一种温敏性聚乳酸高分子材料及其制备方法和应用
本发明涉及高分子材料技术领域,尤其涉及一种温敏性聚乳酸高分子材料及其制备方法和应用。
背景技术:
聚乳酸高分子材料因其良好的生物相容性、可生物降解性、可调节性等,被广泛应用于生物医药研究,例如将通过赋予聚乳酸高分子材料温敏性可将材料应用于药物递送或触发药物释放。
目前文献已报道的制备温敏性聚乳酸高分子材料的方法,包括以下几种:
(1)以聚n-异丙基丙烯酰胺(pnipam)、聚n-乙烯基己内酰胺(pnvcl)等聚酰胺类高分子为原料改性聚乳酸材料,制备得到功能化的温敏性聚乳酸材料[alexandera,khanj,sarafs,etal.eurjpharmbiopharm,2014,88(3):575;zhaohl,taol,yur,etal.jnanoscinanotechnol,2015,15(8):5553;seidif,heshmatip.chinjpolymsci,2015,33(1):192;elashnikovr,
(2)以聚乙烯基醚、聚乙二醇等聚醚类高分子为原料改性聚乳酸材料,制备得到功能化的温敏性聚乳酸材料[yoshimitsuh,kanazawaa,kanaokas,etal.macromolecules,2012,45(23):9427;fusz,liz,fanjm,etal.jbiomednanotechnol,2014,10(3):427;guoqf,caoh,lixh,etal.matertechnol,2015,30(5):294;maohl,shangr,baoyz,etal.softmatter,2016,12(20):4628;chenx,lif,fengll,etal.jbiomednanotechnol,2017,13(11):1357;.sekiy,kanazawaa,kanaokas,etal.macromolecules,2018,51(3):825;yokotad,kanazawaa,aoshimas.macromolecules,2019,52(16):6241]。
(3)以聚(2-异丙基-2-噁唑啉)(pipox)和聚(2-乙基-2-噁唑啉)(petox)等拟聚肽类高分子改性聚乳酸材料,制备得到功能化的温敏性聚乳酸材料[wangx,lix,liy,etal.actabiomater,2011,7(12):4149;poochf,sliepenm,
值得注意的是,这些基于自身具有温敏性的聚合物来改性聚乳酸,要么成本太高,要么温敏性聚合物制备流程复杂,大多不能同时满足成本低廉、工艺简单和材料性能可调等条件,很难有进一步的应用[choh,gaoj,kwongs.jcontrolrelease,2016,240:191]。
技术实现要素:
本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明的第一个目的是提出一种温敏性聚乳酸高分子材料,该材料具有温敏性能,且性能可调。
本发明的第二个目的还提供一种温敏性聚乳酸高分子材料的制备方法,通过一锅法直接熔融缩聚进行共聚反应,制备方法简便。
本发明还提供所述温敏性聚乳酸高分子材料的应用。
具体地,本发明采取如下的技术方案:
本发明的第一方面是提供一种温敏性聚乳酸高分子材料,所述温敏性聚乳酸高分子材料具有如下式i所示的结构式:
其中,所述r为温敏性聚合物单体的聚合链段,所述n表示10~100之间的整数,q表示1~10之间的整数,m分别为表示10~100之间的整数。
