引物和使用了该引物的双链DNA的制造装置以及双链DNA的制造方法与流程
引物和使用了该引物的双链dna的制造装置以及双链dna的制造方法
技术领域
1.本发明涉及一种引物和使用了该引物的双链dna的制造装置以及双链dna的制造方法。
背景技术:
2.在分子生物学等领域,为了进行基因重组或转化等,使用将目标dna整合到宿主中而得的载体。通常,在目标dna的量少的情况下,通过聚合酶链反应(pcr)扩增后使用。在pcr中,使用包含目标dna序列的模板dna,使用与其互补性地结合的引物反复进行多个循环的热变性和退火,从而扩增模板dna。
3.通过pcr扩增的模板dna的扩增产物直接是平滑末端,为了使其与质粒dna等宿主dna结合(连接)而需要进行处理。通常,作为这样的处理,使用切割特定序列的限制酶,但在该方法中由于可结合的dna依赖于限制酶的切割位点的序列,因此存在缺乏通用性的问题。
4.作为不使用限制酶的方法,有以下的技术:以扩增产物的3’末端和5’末端作为粘附末端(也称为粘合末端、突出末端等),在宿主侧也同样地形成粘附末端,连接两者以构建载体。例如,近年来,已知有gibson装配(gibson assembly)法、in-fusion法、slice法等。在这些方法中,均在双链dna片段末端具有15bp左右的相同序列,利用核酸外切酶活性消化双链中的一侧的链使产生粘附末端,之后进行连接。此外,在gibson装配法中,在体外使用taq dna连接酶进行连接,而在in-fusion法中利用大肠杆菌内的修复系统进行连接。
5.在这些方法中,由于使用作为酶的核酸外切酶,所以除了花费成本以外,有时还因反应条件等导致位点特异性变差,难以形成定量的粘附末端,所以存在连接反应的效率低的问题。因此,寻求不使用酶的无缝克隆法。
6.因此,开发了利用化学方法调制具有粘附末端的dna的方法,作为用于此方法的pcr用引物,已知有专利文献1中记载的引物。该文献的引物中相当于非互补性dna部分的核苷酸序列中的3’末端的碱基用保护基修饰。该保护基具有使利用dna聚合酶进行的dna复制停止进行的功能,通过光照射处理或碱处理等可从被修饰碱基脱离。另外,在该文献中,使用用于向生物体分子内引入保护基(取代基)的取代基引入剂,将碱基保护基引入到引物的碱基中。
7.现有技术文献
8.专利文献
9.专利文献1:国际公开第2009/113709号(权利要求1、权利要求2等)。
技术实现要素:
10.发明要解决的技术问题
11.在专利文献1中,虽然是在保护基部分终止进行dna复制,但在碱基部分由于保护基而阻碍聚合酶活性,因此终止效率低,其结果,有时会完全延伸。另外,例如在dna中碱基
有腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶这4种,但在专利文献1中,为了向碱基中引入保护基,需要通过与碱基种类相应的方法引入保护基,在引物的制造上费事且花费成本。
12.本发明的目的在于提供一种终止效率高、并且可廉价地制造的引物。另外,本发明的另一目的还在于:提供一种使用这样的引物的具有粘附末端的双链dna的制造装置和双链dna的制造方法。
13.解决技术问题的方法
14.为了解决上述问题,本发明人反复进行了深入研究。其结果,开发了一种在核苷的糖部分引入了分解性保护基的引物。而且,还发现了:通过分解其保护基,可制作具有粘附末端的双链dna,完成了本发明。
15.即,本发明为一种引物,其特征在于:其是用于核酸的扩增的引物,具有下述式(1)所示的结构。
[0016][0017]
(这里,b表示碱基,r1表示分解性保护基,r2表示氢或羟基。*是指与相邻的核苷酸的糖的键。)
[0018]
这种情况下,优选上述r1为下述式(2a)所示的光分解性保护基。
[0019][0020]
(这里,a1表示碳原子数为1~3的亚烷基,可具有碳原子数为1~20的支链。*是指与磷酸的氧(o)的键。)
[0021]
而且,这种情况下,优选上述r1为下述式(4a)所示的光分解性保护基。
[0022][0023]
(这里,r3表示碳原子数为1~20的烷基。)
[0024]
在上述情况下,更优选上述r3为叔丁基或金刚烷基。特别是r3为叔丁基适合。
[0025]
而且,优选上述r1为下述式(3a)所示的2-硝基苄基。
[0026][0027]
或者,优选上述r1为下述式(2b)所示的还原剂分解性保护基。
[0028][0029]
(这里,a2表示碳原子数为1~3的亚烷基,可具有碳原子数为1~20的支链。*是指与磷酸的氧(o)的键。)
[0030]
这种情况下,优选上述r1为下述式(3b)所示的4-硝基苄基。
[0031][0032]
优选在序列中有2个以上的上述式(1)所示的结构连续。
[0033]
另外,本发明为一种双链dna的制造装置,其是用于使用上述任一项所述的引物制造具有粘附末端的双链dna的双链dna制造装置,其特征在于,具备:正向引物,与作为模板的模板dna的反义链的一部分序列互补,并且具有上述式(1)所示的结构;反向引物,与上述模板dna的有义链的一部分序列互补,并且具有上述式(1)所示的结构;扩增设备,以上述模板dna为模板进行多个循环的聚合酶链反应(pcr),生成上述正向引物延伸的正向侧延伸链和上述反向引物延伸的反向侧延伸链,将上述正向侧延伸链和上述反向侧延伸链退火以生成3’末端凹陷的双链dna;以及脱保护设备,将上述r1脱保护。
[0034]
而且,本发明还为一种双链dna的制造方法,其是用于使用上述任一项所述的引物制造具有粘附末端的双链dna的双链dna制造方法,其特征在于,具有以下工序:准备工序,准备正向引物和反向引物,该正向引物与作为模板的模板dna的反义链的一部分序列互补、且具有上述式(1)所示的结构,该反向引物与上述模板dna的有义链的一部分序列互补、且具有上述式(1)所示的结构;扩增工序,以上述模板dna为模板进行多个循环的聚合酶链反应(pcr),生成上述正向引物延伸的正向侧延伸链和上述反向引物延伸的反向侧延伸链,将上述正向侧延伸链和上述反向侧延伸链退火,生成3’末端凹陷的双链dna;以及脱保护工序,将上述r1脱保护。
