人血清淀粉样蛋白Aβ1-42的制备方法与流程
人血清淀粉样蛋白a
β1‑
42的制备方法
技术领域
1.本发明涉及多肽合成技术领域,尤其涉及人血清淀粉样蛋白aβ1
‑
42的制备方法。
背景技术:
[0002]“阿尔茨海默症”(alzheimer disease),简称ad,是与老龄化有关的、以大脑神经细胞广泛死亡为特点的疾病,其结果是大脑神经细胞逐渐丧失,最终造成记忆力、判断决策力、方位感、注意力和语言能力的损伤。近年来阿茨海默症发病率呈明显上升趋势,我国的发病率为0.71%,约有1000万阿尔茨海默症患者。预计到2050年,我国阿尔茨海默症患者人数将达2700万。由于阿尔茨海默症是与年龄密切相关的疾病,在65岁的人群中约有10%的人患病,而在85岁人群中则约有50%的人患病。
[0003]
阿尔茨海默症的病理特征主要包括大脑皮质和海马区域的细胞外出现以β
‑
淀粉样蛋白(beta
‑
amyloid,aβ)为核心的老年斑、细胞内神经原纤维缠结等病变。aβ是由淀粉样前体蛋白经β
‑
和γ
‑
分泌酶的蛋白水解作用而产生的含有39~43个基酸的多肽,aβ的大量沉积,是导致ad病人脑内老年斑周边神经元变性和死亡的主要原因。aβ在脑内有多种存在形式,如:低分子量aβ单体、aβ寡聚体、不溶性aβ纤维体等。aβ单体是可溶性的低分子物质,但是当胞内环境改变时会诱发aβ单体快速聚集,形成二聚体、三聚体或可溶性的aβ寡聚体,aβ寡聚体的稳定性较差,易向aβ纤维体转化。aβ的种类分为aβ1
‑
39、aβ1
‑
40、aβ1
‑
41、aβ1
‑
42和aβ1
‑
43,其中aβ1
‑
42的毒性最大,具有强水性、易聚集和较强的神经毒性作用,是ad的核心致病物质。因此,研究aβ1
‑
42是当前的热点。
[0004]
aβ1
‑
42的氨基酸序列自n端至c端为:daefrhdsgyevhhqklvffaedvgsnkgaiiglmvggvvia。
[0005]
国内外合成aβ1
‑
42的寡聚体均不稳定,淀粉样肽在体外容易随机形成各种高分子量或低分子量的混合产物,这种组分不均一的混合物直接影响到实验结果的准确性和可靠性。近年来,许多研究者用多种方法制备aβ1
‑
42寡聚体,但收率都较低、中间产物不稳定、成分复杂。aβ1
‑
42由于其高度疏水性以及容易形成聚集,常规固相合成的方式很难制备出aβ1
‑
42。进一步开发固相合成多肽aβ1
‑
42的方法,可为ad科研实验打下良好的基础,也为ad的发病机制、病理过程、寻找和筛选有效药物提供有效依据。
技术实现要素:
[0006]
有鉴于此,本发明要解决的技术问题在于提供人血清淀粉样蛋白aβ1
‑
42的制备方法,该方法以侧链活性基团全保护的氨基酸为原料,降低了合成过程中发生副反应的几率;并且各步骤中,控制合成参数,选择合理的反应试剂,从而提高了产物的收率,减少了副反应的发生。
[0007]
本发明提供的人血清淀粉样蛋白aβ1
‑
42的制备方法,包括:
[0008]
以boc
‑
ser(fmoc
‑
gly)
‑
oh为原料之一,按照人血清淀粉样蛋白aβ1
‑
42的氨基酸序列制备26
‑
氧酰异aβ1
‑
42,经酰基迁移得到人血清淀粉样蛋白aβ1
‑
42;
[0009]
偶联1~25位中任意氨基酸或多肽后,脱除fmoc的步骤中采用8vol%~12vol%哌啶/dmf溶液。
[0010]
本发明中,采用逐个偶联的方式合成26
‑
氧酰异aβ1
‑
42,其中,对第25~26位氨基酸的偶联以boc
‑
ser(fmoc
‑
gly)
‑
oh为原料。对其他氨基酸的偶联采用fmoc保护的氨基酸原料。使用boc
‑
ser(fmoc
‑
gly)
‑
oh为原料,降低了该步骤偶联的困难,能够提高反应效率,提高产物的纯度。但是采用boc
‑
ser(fmoc
‑
gly)
‑
oh为原料合成后,肽链中存在氧酰异基团,为了维持该基团的稳定性,在之后偶联的步骤中都必须对脱除fmoc保护基的条件进行优化。