优选地,所述温敏性聚合物单体的聚合链段为
优选地,所述r1为
优选地,所述r1为
本发明的第二方面是提供一种温敏性聚乳酸高分子材料的制备方法,包括如下步骤:
(1)将乳酸单体和马来酸酐进行混合,在真空下进行预聚,得到预聚体;
(2)在所述预聚体中加入催化剂,在真空下进行催化熔融聚合,得到催化预聚体;
(3)向所述催化预聚体中加入温敏性聚合物单体和引发剂,反应得到温敏性聚乳酸高分子材料。
步骤(1)中,所述马来酸酐和乳酸单体的摩尔投料比范围为1:1~200。
步骤(1)中,所述预聚的温度为110~200℃[在实际操作中,步骤(1)的预聚温度略低于步骤(2)的催化熔融聚合的温度,有利于乳酸酯化缩聚形成预聚体],所述预聚的时间为4~12h,所述预聚的真空度为1~10kpa。
步骤(1)中,在进行所述预聚前,还包括对乳酸单体和马来酸酐的混合物进行脱水的步骤。所述的脱水温度为100~160℃,所述的脱水时间为1~10h。
步骤(2)中,所述催化剂包括tsa(对甲基苯磺酸)、zncl2、sncl2、zno和sno中的至少一种。
步骤(2)中,所述催化剂的添加量为所述预聚体质量的0.1%~1%。
步骤(2)中,所述催化熔融聚合的温度为110~200℃,所述催化熔融聚合的时间为6~12h。
步骤(2)中,所述催化熔融聚合的真空度为1~10kpa。通过持续抽真空,快速的除去缩聚反应产生的水在高温汽化后的水蒸气。
步骤(3)中,所述温敏性聚合物单体与乳酸单体的摩尔投料比范围为1:1~100;所述引发剂的添加量为乳酸单体质量的0.1%~1%。
步骤(3)中,所述温敏性聚合物单体包括n-异丙基丙烯酰胺、n-异丙基甲基丙烯酰胺、n-乙烯基己内酰胺中的至少一种。
步骤(3)中,所述引发剂包括过硫酸盐(如过硫酸钾、过硫酸铵)、过氧化物、偶氮类引发剂中的至少一种。
步骤(3)中,所述反应在非真空下进行。所述反应的温度为110~200℃,所述反应的时间为1~12h。
步骤(3)中,所述反应结束后还包括对反应产物进行提纯的步骤。所述提纯方法可包括如下步骤:采用溶剂将反应产物溶解,然后采用水进行沉析、洗涤,过、干燥。所述溶剂包括但不限于甲醇、三氯甲烷、二氯甲烷、乙醚、乙酸乙酯和石油醚。所述干燥可在真空干燥箱中进行加热真空干燥,所述的真空干燥的真空度为1~10kpa,所述的真空干燥温度为30~45℃。所述的提纯产物操作次数为1~3次,视产物纯度而定。
本发明的第三方面是提供所述温敏性聚乳酸高分子材料的应用。具体地,本发明提供所述温敏性聚乳酸高分子材料制备药物载体、制备药物或制备伤口敷料中的应用。
本发明具有如下有益效果:
本发明基于廉价易得的生物基乳酸单体,以及简单的马来酸酐、温敏性聚合物单体,首先使乳酸单体和马来酸酐分两步进行聚合,减少自由基聚合发生的比例,控制反应朝着既定的开环、酯化缩聚的反应进行,结合后后续的温敏性聚合物单体聚合步骤,得到一种新型的聚乳酸高分子材料,所述聚乳酸高分子材料具有温敏性和生物可降解性,其低临界相转变温度(lcst)为30~32℃(根据透光度测定)。在温度低于lcst时,所述聚乳酸高分子材料具有良好的亲水性;随着温度升高,在30~35℃(根据dls测定)间发生明显相变,粒径收缩,疏水性增加。由于聚乳酸高分子材料的lcst低于人体温度,将其用作小分子药物或抗菌药物载体,对药物进行包封,药物进入人体后或者敷在人体表面后,随着温度变化变成胶体,随着聚合物的降解,药物缓慢从胶体中释放,从而实现药物的缓释。因此,本发明的聚乳酸高分子材料有望用作小分子药物或抗菌药物载体,或可直接以粉末状、与溶剂调和成凝胶状或纺丝制成纤维状作为伤口敷料基材。