[0035]
这种情况下,上述r1为上述式(2a)所示的光分解性保护基,脱保护工序优选通过光照射将上述r1脱保护。
[0036]
或者,上述r1为上述式(2b)所示的还原剂分解性保护基,脱保护工序优选利用还原剂将上述r1脱保护。
[0037]
另外,上述引物优选在序列中有2个以上的上述式(1)所示的结构连续。
[0038]
发明效果
[0039]
根据本发明,可提供一种脱保护效率高、并且可廉价地制造的引物。另外,根据本发明,还可提供一种使用这样的引物的具有粘附末端的双链dna的制造装置和双链dna的制造方法。
附图说明
[0040]
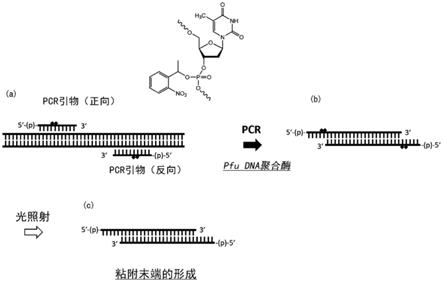
图1是示出具有粘附末端的双链dna的制造方法和制造装置的示意图。
[0041]
图2是在合成寡核苷酸pacgfp_fw2时脱保护后(a)和纯化后(b)的色谱图。
[0042]
图3是示出以包含链延伸终止笼蔽类似物t*的寡核苷酸为模板的链延伸反应终止的图。
[0043]
图4是示出pcr反应的琼脂糖凝胶电泳分析的图。
[0044]
图5是pcr片段的连接反应的示意图。
[0045]
图6是示出使用包含t**的寡核苷酸的粘附末端的制作实验的图。
[0046]
图7是示出使用na2s2o4的1t的脱保护反应的实验结果的图。
[0047]
图8是示出使用na2s2o4的2t的脱保护反应的实验结果的图。
[0048]
图9是示出磷酸修饰基中的取代基r的体积进一步增大后的实验的概要的图。
[0049]
图10是示出修饰寡核苷酸在热循环条件下的稳定性试验的结果的图。
[0050]
图11是示出修饰引物在pcr条件下稳定(耐分解性)的实验结果的图。
[0051]
图12是示出tbu型修饰引物的脱保护反应的反相hplc分析的实验结果的图。
[0052]
图13是示出pcr产物在试管内的连接反应(taq dna连接酶)的实验结果的图。
[0053]
图14是示出以包含链延伸终止笼蔽类似物(序列tt、tbu型)的寡核苷酸为模板的链延伸反应的终止的图。
[0054]
图15是示出以包含链延伸终止笼蔽类似物(序列tt、金刚烷基型)的寡核苷酸为模板的链延伸反应的终止的图。
[0055]
图16是示出以包含链延伸终止笼蔽类似物(序列aa和ta、tbu型)的寡核苷酸为模板的链延伸反应的终止的图。
[0056]
图17是示出以包含链延伸终止笼蔽类似物(序列cc和gc、tbu型)的寡核苷酸为模板的链延伸反应的终止的图。
具体实施方式
[0057]
1.引物
[0058]
以下,对本发明的引物进行说明。本发明的引物是用于核酸的扩增的引物,具有下述式(1)所示的结构。
[0059]
[0060]
(这里,b表示碱基,r1表示分解性保护基,r2表示氢或羟基。*是指与相邻的核苷酸的糖的键。)。此外,键*中,式(1)的3’侧的键是在3’侧与相邻的核苷酸的糖的5’碳结合,5’侧的键是在5’侧与相邻的核苷酸的糖的3’碳结合。另外,在dna引物的情况下r2为氢,在rna引物的情况下r1为羟基。
[0061]
b为碱基,具体而言,在dna引物情况下,b选自腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶,在rna引物的情况下,b选自腺嘌呤、鸟嘌呤、胞嘧啶、尿嘧啶。
[0062]
r1的分解性保护基是指通过任一种处理而分解的保护基(取代基)。这里所说的处理可列举:光照射处理、还原处理、碱处理、酸处理、氧化处理、脱甲硅烷基化处理、热处理、酯酶处理、磷酸酶处理等。由于聚合酶更强地识别核酸的磷酸基的负电荷,所以推测与像专利文献1那样向碱基中引入保护基相比,像式(1)那样用保护基屏蔽磷酸基更可提高聚合酶的终止效率。
[0063]
(1)光分解性保护基
[0064]
在处理为光照射的情况下,优选r1为下述式(2a)所示的光分解性保护基。
[0065][0066]
(这里,a1表示碳原子数为1~3的亚烷基,可具有碳原子数为1~3的支链、或碳原子数为1~20的支链。*是指与磷酸的氧(o)的键。)
[0067]
作为碳原子数为1~20的亚烷基,可列举:亚甲基、亚乙基、亚丙基、叔丁基、仲丁基、环己基、金刚烷基等。
[0068]
作为r1,优选为下述式(4a)所示的光分解性保护基。
[0069][0070]
(这里,r3表示碳原子数为1~20的烷基。)
[0071]
特别是,r3优选为叔丁基或金刚烷基这样的大体积的基团。如后述的实施例所示,r3的体积越大,修饰位点的化学稳定性和复制反应的阻碍效果就越高。因此,r3的碳原子数为3以上、优选为4以上、更优选为7以上、特别优选为10以上。在实施例所记载的取代基(r3为甲基、叔丁基、金刚烷基)中,按照化学稳定性低的顺序依次为甲基<叔丁基<金刚烷基。
[0072]
作为r1,可列举:下述式(3a)所示的2-硝基苄基。
[0073][0074]
(2)还原剂分解性保护基
[0075]
在处理为还原处理的情况下,优选r1为下述式(2b)所示的还原剂分解性保护基。
[0076][0077]
(这里,a2表示碳原子数为1~3的亚烷基,可具有碳原子数为1~20的支链。*是指与磷酸的氧(o)的键。)
[0078]
作为碳原子数为1~20的亚烷基,可列举:亚甲基、亚乙基、亚丙基、叔丁基、仲丁基、环己基、金刚烷基等。
[0079]
作为r1,可列举:下述式(3b)所示的4-硝基苄基。
[0080][0081]
(3)其他的分解性保护基
[0082]