[0011]
本发明实施例中,所述制备方法为固相合成法。本发明中,所述固相合成的载体为ctc树脂或wang树脂。采用2
‑
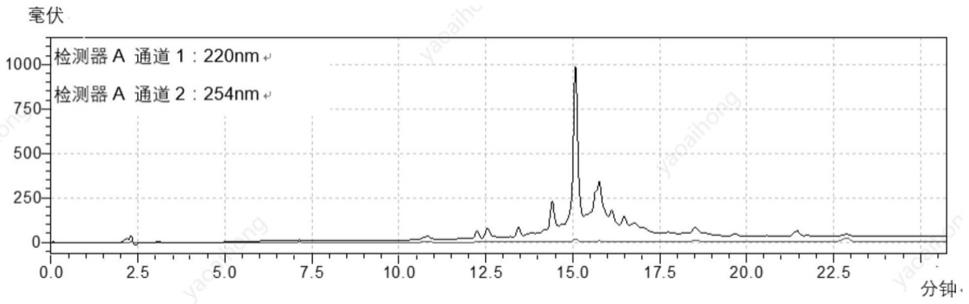
氯三苯甲基氯树脂为固相载体,避免c端氨基酸与树脂酯化发生消旋的风险;一些实施例中,载体为ctc树脂。
[0012]
一些实施例中,本发明多肽的合成具体包括如下步骤:
[0013]
步骤1:制备fmoc
‑
ala
‑
ctc树脂;
[0014]
步骤2:在fmoc
‑
ala
‑
ctc树脂的基础上,依次偶联氨基酸,其中第25~26位氨基酸的偶联以boc
‑
ser(fmoc
‑
gly)
‑
oh为原料,制得26
‑
氧酰异aβ1
‑
42
‑
肽树脂;
[0015]
步骤3:切割肽树脂后,26
‑
氧酰异aβ1
‑
42粗品经纯化、氨基迁移酰基迁移得到人血清淀粉样蛋白aβ1
‑
42。
[0016]
本发明中,偶联第41位氨基酸的步骤中,偶联剂为hatu/diea,或pybop/diea。
[0017]
一些实施例中,所述hatu和diea的摩尔比为1:3;所述pybop和diea的摩尔比为1:3。
[0018]
在偶联boc
‑
ser(fmoc
‑
gly)
‑
oh的步骤中,依次以dic/hobt和hatu/hobt/diea为偶联试剂,经过两次偶联反应。
[0019]
在偶联32位和31位的步骤中,依次以dic/hobt和hatu/hobt/diea为偶联试剂,经过两次偶联反应。
[0020]
本发明中,除第41位、32位、31位、25
‑
26位氨基酸外,其他偶联步骤的偶联剂选自dic/hobt、hatu/hobt/diea、hbtu/hobt/diea或pybop/hobt/diea。
[0021]
一些实施例中,其他偶联步骤的偶联剂为dic/hobt。
[0022]
本发明中,偶联第32位和第31位氨基酸的步骤中,氨基酸的投料量为5eq。
[0023]
本发明中,除偶联第32位和第31位氨基酸外,其他偶联步骤中,氨基酸的投料量为3eq。
[0024]
本发明中,所述偶联的溶剂选自dmf、ch2cl2、nmp。
[0025]
一些实施例中,所述偶联的溶剂为dmf。
[0026]
本发明中,偶联1~25位中任意氨基酸或多肽后,所述脱除fmoc的试剂为10vol%哌啶/dmf溶液,条件为20~25℃,5min,脱除2次;
[0027]
偶联27~42位中任意氨基酸或多肽后,脱除fmoc的试剂为20vol%哌啶/dmf溶液,条件为20~25℃,5~30min。
[0028]
一些实施例中,所述固相合成后,切割树脂的裂解剂为苯甲硫醚、苯酚、edt、碘化铵和水中的至少一种与tfa的混合液。
[0029]
所述裂解剂中tfa的浓度不小于80%。在切割试剂中加入碘化铵,能够避免了氧化杂质met(o)的产生。一些具体实施例中,所述裂解剂由tfa、苯甲硫醚、苯酚、edt、水和碘化
胺组成。其中tfa:苯甲硫醚:苯酚:edt:水=87.5:2.5:2.5:5:2.5;碘化胺的浓度为1mg/ml。
[0030]
所述裂解的步骤为:26
‑
氧酰异aβ1
‑
42
‑
树脂复合物中加入裂解剂进行反应,反应温度为0℃~40℃,反应时间为1小时~4小时。
[0031]
本发明中,所述酰基迁移为将26
‑
氧酰异aβ1
‑
42溶解于乙腈水溶液中,以氨水调节ph值为7.4,25℃反应4小时,制得aβ1
‑
42。
[0032]
本发明提供的人血清淀粉样蛋白aβ1
‑
42的制备方法,以boc
‑
ser(fmoc
‑
gly)
‑
oh为原料之一,按照人血清淀粉样蛋白aβ1
‑
42的氨基酸序列制备26
‑
氧酰异aβ1
‑
42,经酰基迁移得到人血清淀粉样蛋白aβ1
‑