本发明的制备方法不再采用传统的聚合物与聚合物化学键合或物理共混等方式来制备温敏性聚乳酸高分子材料,而是采用全新的制备思路和方法,通过一锅法使乳酸单体与马来酸酐、n-异丙基丙烯酰胺直接熔融缩聚进行共聚反应,不需要使用价格高昂的试剂、不需要使用定制的合成设备,反应工艺简单且容易控制,产物提纯操作简便,产率高。
附图说明
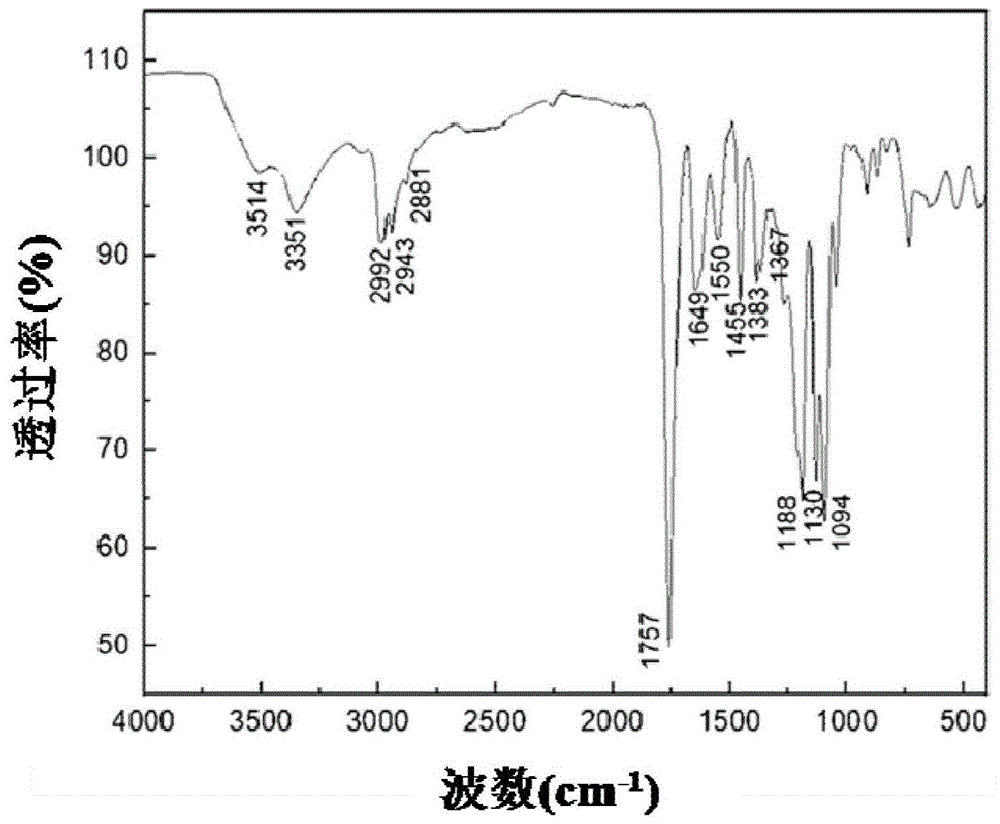
图1为plamn7的ft-ir谱图;
图2为plamn7的1hnmr谱图;
图3为以plamn4配制的不同浓度的高分子胶束溶液在不同温度、搅拌时间时的外观变化情况;
图4为plamn4、plamn7、plamn12的胶束溶液的dls测试结果;
图5为plamn4、plamn7、plamn12的胶束溶液的透光度测试结果;
图6为不同浓度的胶束溶液的荧光激发光谱;
图7为不同浓度胶束溶液在345nm处和339nm处的荧光强度比值i345/i339与浓度的关系曲线。
具体实施方式
本发明提供一种温敏性聚乳酸高分子材料,其制备方法如下:
首先,采用一次投料法制备预聚物,将磁力搅拌子置入充分干燥的反应瓶中,将乳酸单体与马来酸酐加入反应瓶,开启电磁搅拌。先常压脱水,之后抽真空预聚,得到预聚体。
然后,在预聚体中加入催化剂,催化直接熔融聚合。接着,撤除真空,在上述反应体系中加入温敏性聚合物单体、引发剂,开启电磁搅拌进行反应。
最后,对根据产物进行提纯,先用溶剂把产物溶解,必要时加热助溶产物,再用去离子水沉析和洗涤,过滤后,滤渣置于真空干燥箱中加热干燥,研磨成粉末,制得共聚物。所述的提纯产物操作次数为1~3次,视产物纯度而定。
以下以温敏性聚合物单体n-异丙基丙烯酰胺为例,详细介绍该温敏性聚乳酸高分子材料的制备方法以及性能。
聚(乳酸-马来酸酐-n-异丙基丙烯酰胺)(共聚物plamn)反应路线为:
其中,n表示10~100之间的整数,p表示1~100之间的整数,q表示10~10之间的整数,m分别为表示10~100之间的整数。
实施例1
共聚物plamn4的合成:
将磁力搅拌子置入反应瓶中,将约3.6g(40mmol)乳酸单体与0.2g(2mmol)马来酸酐加入反应瓶,开启搅拌。常压下在140℃温度脱水4h,之后用循环水真空泵抽真空,预聚4h。预聚结束后,加入0.01g催化剂sno,用旋片式真空油泵抽真空,在160℃温度聚合6h。解除真空,加入0.9g(8mmol)n-异丙基丙烯酰胺和0.02g的过硫酸钾,120℃温度聚合2h。聚合结束后,用甲醇溶解产物,再用去离子水沉析、洗涤,过滤后,滤渣置于45℃真空干燥箱中干燥24h,得到共聚物plamn4[plamn4中的4表示n-异丙基丙烯酰胺与马来酸酐的投料比(摩尔比),即p/q;以下实施例2中的plamn7和实施例3中的plamn12同理],产率为57%。
实施例2
共聚物plamn7的合成:
将磁力搅拌子置入反应瓶中,将约3.6g(40mmol)乳酸单体与0.2g(2mmol)马来酸酐加入反应瓶,开启搅拌。常压下在130℃温度脱水6h,之后用循环水真空泵抽真空,预聚8h。预聚结束后,加入0.01g催化剂sno,用旋片式真空油泵抽真空,在140℃温度聚合3h。解除真空,加入1.6g(14mmol)n-异丙基丙烯酰胺和0.02g的过硫酸钾,100℃温度聚合4h。聚合结束后,用甲醇溶解产物,再用去离子水沉析、洗涤,过滤后,滤渣置于45℃真空干燥箱中干燥24h,得到共聚物plamn7,产率为49%。