作为通过碱处理可由被修饰碱基脱离的分解性保护基,可列举:异丁酰基、苯甲酰基、乙酰氧基甲基等。作为通过酸处理可由被修饰碱基脱离的分解性保护基,可列举:三苯甲基。作为通过氧化处理可由被修饰碱基脱离的分解性保护基,可列举:烯丙氧基甲基、二甲氧基苄氧基甲基、三甲氧基苄氧基甲基等。作为通过脱甲硅烷基化处理可由被修饰碱基脱离的分解性保护基,可列举:叔丁基二甲氧基甲硅烷氧基甲基、叔丁基二苯基甲硅烷氧基甲基等。作为通过热处理可由被修饰碱基脱离的分解性保护基,可列举:异氰酸酯基。作为通过酯酶处理可由被修饰碱基脱离的分解性保护基,可列举:乙酰氧基甲基。作为通过磷酸酶处理可由被修饰碱基脱离的分解性保护基,可列举:磷酸甲酯基。
[0083]
本发明的引物是特别适合在pcr中使用的单链dna或单链rna,是分子内具有上述式(1)所示的结构的寡核苷酸或多核苷酸。引物的碱基对的数目可根据目标dna等的序列等适当设定,通常在寡核苷酸中例如是20个碱基对以下、在多核苷酸中例如超过20个碱基对。对多核苷酸的碱基对的数目的上限没有特别限定,作为通常使用的引物,例如优选40个碱基对以下。另外,对寡核苷酸的碱基对的数目的下限也没有特别限定,只要是可用作引物的长度即可,作为通常使用的引物,例如优选5个碱基对以上。
[0084]
在引物中,与序列中仅存在1个上述式(1)所示的结构相比,优选2个以上的该结构连续。在序列中仅存在1个式(1)的结构的情况下,在其结构部分,基于dna聚合酶的dna复制未终止的情况下无法形成粘附末端。然而,由于2个以上的式(1)的结构连续,所以在概率上更易终止dna的复制,粘附末端的形成效率提高。
[0085]
2.引物的制造方法
[0086]
本发明的引物可通过合成具有上述式(1)的结构的修饰核苷酸(以下,有时称为“核苷酸衍生物”),并通过亚磷酰胺法等固相合成法在其上连接未修饰的核苷酸来制造。
[0087]
作为引物的合成方法的概要,首先,保护核苷的5’羟基,使n,n-双(二异丙基氨基)氯膦与3’羟基反应。接下来,使硝基苄醇反应以引入分解性保护基。之后,使亚磷酰胺等反应,通过固相合成法连接未修饰的核苷酸以合成引物。以下,对实施例中登载的引物的具体的制造方法进行详细说明。
[0088]
(a)具有还原剂分解性保护基的核苷衍生物(
※
式(1)中,r1为式(2b)、r2为氢的化
合物)和引物的合成
[0089][0090]
按照上述的合成图解进行说明。在以下说明的合成图解中,数字表示化合物的编号。首先,准备脱氧核糖核苷(在下述图解中为胸苷)作为起始物质。向其中加入4,4’-二甲氧基三苯甲基氯(dmtrcl)、吡啶,使4,4’-二甲氧基三苯甲基氯基与核糖的5’羟基结合(化合物24)。接下来,将n,n-双(二异丙基氨基)氯膦添加到三乙胺(tea)、四氢呋喃(thf)中,使亚磷酰胺与脱氧核糖的3’羟基结合(化合物25)。接下来,在反应混合物中加入4-硝基苄醇,然后添加5-(甲硫基)-1h-四唑以得到化合物26。之后,利用常规方法,以形成所期望的序列的方式固相合成核苷酸以合成引物。
[0091]
3.具有粘附末端的双链dna的制造方法和制造装置
[0092]
接着,对具有粘附末端的双链dna的制造方法和制造装置进行说明。本发明的双链dna的制造装置是用于使用本发明的引物以具有粘附末端的装置。另外,本发明的双链dna的制造方法是使用包含目标dna序列的模板dna,使用本发明的引物以具有粘附末端的方法。以下,参照图1进行说明。此外,在本实施方式中,虽然在引物中使用连续存在2个式(1)的结构的引物,但在仅有1个式(1)的情况下也可使用同样的方法/装置。
[0093]
首先,准备包含正向引物和反向引物的pcr扩增用引物对作为试剂(准备工序)。正向引物与模板dna的反义链的一部分序列互补,并且具有式(1)所示的结构。另外,反向引物与模板dna的有义链的一部分序列互补,并且具有式(1)所示的结构。
[0094]
如图的(a)所示,正向引物和反向引物以夹持要扩增的目标dna序列的方式确定序列。另外,引物中的式(1)的具有分解性保护基的核苷酸的位置设计成在作为目标的粘附末端的序列(图的(c))中与3’凹陷侧的最3’末端侧的核苷酸的3’侧相邻的位置互补的位置。在2个以上的式(1)的结构连续的情况下,使式(1)的结构的核苷酸中位于最3’侧的核苷酸成为上述的位置。其他的试剂为用于pcr的聚合酶(taq聚合酶等)、缓冲剂、dntp等。
[0095]
接下来,使用pcr装置(扩增设备)扩增模板dna的序列(扩增工序)。pcr装置以模板dna为模板进行多个循环的聚合酶链反应(pcr),生成正向引物延伸的正向侧延伸链和反向引物延伸的反向侧延伸链,将正向侧延伸链和反向侧延伸链退火,生成3’末端凹陷的双链dna(图的(b))。
[0096]
在pcr中,反复进行热变性、退火、延伸反应,以扩增模板dna的序列。虽然还取决于pcr的条件,但热变性在约95℃下进行1~3分钟,退火是在引物的tm
±
5℃下进行,延伸反应进行1~10分钟。对pcr的循环数没有特别限定,通常为24~40个循环左右。
[0097]
如图所示,在pcr扩增产物中包含3’末端凹陷的双链dna。这是由于:在以引物为模板合成互补链时,式(1)的分解性保护基阻碍聚合酶反应,使反应终止。
[0098]
之后,如图的(c)所示,通过规定的处理将r1脱保护,形成5’末端突出的粘附末端(脱保护工序)。规定的处理是用于将r1脱保护的处理,可列举上述的光照射处理、还原处理等。
[0099]
以下,对脱保护机制进行说明。如下式所示,若施行规定的处理,则式(1)的分解性保护基r1从核苷酸的磷酸上脱离。
[0100][0101]
作为光照射处理,例如可列举:使用光源装置(脱保护设备)照射300~400nm的波长的光1~30分钟的方法。另外,作为还原处理,例如可列举:使用连二亚硫酸钠(na2s2o4)等还原剂,例如进行70~80℃、1~30分钟的处理的方法。由此,引物的式(1)的核苷的分解性保护基被脱保护,可合成具有5’突出末端(3’凹陷末端)的双链dna。关于其他的处理,也同样使用将分解性保护基脱保护的装置(脱保护装置)进行脱保护。