42;该方法以侧链活性基团全保护的氨基酸为原料,降低了合成过程中发生副反应的几率;并且各步骤中,控制合成参数,从而提高了产物的收率,减少了副反应的发生。
附图说明
[0033]
图1示26
‑
氧酰异aβ1
‑
42粗品高效液相色谱图;
[0034]
图2示aβ1
‑
42纯品高效液相色谱图;
[0035]
图3示aβ1
‑
42纯品质谱图;
[0036]
图4示aβ1
‑
42纯品高效液相色谱图;
[0037]
图5示aβ1
‑
42纯品质谱图;
[0038]
图6示aβ1
‑
42纯品高效液相色谱图;
[0039]
图7示aβ1
‑
42纯品质谱图;
[0040]
图8示aβ27
‑
42质谱图;
[0041]
图9示26
‑
氧酰异aβ1
‑
42粗品质谱图;
[0042]
图10示26
‑
氧酰异aβ1
‑
42粗品高效液相色谱图。
具体实施方式
[0043]
本发明提供了人血清淀粉样蛋白aβ1
‑
42的制备方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
[0044]
本发明采用的试材皆为普通市售品,皆可于市场购得。
[0045]
除非另有定义,本文使用的所有科技术语具有本领域普通技术人员所理解的相同含义。
[0046]
本发明中,所述“多肽”或“肽”具有其在本领域中的普通含义并且可指通过由一个氨基羧酸分子的羰基碳与另一个氨基羧酸分子的氮原子形式上失去水而形成共价键之来自两个或更多个氨基羧酸分子(相同或不同)的酰胺。“氨基酸残基”也具有其在本领域中的普通含义并且指氨基酸(作为单个氨基酸或作为肽的一部分)在其与肽、另一个氨基酸或氨基酸残基组合之后的组合物。通常,当氨基酸与另一个氨基酸或氨基酸残基组合时,除去水,并且保留下来的氨基酸被称为氨基酸残基。
[0047]
所述“氨基酸”也具有其在本领域中的普通含义并且可包括蛋白质氨基酸和非蛋
白质氨基酸。本发明中氨基酸残基的缩写是本领域中所用的指代20个常用l
‑
氨基酸之一的标准3字母和/或1字母代码。
[0048]
本发明所述“保护基团”或“保护基”具有其在本领域中的普通含义。保护基团包括连接至或被配置成连接至分子(例如,肽)内的反应性基团(即,受保护的基团)以使得所述保护基团阻止或以其他方式抑制受保护的基团参与反应的化学部分。可通过将保护基团连接至分子来进行保护。当将保护基团从分子中除去时,例如,通过除去保护基团的化学转化,可发生去保护。
[0049]
本发明合成多肽采用的氨基酸原料中,所有氨基酸的α氨基均为fmoc保护基,丝氨酸除了第26位氨基酸(ser
26
)之外,其余侧链羟基保护基为叔丁基,天冬氨酸和谷氨酸侧链羧基保护基为叔丁基,谷氨酰胺和天冬酰胺侧链酰胺保护基为三苯甲基,组氨酸侧链咪唑保护基为三苯甲基或叔丁氧羰基,赖氨酸和色氨酸侧链保护基为叔丁氧羰基,精氨酸侧链胍基保护基为2,2,4,6,7
‑
五甲基二氢苯并呋喃
‑5‑
磺酰基。具体的,本发明采用的氨基酸原料包括:甘氨酸(gly,g):fmoc
‑
gly
‑
oh;精氨酸(arg,r):fmoc
‑
arg(pbf)
‑
oh;缬氨酸(val,v):fmoc
‑
val
‑
oh;亮氨酸(leu,l):fmoc
‑
leu
‑
oh;色氨酸(trp,w):fmoc
‑
trp(boc)
‑
oh;丙氨酸(ala,a):fmoc
‑
ala
‑
oh;异亮氨酸(ile,i):fmoc
‑
ile
‑
oh;苯丙氨酸(phe,f):fmoc
‑
phe
‑
oh;谷氨酸(glu,e):fmoc
‑
glu(otbu)
‑
oh;丙氨酸(ala,a):fmoc
‑
ala
‑
oh;谷氨酰胺(gln,q):fmoc
‑
gln(trt)
‑
oh;甲硫氨酸(met,m):fmoc
‑
met
‑
oh;丝氨酸(ser,s):fmoc
‑
ser(tbu)
‑
oh;亮氨酸(leu,l):fmoc
‑
leu
‑
oh;苏氨酸(thr,t):fmoc
‑
thr(tbu)
‑
