第二发泡气体he使压力平衡,使总压力始终保持等于150巴;
[0267]
·
在步骤4中,将发泡气体压力保持在150巴持续4.5分钟。
[0268]
在步骤4结束时,以1000巴/秒的最大速度释放压力以进行发泡。
[0269]
图14a示出了所得发泡珠粒的照片,以及在图14b和图14c中示出了焊接区的两种细节。与实施例11的情况相比,其示出了在表皮上具有更高密度的界面处的梯度形态。
[0270]
图15示出了对根据表12中描述的步骤发泡的所得珠粒在n2吹扫下以10℃/分钟进行的dsc测试的图。特别地,从发泡珠粒的外部切下数个 100微米的切片,并且共同地包括(作为“壳”)在dsc盘中以进行分析。此外,还从剩余珠粒的中心区域切割下珠粒的核,并包括(作为“核”)在 dsc盘中以进行分析。如图15所报道的,取自同一发泡珠粒的两个样品 (即“壳”和“核”)的dsc图在总体结晶度和曲线形状二者方面显示不同,证明分等级的发泡样品的结晶结构不同,这是由于不同的发泡程度和用不同发泡剂的处理。
[0271]
实施例13-比较
[0272]
在该实施例中使用的聚合物为由total corbion供应的聚(乳酸)(pla) l175。在室温下,将总固定重量为2g的一些颗粒安置在图1所示且以上描述的分批膨胀设备的反应
器内部。然后关闭反应器并使其保持在20℃的室温。
[0273]
然后使用表13中描述的压力分布使系统经历发泡气体(co2)溶解步骤。
[0274]
表13
[0275][0276]
如表13所示,在释放步骤3之前,压力分布包括两个步骤:
[0277]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体压力从大气压达到20巴;
[0278]
·
在步骤2中,将发泡气体压力保持在20巴持续900分钟;
[0279]
在步骤2结束时,以10巴/分钟的最大速度释放压力。在这些低温条件下,负载有co2的颗粒不发泡。为了发泡,将颗粒通过温度枪在110℃下加热5秒,然后在空气中冷却。
[0280]
在图16中,示出根据表13中描述的步骤制成并且其中可以看出均匀形态的单个膨胀颗粒。
[0281]
实施例14-发明
[0282]
在室温下将在几何形状上与实施例13中描述的样品类似的pla样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其保持在20℃的室温。然后使用表14中描述的压力分布使系统经历发泡气体(co2)溶解步骤。
[0283]
表14
[0284][0285]
如表14所示,在释放步骤5之前,压力分布包括四个步骤:
[0286]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内将发泡气体压力从大气压达到20巴;
[0287]
·
在步骤2中,将发泡气体压力保持在20巴持续900分钟;
[0288]
·
在步骤3中,将发泡气体压力在0.2分钟内从20巴增加至40巴;
[0289]
·
在步骤4中,将发泡气体压力保持在40巴持续60分钟。
[0290]
在步骤4结束时,以20巴/分钟的最大速度释放压力。在这些低温条件下,负载有co2的颗粒不发泡。为了发泡,将颗粒通过温度枪在110℃下加热5秒,然后在空气中冷却。
[0291]
在图17中,示出根据表14中描述的步骤制成的并且其中可以看出分等级的形态的单个膨胀颗粒,其特征在于外层中的泡沫较大。
[0292]
实施例15-发明
[0293]
在室温下将在几何形状上与实施例13中描述的样品类似的pla样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其保持在20℃的室温。
[0294]
然后使用表15中描述的压力分布使系统经历发泡气体(co2)溶解步骤。
[0295]
表15
[0296][0297]
如表15所示,在释放步骤5之前,压力分布包括四个步骤:
[0298]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使发泡气体压力从大气压达到20巴;
[0299]
·
在步骤2中,将发泡气体压力保持在20巴持续900分钟;
[0300]
·
在步骤3中,使压力在1分钟的时间内从20巴达到5巴;同时使压力容器的温度变为40℃;
[0301]
·
在步骤4中,将发泡气体压力保持在5巴持续60分钟;
[0302]
在步骤4结束时,以20巴/分钟的最大速度释放压力。在这些低温条件下,负载有co2的颗粒不发泡。当加热颗粒以进行发泡(通过温度枪在 110℃下持续5秒以进行发泡,然后在空气中冷却)时,所得泡沫的特征在于外部致密层和内部发泡层,如图18所示。
[0303]
实施例16-发明
[0304]
在室温下将在几何形状上与实施例13中描述的样品类似的pla样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其保持在20℃的室温。