[0102]
在本发明中,无论限制酶等如何,均可自由自在地设计粘附末端,因此可自由地连接具有所期望的序列的dna。例如,可设计目标dna和载体两者的序列,通过脱保护处理在两者中形成通用的粘附末端以进行连接,调制重组dna,将其用于克隆或制作文库、构建大量表达系统等。另外,通过连接具有粘附末端的多个基因组序列,可在试管内进行基因组叠合反应。或者,还可在平滑末端的状态下向细胞内引入双链dna,在细胞内进行脱保护处理,从而在细胞内进行基因组叠合反应。
[0103]
实施例
[0104]
以下,根据实施例,具体地说明本发明,但这些实施例并不限定本发明的目的。另外,在以下的实施例中,只要没有特别规定,则“%”的表述为质量基准(质量百分比)。
[0105]
1.链延伸终止笼蔽类似物t*
[0106]
(1)包含链延伸终止笼蔽类似物t*的寡核苷酸的合成
[0107]
包含链延伸终止笼蔽类似物t*的寡核苷酸(表1)根据亚磷酰胺法使用核酸自动合成仪(nr-2a 7mx、日本techno service公司制造)来合成。链延伸终止笼蔽类似物t*的亚酰胺化合物(化学式13)按照已有报道(wu,l等人,chem.eur.j.2014,20,12114-12122)进行合成。5’末端的磷酸化使用市售的亚酰胺试剂5’-磷酸盐-on试剂(chemgenes公司制造)。合成后的寡核苷酸按照常规方法进行脱保护,之后通过反相hplc纯化[系统:日立high-tech science公司制造的lachrom elite;色谱柱:ymc公司制造的hydrosphere c18(250
×
10mm i.d.);洗脱液a:含有5%乙腈的50mm的三乙基乙酸铵(ph7.0);洗脱液b:乙腈;梯度条件、0~60%洗脱液b/20分钟;洗脱液量:3ml/分钟;根据波长260nm的吸光度进行检测](图2)。
[0108][0109]
下表示出所合成的包含链延伸终止笼蔽类似物t*的寡核苷酸序列。p表示5’末端的羟基被磷酸化。
[0110]
[表1]
[0111]
序列名称序列1t5
′
·
acgactcact*atagggcgaattcgagctcggt
·3′
2t5
′
·
acgactcat*t*atagggcgaattcgagctcggt
·3′
pacgfp_fw25
′
·
paaagaaggagat*t*aaccatggtgagcaagggcgcc
·3′
pacgfp_rev25
′
·
paagcagccggt*t*ctcacttgtacagctcatccat
·3′
pet21d_fw25
′
·
paaccggctgct*t*ccaaagcccgaaaggaa
·3′
pet21d_rev25
′
·
paatctccttctt*t*aagttaaacaaaattatttctagag
·3′
[0112]
图2是示出合成寡核苷酸pacgfp_fw2时的脱保护后(a)和纯化后(b)的色谱图的图。纯化前的色谱图(a)所示的14.1分钟附近的峰是来自目标物的峰。对其进行分提、纯化(b)。
[0113]
(2)以包含链延伸终止笼蔽类似物t*的寡核苷酸为模板的复制反应
[0114]
q5(注册商标)高保真dna聚合酶、deep vent(注册商标)dna聚合酶、phusion(注册商标)高保真dna聚合酶从new england biolabs公司购入。pfu dna聚合酶从promega公司购入。酶反应的反应液[包含1μm的引物(5’荧光素-accgagctcgaattcgcc 3’)、1μm的模板(0t、1t或2t、表1)、0.2mm dntps、0.02单位/μl的聚合酶]是使用各酶所附带的缓冲液,按照推荐条件进行制作。使用applied biosystems 2720热循环仪将反应液按照95℃下1分钟、55℃下30秒、然后是72℃下10、30或60分钟进行加热。在加热后的10μl反应液中加入10μl的2
×
甲酰胺加样溶液,在90℃下加热3分钟后,通过包含7.5m尿素的20%改性page进行分析(图3)。根据来自在引物链的5’末端修饰的荧光素基的荧光,利用chemidoc xrs+成像系统检测电泳后的凝胶中所含的寡核苷酸链。
[0115]
图3是示出以包含链延伸终止笼蔽类似物t*的寡核苷酸为模板的链延伸反应的终止的图。(a)为使用的寡核苷酸的序列。(b)~(e)示出使用市售的耐热性聚合酶的链延伸反应的改性page分析结果,(f)示出使用pfu dna聚合酶的、以2t为模板的反应的maldi-tof分子量分析结果。下图示出原料(引物)的分析结果(对照实验)。
[0116]
图3(f)所示的链延伸产物的分子量分析如下进行。在上述条件下,进行以包含pfu dna聚合酶的2t为模板的酶反应,按照95℃下1分钟、55℃下30秒、然后是72℃下40分钟进行加热。将50μl反应液用te饱和苯酚与氯仿的等量混合液提取,在乙酸铵盐的存在下进行醇沉淀,回收dna。使用质谱仪ultraflextreme(bruker daltonics)进行maldi-tof分子量分析。
[0117]
(3)利用了通过链延伸终止进行的粘附末端形成的克隆反应
[0118]
图5是pcr片段的连接反应的示意图。按照该图所示的方法进行pcr片段的连接。首先,载体侧片段(5.3kb)如下操作进行调制(图4(a))。使用applied biosystems miniamp plus热循环仪,将反应液[0.5μm pet21d_fw2、0.5μm pet21d_rev2、0.8ng/μl pet21d(novagen)、20mm tris-hcl(ph8.8、25℃下)、10mm kcl、10mm(nh 4
)2so4、2mm mgso4、0.1%triton(注册商标)x-100、0.1mg/ml bsa、0.2mm dntps、0.02u/μl pfu dna聚合酶]在以下的加热条件下进行加热[(95℃、15秒