实施例3
共聚物plamn12的合成:
将磁力搅拌子置入反应瓶中,将约3.6g(40mmol)乳酸单体与0.2g(2mmol)马来酸酐加入反应瓶,开启搅拌。常压下在160℃温度脱水2h,之后用循环水真空泵抽真空,预聚12h。预聚结束后,加入0.01g催化剂sno,用旋片式真空油泵抽真空,在180℃温度聚合4h。解除真空,加入2.7g(24mmol)n-异丙基丙烯酰胺和0.02g的过硫酸钾,150℃温度聚合6h。聚合结束后,用甲醇溶解产物,再用去离子水沉析、洗涤,过滤后,滤渣置于45℃真空干燥箱中干燥24h,得到共聚物plamn12,产率为53%。
结构表征:
以实施例2的plamn7为例,其ft-ir、1hnmr测试表征结果分别如图1和2所示。
图1的ft-ir分析(kbr,ν,cm-1):3514,o-h基团伸缩振动峰;2992、2943、2881,乳酸单元中饱和c-h基团的伸缩振动吸收峰;1188、1130、1094,乳酸单元中c-o-c基团的强伸缩振动吸收峰;1757,乳酸单元中c=o基团的强伸缩振动吸收峰;3351,pnipam中-nh-基团伸缩振动吸收峰;1383和1367,pnipam单元中异丙基上的两个-ch3特征吸收峰;1550和1649,pnipam单元中酰胺基的特征吸收峰。由此可知,乳酸单体与马来酸酐、n-异丙基丙烯酰胺等反应,成功地制备生成共聚物聚(乳酸-马来酸酐-n-异丙基丙烯酰胺)。
图2的1hnmr分析(δ,ppm):1.46~1.50(乳酸链段末端中-ch3,ha’),1.53~1.61(乳酸单元中的-ch3,ha),4.34~4.39(乳酸链段末端中-ch-,hb’),5.15~5.20(乳酸单元中的-ch-,hb);1.18~1.20(nipam单元中的-ch3,hg),1.68~1.94(nipam单元中的-ch-ch2-,hd),2.02~2.14(nipam单元中的-ch-ch2-,he),4.16-4.19(nipam中的-ch-,hf);2.24~2.40(mah单元中的-ch-,hc)。由此可进一步证实,聚(乳酸-马来酸酐-n-异丙基丙烯酰胺)如预期合成。
实施例1的plamn4和实施例3的plamn12具有相同的ft-ir、1hnmr测试表征结果,在此不再赘述。
性能测试:
(1)外观
以实施例1获得的plamn4为例,称取约5mgplamn4溶于0.5ml丙酮中,得到plamn4的丙酮溶液。另取25ml圆底烧瓶,加入约5ml蒸馏水,将上述丙酮溶液缓慢滴加到蒸馏水中,搅拌(1h或6h,搅拌过程中丙酮挥发掉),得1mg/ml的胶束溶液。
称取约2.5mgplamn4溶于0.5ml丙酮中。另取反应瓶加入约5ml蒸馏水,将上述丙酮溶液缓慢滴加到蒸馏水中,搅拌(6h),得0.5mg/ml的胶束溶液。
观察上述胶束溶液随温度、搅拌时间等变化时的变化情况,具体地,观察不同搅拌时间下制得的不同浓度的胶束溶液在25℃、40℃的外观,实验结果如图3所示。
从图3中不同温度、不同浓度梯度以及不同搅拌时间等影响的结果可见,plamn4的胶束溶液受浓度和温度因素的影响显著:在25℃下,搅拌不同时间后的不同浓度的胶束溶液呈现均匀的溶液形态,当温度升高时,plamn4疏水性能增加,溶解性变差后溶液开始变得浑浊(④相较①变浑浊,⑤相较②变浑浊,⑥相较③变浑浊),表现出明显的温敏特性。
同时,高浓度的胶束溶液在升温后会表现出更大的浊度(⑤比⑥更浑浊);相应地。受溶剂蒸发法制备过程中搅拌时间的影响,搅拌时间长的胶束溶液由于胶束分散得更均匀(②比①澄清,胶束分散更均匀),在温度升高后,沉淀的析出也更均匀,相对透光度更大(⑤比④的透光度更大)。