[0305]
然后使用两种发泡气体(第一种为co2;第二种为n2)使用表16中描述的压力分布使系统经历溶解步骤。
[0306]
表16
[0307][0308]
如表16所示,在释放步骤5之前,压力分布包括四个步骤:
[0309]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使co2气体压力从大气压达到20巴;
[0310]
·
在步骤2中,将发泡气体压力保持在20巴持续900分钟;
[0311]
·
在步骤3中,使第一发泡气体(co2)的压力在1分钟的时间内从 20巴降低至0巴;同时,在同一步骤中,使用第二发泡气体n2增加压力;
[0312]
·
在步骤4中,将发泡气体压力保持在100巴持续60分钟。
[0313]
在步骤4结束时,以50巴/分钟的最大速度释放压力。在这些低温条件下,负载有co2和负载有n2的颗粒不发泡。为了发泡,将颗粒通过温度枪在110℃下加热5秒,然后在空气中冷却。
[0314]
在图19中,示出了根据表16中描述的步骤制成的并且其中可以看出分等级的形态的单个膨胀颗粒。特别地,负载有n2的外层显示出较精细的形态,这是n2发泡聚合物的特征,而在内层中,较粗糙的形态是明显的,这是co2发泡聚合物的特征。
[0315]
实施例17-发明
[0316]
在室温下将在几何形状上与实施例13中描述的样品类似的pla样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其保持在20℃的室温。
[0317]
然后使用两种发泡气体(第一种为co2;第二种为n2)使用表17中描述的压力分布使系统经历溶解步骤。
[0318]
表17
[0319]
[0320]
如表17所示,在释放步骤5之前,压力分布包括四个步骤:
[0321]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使co2气体压力从大气压达到20巴;
[0322]
·
在步骤2中,将发泡气体压力保持在20巴持续900分钟;
[0323]
·
在步骤3中,使第一发泡气体(co2)的压力在1分钟的时间内从 20巴降低至0巴;同时,在同一步骤中,使用第二发泡气体n2增加压力。
[0324]
·
在步骤4中,将发泡气体压力保持在100巴持续120分钟。
[0325]
在步骤4结束时,以50巴/分钟的最大速度释放压力。在这些低温条件下,负载有co2和负载有n2的颗粒不发泡。为了发泡,将颗粒通过温度枪在110℃下加热5秒,然后在空气中冷却。
[0326]
在图20中,示出了根据表17中描述的步骤制成的并且其中可以看出分等级的形态的单个膨胀颗粒。特别地,负载有n2的外层显示出较精细的形态,这是n2发泡聚合物的特征,而在内层中,较粗糙的形态是明显的,这是co2发泡聚合物的特征。
[0327]
实施例18-发明
[0328]
在室温下将在几何形状上与实施例13中描述的样品类似的pla样品安置在图1所示且以上描述的分批膨胀系统的反应器中。然后关闭反应器并使其保持在20℃的室温。
[0329]
然后使用表18中描述的压力分布使系统经历发泡气体(co2)溶解步骤。
[0330]
表18
[0331][0332]
如表18所示,在释放步骤5之前,压力分布包括四个步骤:
[0333]
·
在步骤1中,以线性斜坡在0.2分钟(12秒)的时间内使co2气体压力从大气压达到20巴;
[0334]
·
在步骤2中,将发泡气体压力保持在20巴持续900分钟;
[0335]
·
在步骤3中,使压力在1分钟的时间内从20巴达到5巴。
[0336]
·
在步骤4中,将发泡气体压力保持在5巴持续60分钟。
[0337]
在步骤4结束时,以20巴/分钟的最大速度释放压力。在这些低温条件下,负载有co2的颗粒不发泡。为了发泡,将颗粒通过温度枪在110℃下加热5秒,然后在空气中冷却。
[0338]
在图21中,示出了根据表18中描述的步骤制成的并且其中可以看出分等级的形态的单个膨胀颗粒。特别地,由于发泡剂的量减少,其中co2从溶液中扩散溶出的外层相对于内层显示出高密度层。
[0339]
图22示出了对根据实施例13、16和18中描述的步骤发泡的所得珠粒在n2吹扫的情况下以10℃/分钟进行的dsc测试的图。还在表19中收集了所得dsc数据。
[0340]
特别地,与图15的情况不同的是,测试了整个样品,而没有切片得到核和壳。通过评估峰面积,可以评估不同发泡珠粒的结晶度。可以观察到,由于不同的处理,相对于实施例13(均匀泡沫),当采用实施例16 (分等级的泡沫)中描述的步骤时结晶度可以下降21%,而当采用步骤 18时结晶度可以下降61%。
[0341]
表19
[0342]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1