→
50℃、30秒
→
72℃、7.5分钟)/循环
×
30个循环]。
[0119]
插入物侧片段(0.75kb)如下操作进行调制(图4(b))。使用applied biosystems miniamp plus热循环仪,将反应液[0.5μm pacgfp1_fw2、0.5μm pacgfp1_rev2、0.8ng/μl pacgfp1(takara)、20mm tris-hcl(ph8.8、25℃下)、10mm kcl、10mm(nh 4
)2so4、2mm mgso4、0.1%triton(注册商标)x-100、0.1mg/ml bsa、0.2mm dntps、0.02u/μl pfu dna聚合酶]在以下的加热条件下进行加热[(95℃、15秒
→
55℃、15秒
→
72℃、1分钟)/循环
×
30个循环]。
[0120]
在pcr反应后的50μl反应液中分别加入100μl te饱和苯酚(nacalai tesque)与氯仿的等量混合液,剧烈混合后离心(14,000
×
g、3分钟),分离水层。同样用100μl氯仿提取反应液,之后在水层中加入5μl的3m的naoac(ph5.2)和60μl异丙醇。在-30℃下冷却1小时后离心(20,000
×
g、20分钟),以颗粒的形式回收dna。为了分解pcr反应的模板质粒dna,在37℃下使2种反应产物分别与限制酶dpni(toyobo)反应1小时(0.8u/μl dpni的33mm的tris-乙酸盐溶液(ph7.9)、10mm mg(oac)2、66mm koac、0.5mm二硫苏糖醇、反应液量为20μl)。在反应液中添加80μl水,加入100μl te饱和苯酚与氯仿的等量混合液,剧烈混合后离心(14,000
×
g、3分钟),分离水层。同样用100μl氯仿提取反应液后,加入10μl 3m的naoac(ph5.2)和110μl异丙醇。在-30℃下冷却1小时后离心(20,000
×
g、20分钟),以颗粒的形式回收dna。将dna颗粒溶解于水,通过琼脂糖凝胶电泳(包含gelred(和光纯药工业)的0.8%agarose s(和光纯药工业))进行分析,通过与dna尺寸标志物(quick-load purple 1kb plus dna ladder、new england biolabs)的谱带强度作对比,算出所含的目标dna的浓度[5μl 13ng/μl的载体dna片段;50μl 23ng/μl的插入物dna片段]。
[0121]
在96孔多孔板的孔中加入26.5ng载体dna片段和26.5ng插入物片段的5μl混合液,使用光照射装置max-305(朝日分光),以约4mw/cm2照射波长365nm的光10分钟。将该溶液添加到25μl大肠杆菌感受态细胞溶液(neb 5-α感受态大肠杆菌(高效)、new england biolabs)中进行转化。将其涂布于含有50μg/ml氨苄西林钠的lb琼脂培养基,在37℃下培养一夜。从产生的476个集落中选择20个,进行集落pcr,判别载体中是否有目标插入物的插入。通过琼脂糖凝胶电泳分析pcr反应液,可知在20个克隆中有18个含有目标连接反应产物(pcr引物、5’taatacgactca ctataggg 3’、5’gctagttattgctcagcgg 3’;集落阳性率为90%)。对于10个克隆的连接反应产物,将包含其的菌体进行液体培养,提取质粒dna。使用2种引物序列(5’ggtgatgtcggcgatatagg 3’、5’gccaatccggatatagttcct 3’),利用dna测序仪abi prism 3500xl基因分析仪对得到的质粒dna的序列进行分析。对来自引物dna的4处位点、其中的2处重叠位点、以及它们所夹持的范围的核苷酸序列进行分析,结果在10个克隆中均含有如设计的序列,没有观察到突变。
[0122]
图4是示出pcr反应的琼脂糖凝胶电泳分析的图。泳道1:尺寸标志物,泳道2:反应
液。(a):载体侧片段的调制。(b):插入物侧片段的调制。
[0123]
2.在还原条件下脱保护的类似物(t**)
[0124]
(1)在还原条件下脱保护的类似物(t**)的合成
[0125]
以下示出在还原条件下脱保护的类似物(还原脱保护类似物)的合成图解。以下,按照该合成图解,对还原脱保护类似物的合成顺序进行说明。
[0126][0127]
3-o-[2-(4-硝基苄氧基)(二异丙基氨基)膦烷基]-5-o-(4,4’-二甲氧基三苯甲基)-2-脱氧-胸苷(化合物26)的合成
[0128]
室温下向0.50g(1.32mmol)化合物24和200μl(1.45mmol)et3n的混合物的7ml thf溶液中加入0.39g(1.45mmol)n,n-双(二异丙基氨基)氯膦。将生成的混合物在室温下搅拌25分钟直至反应完成(化合物25)。无需纯化继续进行后续工序。将0.22g(1.45mmol)4-硝基苄醇加入到反应混合物中,然后加入0.17g(1.45mmol)5-(甲硫基)-1h-四唑。在室温下20分钟后完成反应。将反应混合物用etoac稀释,用饱和nahco3、h2o和盐水洗涤。蒸发后,混合物通过中性闪蒸硅胶层析(2%三乙胺的己烷溶液/etoac=2∶1~1∶1)纯化,得到了0.46g(0.56mmol)化合物26(42%)。
[0129]
(2)寡核苷酸的合成
[0130]
将所合成的切割类似物t**的亚酰胺制成终浓度为50mm的乙腈溶液,根据亚磷酰胺法,使用dna合成仪合成dna寡聚物。脱保护按照常规方法进行,通过反相hplc[日立high-tech science公司制造的lachrom elite;色谱柱:ymc公司制造的hydrosphere c18(250
×
10mm)]纯化,使用maldi-tof/ms(bruker)来确认结构。合成的dna的序列见下表。