(2)dls测试
对实施例1~3中对应获得的共聚物plamn4、plamn7、plamn12进行dls的测试。具体地,分别称取2.0mg共聚物溶于1ml丙酮中。另取反应瓶加入10ml蒸馏水,将上述丙酮溶液缓慢滴加到蒸馏水中,搅拌24h。搅拌结束后再将反应瓶中的溶液转移至25ml圆底烧瓶中进行搅拌,约12h,使胶束分散更均匀,制备得到0.2mg/ml胶束溶液,随后进行dls测试。测试时,以2℃为一间隔,从20℃升温至48℃,得到不同温度下胶束溶液的粒径,实验结果如图4所示。
根据dls测试结果可得,plamn4的相变温度在30~32℃之间,plamn7的相变温度在28~32℃之间,plamn12的相变温度在28~32℃之间。发明人推测,共聚物plamn发生相变是由于亲水的pniam部分(n-异丙基丙烯酰胺聚合链段)作为壳,在水溶液中自行聚集成为一个胶束结构;随着温度的升高,壳上亲水的pnipam部分变得疏水,倾向于进入胶束的中心,因而导致粒径减小。dls测试结果进一步表明共聚物plamn具有一定的温敏特性。
(3)透光度
对实施例1~3中对应获得的共聚物plamn4、plamn7、plamn12进行透光度的测试。具体地,分别称取2.0mg共聚物溶于1ml丙酮中。另取25ml圆底烧瓶加入10ml蒸馏水,将上述丙酮溶液缓慢滴加到蒸馏水中,搅拌24h,制备得到0.2mg/ml胶束溶液,随后利用紫外可见分光光度计进行透光度测试。
测试时,以2℃为一间隔,从20℃升温至48℃,扫描不同波长时的胶束溶液的透光度,并取用500nm处的透光度值进行作图比较,实验结果如图5所示。
从图5不同温度胶束溶液透光度可见,plamn4、plamn7、plamn12的胶束溶液的初始透光度值在95%附近(plamn4约98%,plamn7约97%,plamn12约95%),透光度值随着温度的升高而减小,最终稳定在75%左右(plamn4约70%,plamn7约70%,plamn12约75%)。
参考文献定义[daix-h,jinh,caim-h,etal.reactfunctpolym,2015,89:9-17;zhangm,jinxq,gougj.jbiomatsci-polyme,2016,27(8):773-791],取透光度在升温前后变化50%时对应的温度为低临界溶解温度(lcst),得到的plamn4、plamn7、plamn12的lcst分别为30.0℃、30.2℃、30.4℃,由此得到共聚物plamn温敏特性的基本参数。
(4)cmc测试
通过荧光探针法测试实施例1~3中对应获得的共聚物plamn4、plamn7、plamn12的cmc(临界胶束浓度)。具体地,称取5.0mg聚合物溶于1ml丙酮中。另取25ml圆底烧瓶加入10ml蒸馏水,将上述丙酮溶液缓慢滴加到蒸馏水中,搅拌24h,制备得到0.5mg/ml的胶束溶液。类似地,依次配置0.001~0.5mg/ml的胶束溶液。
称取10mg芘于10ml干净的玻璃瓶中,加入5ml甲苯作为溶剂配制成母液,稀释1000倍后得到芘溶液。分别向若干个10ml的塑料管中加入5μl芘溶液,并使溶剂甲苯挥发。然后,在这些管中加入一系列5ml的浓度为0.001~0.5mg/ml的胶束溶液,摇匀静置2h,进行荧光激发光谱测试,实验结果如图6所示。
从图6不同浓度的胶束溶液的荧光激发光谱结果可知,当胶束溶液浓度增加时,可观察到吸收带从345nm附近蓝移到339nm附近,这种蓝移是由于芘分子从水环境转移到疏水性胶束核心而引起的,表明了胶束的形成。
进一步地,由不同浓度胶束溶液在345nm处和339nm处的荧光强度比值的突变,可以得到plamn的cmc。根据i345/i339的值与浓度作图,得到的结果如图7所示。由图可知,plamn4、plamn7、plamn12的cmc分别为7.76、4.17、5.62mg/l。
药物包封:
以实施例1的plamn4为例,将其用作药物载体,对具有消炎、抗菌的药物布洛芬进行包封,包封方法可分为物理方法和化学方法。物理方法:称取0.2gplamn4溶于1ml丙酮中。另取25ml圆底烧瓶加入10ml蒸馏水,将上述丙酮溶液缓慢滴加到蒸馏水中,搅拌24h,制备得到20mg/ml的胶束溶液;称取0.2g的缓释药物布洛芬,加入甲醇溶液中,配置成20mg/ml溶液,取100μl布洛芬甲醇溶液,静置挥发后,加入上述胶束溶液,制备物理负载的高分子缓释药物aa。
通过紫外光谱测定高分子缓释药物aa的吸光度值为2.343,根据布洛芬浓度与吸光度的标准曲线y=0.0253+1.2696x,r2=0.9998计算可得,药物含量为1.82mg/ml,载药量为9.1%。
化学方法:称取2gplamn4加入反应瓶中,继续加入0.2g的缓释药物布洛芬,升温至120℃,反应4h,反应结束后,用甲醇抽提、纯化,得化学键载布洛芬药物的高分子缓释药物bb。
将高分子缓释药物bb配制成16.5mg/ml的溶液,通过紫外光谱测定高分子缓释药物a的吸光度值为1.979,根据布洛芬浓度与吸光度的标准曲线y=0.0253+1.2696x,r2=0.9998计算可得,药物含量为1.54mg/ml,载药量为9.3%。
除了布洛芬外,本发明的聚乳酸高分子材料还可以负载其他药物,同样具有高载药量;此外,若聚乳酸高分子材料进一步纺丝制成纤维状等加工后可作为伤口敷料。
对比例1
本对比例与实施例3的不同之处在于:将乳酸单体替换成壳聚糖。
将磁力搅拌子置入反应瓶中,将约3.6g壳聚糖与0.2g马来酸酐加入反应瓶,开启搅拌。常压下在160℃温度脱水2h,之后用循环水真空泵抽真空,预聚12h。预聚结束后,加入0.01g催化剂sno,用旋片式真空油泵抽真空,在180℃温度聚合4h。解除真空,加入2.7g(24mmol)n-异丙基丙烯酰胺和0.02g的过硫酸钾,150℃温度聚合6h。聚合结束后,用甲醇溶解产物,再用去离子水沉析、洗涤,过滤后,滤渣置于45℃真空干燥箱中干燥24h,得到块状固体a,产率为57%。
经检测,本对比例的产品无法获得类似于以乳酸为原料时玻璃化转变温度的热性能以及结晶度的结晶性能结果,可能与壳聚糖中氨基干扰反应、而小分子单体自聚后水洗去掉有关,从而无法获得与实施例3相似的效果。
对比例2
本对比例与实施例3的不同之处在于:使乳酸、马来酸酐和n-异丙基丙烯酰胺进行一步法聚合。将3.6g乳酸与0.2g马来酸酐加入反应瓶中,同时加入0.01g催化剂sno,油浴升温至130℃反应2h,再升温至180℃反应4h,结束后降至室温,继续加入2.7g(24mmol)n-异丙基丙烯酰胺和0.02g的过硫酸钾,升温至65℃进行聚合,反应结束后得到胶黏剂共聚物b,产率为21%。
产品为粘稠状固体,粘性高,但无法获得类似于以乳酸等为原料分步聚合时玻璃化转变温度的热性能以及结晶度的结晶性能结果,可能与分子量太低等因素有关,无法获得与实施例3相似的效果。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!