[0131]
[表2]
[0132]
序列名称序列0t5
’‑
acgactcactatagggcgaattcgagctcggt-3’1t5
’‑
acgactcact**atagggcgaattcgagctcggt-3’2t5
’‑
acgactcact**t**tagggcgaattcgagctcggt-3’[0133]
(3)引物延伸实验
[0134]
q5高保真dna聚合酶从new england biolabs公司购入。pfu dna聚合酶从promega公司购入。酶反应的反应液[包含1μm的引物(5’荧光素-accgagctcgaattcgcc 3’)、1μm的模板(0t、1t或2t、表2)、0.2mm的dntps、0.02单位/μl的聚合酶]是使用各酶所附带的缓冲液按照推荐条件进行制作。使用applied biosystems 2720热循环仪,将反应液按照95℃下1分钟、55℃下30秒、然后是72℃下10、30或60分钟进行加热。在加热后的9μl反应液中加入9μl的2
×
甲酰胺加样溶液,在90℃下加热3分钟,之后通过包含7.5m尿素的20%改性page进行分析(图6)。根据来自在引物链的5’末端修饰的荧光素基的荧光,利用chemidoc xrs+成像系统检测电泳后的凝胶中所含的寡核苷酸链。
[0135]
(4)包含1个t**的寡核苷酸(1t)的脱保护反应
[0136]
将10μm的寡核苷酸(5
’‑
acgactcact**atagggcgaattcgagctcggt-3’)溶解于20mm的tris-hcl(ph7.4),加入na2s2o4使终浓度达到1mm,在室温下静置30分钟。之后,通过反相hplc[日立high-tech science公司制造的lachrom elite;色谱柱:ymc公司制造的hydrosphere c18(250
×
10mm)]分析脱保护反应的进行(图7)。
[0137]
(5)包含2个t**的寡核苷酸(2t)的脱保护反应
[0138]
将10μm的寡核苷酸(5’-acgactcact**t**tagggcgaattcgagctcggt-3’)溶解于20mm的tris-hcl(ph7.4),加入na2s2o4使终浓度达到10mm,在室温下静置30分钟。之后,通过反相hplc[日立high-tech science公司制造的lachrom elite;色谱柱:ymc公司制造的hydrosphere c18(250
×
10mm)]分析脱保护反应的进行(图8)。
[0139]
3.磷酸修饰基中的取代基r的体积进一步增大后的实验
[0140]
以下,在pcr终止引物中进一步增大磷酸修饰基中的取代基r的体积时,评价修饰位点的化学稳定性和复制反应阻碍效果。图9是示出该实验的概要的图。
[0141]
(1)亚酰胺试剂(dt、r=me、tbu、金刚烷基)的合成
[0142]
进一步增大取代基r的体积,以提高复制反应阻碍效果、化学稳定性为目标。
[0143]
(1-1)甲基型dt亚磷酰胺的合成
[0144][0145]
参照)chem.eur.j.2014,20,12114-12122
[0146]
在氩气中、0℃下,向343mg(0.63mg)5’-dmtr-胸苷的1.7ml ch2cl2悬浮液中加入300mg(0.75mmol)光保护亚酰胺试剂和53mg(0.75mmol)1h-四唑,在室温下搅拌4小时。将该混合物直接加载到硅胶柱(含有3%tea的ch2cl2/acoet=3/1)上,得到粗产物。将粗产物通过硅胶柱层析(含有3%tea的hex/acoet=3/1)纯化,得到了309mg me类似物亚酰胺(58%)。
[0147]
31
p-nmr(159mhz,cdcl3):δ148.8,147.8,147.7;hr-esi-ms(m/z)calcd.863.3392[m+na]
+
、found 863.3415。
[0148]
(1-2)金刚烷基型dt亚磷酰胺的合成
[0149][0150]
在氩气中、0℃下,向258mg(0.47mmol)5’-dmtr-胸苷的4ml ch2cl2溶液中加入107μl(0.61mmol)dipea和163mg(0.61mmol)双(二异丙基氨基)-cl-膦,在室温下搅拌2小时。然后,在上述反应混合物中加入150mg(0.52mmol)2-硝基-α-金刚烷基苄醇和50mg(0.71mmol)1h-四唑,在室温下搅拌2小时。在反应混合物中加入50mg(0.71mmol)1h-四唑,再搅拌1小时。加入h2o后,用acoet提取反应混合物,用盐水洗涤,在na2so4上干燥,使其蒸发。将粗产物通过硅胶柱层析(含有0.5%tea的hex/acoet=1/1)纯化,得到了169mg ad类似物亚酰胺(37%)。
[0151]
31
p-nmr(159mhz,cdcl3):δ153.1,151.9,150.2,148.1;hr-esi-ms(m/z)calcd.983.4331[m+na]
+
、found 983.4336。
[0152]
(1-3)tbu取代类似物的4种碱基(a、g、c、t)的亚酰胺试剂合成图解(汇总)
[0153]
以下,示出在使用4种碱基的情况下亚酰胺试剂的合成图解的概要。
[0154][0155]
(1-4)pac-da亚磷酰胺的合成
[0156][0157]
在氩气中、0℃下,向2.0g(2.9mmol)5’-dmtr-pac-da的7.5ml ch2cl2悬浮液中加入2.6g(5.8mmol)光保护亚酰胺试剂和1.0g(8.7mmol)5(甲硫基)-1h-四唑,在室温下搅拌6小时。将溶液注入200ml acoet中,用200ml水、200ml饱和碳酸氢钠水溶液、200ml食盐水洗涤。将有机相在硫酸钠上干燥,过滤,通过硅胶柱层析(hex/acoet=2/1、含有1%tea)纯化,得到了1.1g t-bu dt类似物亚酰胺(37%)。
[0158]
hr-esi-ms(m/z)calcd.1048.4350[m+na]
+
、found 1048.4437。
[0159]
(1-5)dt亚磷酰胺的合成
[0160][0161]
在氩气中、0℃下,向1.5g(2.8mmol)5’-dmtr-pac-da的10ml ch2cl2悬浮液中添加2.4g(5.5mmol)光保护亚酰胺试剂和0.64g(5.5mmol)5-(甲硫基)-1h-四唑,在室温下搅拌7小时。将溶液注入200ml acoet中,用200ml水、200ml饱和碳酸氢钠水溶液、200ml食盐水洗涤。将有机相在硫酸钠上干燥,过滤,通过硅胶柱层析(hex/acoet=2/1、含有1%tea)纯化,得到了2.0g t-bu da类似物亚酰胺(81%)。
[0162]
hr-esi-ms(m/z)calcd.905.2267[m+na]
+
、found 905.3978。
[0163]
(1-6)pac-dg亚磷酰胺的合成
[0164][0165]
在氩气中、0℃下,向2.0g(2.8mmol)5’-dmtr-pac-dg的7.5ml ch2cl2悬浮液中加入2.5g(5.7mmol)光保护亚酰胺试剂和0.99g(8.5mmol)5-(甲硫基)-1h-四唑,在室温下搅拌6小时。将溶液注入200ml acoet中,用200ml水、200ml饱和碳酸氢钠水溶液、200ml食盐水洗涤。将有机相在硫酸钠上干燥,过滤,通过硅胶柱层析(hex/acoet=2/1、含有1%tea的acoet/meoh=40/1)纯化,得到了1.2g t-bu dg类似物亚酰胺(41%)。hr-esi-ms(m/z)calcd.1143.5678[m+tea]
+
、found 1143.5867。
[0166]
(1-7)ac-dc亚磷酰胺的合成
[0167][0168]
在氩气中、0℃下,向1.8g(3.2mmol)5’-dmtr-ac-dc的8.0ml ch2cl2悬浮液中加入2.8g(6.3mmol)光保护亚酰胺试剂和0.73mg(6.3mmol)5(甲硫基)-1h-四唑,在室温下搅拌6小时。将溶液注入200ml acoet中,用200ml水、200ml饱和碳酸氢钠水溶液、200ml食盐水洗涤。将有机相在硫酸钠上干燥,过滤,通过硅胶柱层析(hex/acoet=1/1、含有1%tea)纯化,得到了2.1g t-bu dc类似物亚酰胺(74%)。
[0169]
hr-esi-ms(m/z)calcd.1011.5355[m+tea]
+
、found 1011.5503。
[0170]
(2)修饰寡核苷酸在热循环条件(pcr条件)下的稳定性(耐分解性)试验
[0171]
(反应条件)
[0172]
将组成为10μm寡核苷酸(me1t或tbu1t)的20mm的tris-hcl溶液、10mm(nh 4
)2so4、10mm kcl、2mm mgso4、ph8.8的溶液在下述条件[(95℃、1分钟
→
50℃、30秒
→
72℃、3分钟)
×
30个循环]下加热,之后通过反相hplc进行分析。
[0173]
(分析条件)
[0174]
系统;日立high-tech science公司制造的lachrom elite;
[0175]
色谱柱;ymc公司制造的hydrosphere c18(250
×
4.6mm);
[0176]
流动相;a液:含有5%乙腈的50mm的三乙基乙酸铵(teaa、ph7.0)
[0177]
b液:乙腈;
[0178]
(将b液含量分成20份,从0%增加到60%(线性梯度))
[0179]
流动相流量;1ml/分钟;
[0180]
检测波长;260nm。
[0181]
(结果)
[0182]
将寡核苷酸me1t进行热循环后,在约50%的基质中看到了修饰基的脱离(图10的(d):确认到所观察到的峰与不含修饰的该序列寡核苷酸的洗脱时间一致)。相对于此,在tbu1t的情况下,即使进行热循环,也没有观察到保护基的脱离(图10的(e))。因此,将me、tbu型修饰的稳定性作对比,结果显示了后者的优越性。
[0183]
(3)显示修饰引物在pcr条件下稳定(耐分解性)的实验(tbu类似物引入数为1、2、3的比较)
[0184]
(反应条件)
[0185]
将组成为10μm寡核苷酸(tbu1t、tbu2t或tbu3t)的20mm tris-hcl溶液、10mm(nh4)2so4、10mm kcl、2mm mgso4、ph8.8的溶液在下述条件下[(95℃、1分钟
→
50℃、30秒
→
72℃、3分钟)
×
30个循环]加热,之后通过反相hplc进行分析。
[0186]
(分析条件)
[0187]
系统:日立high-tech science公司制造的lachrom elite;
[0188]
色谱柱:ymc公司制造的hydrosphere c18(250
×
4.6mm);
[0189]
流动相:a液:含有5%乙腈的50mm三乙基乙酸铵(teaa、ph7.0);
[0190]
b液:乙腈;
[0191]
(将b液含量分成20份,从0%增加到60%(线性梯度));
[0192]
流动相流量:1ml/分钟;
[0193]
检测波长:260nm。
[0194]
(结果)
[0195]
如图11所示,即使将修饰的引入数增加至2个、3个,在该条件下也未见保护基的脱离。
[0196]
(4)tbu型修饰引物的脱保护反应的反相hplc分析
[0197]
(脱保护反应条件)
[0198]
光照射(图12的(b)):10μm寡核苷酸、10mm tris-hcl(ph8.5);
[0199]
以4mw/cm2对溶液照射365nm的光10分钟;
[0200]
还原反应(图12的(c)):5μm寡核苷酸、10mm na2s2o4、20mm tris-hcl(ph8.5);
[0201]
将溶液在25℃下培养30分钟。
[0202]
(分析条件)
[0203]
系统:日立high-tech science公司制造的lachrom elite;
[0204]
色谱柱:ymc公司制造的hydrosphere c18(250
×
4.6mm);
[0205]
流动相:a液:含有5%乙腈的50mm三乙基乙酸铵(teaa、ph7.0);
[0206]
b液:乙腈;
[0207]
(将b液含量分成20份,从0%增加到60%(线性梯度));
[0208]
流动相流量:1ml/分钟;
[0209]
检测波长:260nm。
[0210]
已确认过:即使在以下的还原条件下也同样可以脱保护。
[0211]
·
10μm寡核苷酸、10mm b2(oh)4、50mm naoh、30%etoh/水;25℃、2小时;
[0212]
·
10μm寡核苷酸、1.5mm ticl3、20mm柠檬酸盐缓冲液(ph6.0);25℃、2小时;
[0213]
如图12所示,在365nm光照射和连二亚硫酸钠的还原反应下均确认到进行定量的反应。
[0214]
(5)pcr产物在试管内的连接反应(taq dna连接酶)
[0215]
使用图13中记载的耐热性聚合酶,通过pcr反应调制出双链dna片段。用于调制1.0kb片段的模板为pet21质粒dna,引物序列为以下的2条序列。
[0216]
下划线部t(t)示出在磷酸部含有me、tbu型修饰基。p示出5’末端被磷酸化。
[0217]
fw(21-nt):5’cgccgagacagaacttaatgg 3’;
[0218]
rev(38-nt):5’paatctccttct ttaagttaaacaaaattatttctagag 3’;
[0219]
用于调制0.74kb片段的模板为pacgfp1质粒dna,引物序列为以下的2条序列。
[0220]
下划线部t(t)示出在磷酸部含有me、tbu型修饰基。p示出5’末端被磷酸化。
[0221]
fw(35-nt):5’paaagaaggagattaaccatggtgagcaagggcgcc 3’;
[0222]
rev(34-nt):5’gcaaccaagcttctcacttgtacagctcatccat 3’。
[0223]
使用图中记载的市售的耐热性聚合酶,按照推荐条件实施pcr反应,分别调制1.0kb片段和0.74kb片段。通过琼脂糖凝胶电泳(含有gelred(和光纯药工业)的1.5%agarose s(和光纯药工业))确认反应的进行。pcr反应后,在200μl反应液中加入200μl te饱和苯酚(nacalai tesque)与氯仿的等量混合液,剧烈混合后离心(20,000
×
g、1分钟),分离水层。同样用200μl氯仿提取反应液,之后加入20μl 3m的naoac(ph5.2)和220μl异丙醇。在-30℃下冷却1小时后离心(20,000
×
g、20分钟),从而回收dna。将目标dna产物通过琼脂糖凝胶电泳(含有gelred(和光纯药工业)的1.5%agarose s(和光纯药工业))纯化。使用wizard sv凝胶和pcr净化系统(promega),由切取的凝胶片提取dna。
[0224]
将如此操作而调制的1.0kb片段和0.74kb片段用taq dna连接酶连接,通过琼脂糖凝胶电泳评价其效率。最初,为了去除磷酸部的保护基,以4mw/cm2的强度对dna溶液(5μl;24nm 1.0kb片段、24nm 0.74kb片段、1mm tris-hcl(ph8.5))照射365-nm的光5分钟。然后,在含有这2个dna片段的溶液中加入taq dna连接酶(new england biolabs),在37℃下培养2小时。反应液组成如下。2.8nm 1.0kb片段、2.8nm 0.74kb片段、1.6u/μl taq dna连接酶、20mm tris-hcl、25mm乙酸钾、10mm乙酸镁、1mm nad 1、10mm dtt、0.1%triton x-100、ph7.6。在反应液中加入80μl水,使总量达到约100μl,加入100μl te饱和苯酚(nacalai tesque)与氯仿的等量混合液,剧烈混合后离心(20,000
×
g、1分钟),分离水层。同样用100μl氯仿提取反应液后,加入10μl 3m的naoac(ph5.2)和110μl异丙醇、1μl 20mg/ml的糖原。在-30℃下冷却15分钟后离心(20,000
×
g、20分钟),从而回收dna。所回收的dna产物通过琼脂糖凝胶电泳(1.5%agarose s(和光纯药工业))进行电泳。将电泳后的凝胶用gelred水溶液染色,使谱带可视化(biorad chemidoc xrs+系统)。
[0225]
如图所示,将me、tbu引物作对比,确认在后者中得到了更高的连接效率。
[0226]
(6)以包含链延伸终止笼蔽类似物的寡核苷酸为模板的复制反应
[0227]
与上述“(2)以包含链延伸终止笼蔽类似物t*的寡核苷酸为模板的复制反应”同样地进行复制反应的实验。
[0228]
酶反应的反应液[含有1μm的引物(5’荧光素-accgagctcgaattcgcc 3’)、1μm的模板(各图中记载序列和结构)、0.2mm的dntps、0.02单位/μl的聚合酶]是使用各酶所附带的缓冲液按照推荐条件进行制作。使用applied biosystems 2720热循环仪将反应液按照95℃下1分钟、55℃下30秒、然后是72℃下30分钟进行加热。在加热后的10μl反应液中加入10μl的2
×
甲酰胺加样溶液,在90℃下加热3分钟后,通过含有7.5m尿素的20%改性page进行分析(图14~图17)。根据来自在引物链的5’末端修饰的荧光素基的荧光,利用chemidoc xrs+成像系统检测到电泳后的凝胶中所含的寡核苷酸链。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1