oh;赖氨酸(lys,k):fmoc
‑
lys(boc)
‑
oh;组氨酸(his,h):boc
‑
his(trt)
‑
oh;gly
25
‑
ser
26
:boc
‑
ser(fmoc
‑
gly)
‑
oh;酪氨酸(tyr,y):fmoc
‑
tyr(tbu)
‑
oh;天冬氨酸(asp,d):fmoc
‑
asp(otbu)
‑
oh。
[0050]
本发明中所述“偶联”是指将新的氨基酸添加至结合的氨基酸或肽的过程。多肽的合成包括固相合成法、液相合成法,也有报道采用固相和液态结合的方式进行。本领域中,所述偶联可采用逐一氨基酸顺序偶联的方式进行,也可根据肽序采用分段偶联的方式对多个多肽片段进行偶联,本发明实施例中,所述制备采用固相法,偶联为逐个偶联。
[0051]
本发明提供的人血清淀粉样蛋白aβ1
‑
42的制备方法,包括:
[0052]
以boc
‑
ser(fmoc
‑
gly)
‑
oh为原料之一,按照人血清淀粉样蛋白aβ1
‑
42的氨基酸序列制备26
‑
氧酰异aβ1
‑
42,经酰基迁移得到人血清淀粉样蛋白aβ1
‑
42;偶联1~25位中任意氨基酸或多肽后,脱除fmoc的步骤中采用10vol%哌啶/dmf溶液。
[0053]
一些实施例中,本发明多肽的合成具体包括如下步骤:
[0054]
步骤1:制备fmoc
‑
ala
‑
ctc树脂;
[0055]
步骤2:在fmoc
‑
ala
‑
ctc树脂的基础上,依次偶联氨基酸,其中第25~26位氨基酸的偶联以boc
‑
ser(fmoc
‑
gly)
‑
oh为原料,制得26
‑
氧酰异aβ1
‑
42
‑
肽树脂;
[0056]
步骤3:切割肽树脂后,26
‑
氧酰异aβ1
‑
42粗品经纯化、氨基迁移酰基迁移得到人血清淀粉样蛋白aβ1
‑
42。
[0057]
具体的,在本发明实施例中,所述依次偶联是指,依照人血清淀粉样蛋白aβ1
‑
42的肽序,首先合成fmoc
‑
ala
‑
ctc,在此基础上,ile、val、val、gly、gly、val、met、leu、gly、ile、ile、ala、gly、lys、asn、gly
‑
ser、val、leu、lys、gln、his、his、val、gln、tyr、gly、ser、asp、his、arg、phe、glu、ala和asp,制成26
‑
氧酰异aβ1
‑
42
‑
ctc树脂。
[0058]
本发明中,每个所述的偶联反应包括:脱除fmoc保护和进行缩合反应两个步骤。对
于脱除fmoc保护的步骤,在偶联第25~26位氨基酸前,即偶联boc
‑
ser(fmoc
‑
gly)
‑
oh前,因不含有氧酰异保护基团,因此,各步骤的fmoc脱除试剂采用20vol%的哌啶/dmf溶液,脱除fmoc的条件为20~25℃,5min,脱除2次;而在偶联boc
‑
ser(fmoc
‑
gly)
‑
oh后的脱除fmoc步骤中,脱除试剂为10vol%哌啶/dmf溶液,脱除条件为20~25℃,5~30min。
[0059]
本发明研究发现,第42位氨基酸与第41位氨基酸的偶联反应非常不稳定,反应效率较低且容易产生副产物。目前,尚未探明副产物的具体结构,经质谱检测其分子量要大于目标分子量。在改进了该步骤的偶联条件后,解决了这种不稳定的问题,获得产物中不存在副产物。本发明在偶联第41位氨基酸的步骤中,采用的偶联剂为hatu/diea,或pybop/diea。实验表明,采用这两种偶联剂可以维持反应的正常进行。一些实施例中,所述hatu和diea的摩尔比为1:3;所述pybop和diea的摩尔比为1:3。在偶联第41位氨基酸的反应中,氨基酸与diea的摩尔比为1:3。
[0060]
在偶联boc
‑
ser(fmoc
‑
gly)
‑
oh的步骤中,依次以dic/hobt和hatu/hobt/diea为偶联试剂,经过两次偶联反应;第一次偶联的溶剂为dmf,第二次偶联的溶剂为dmf和ch2cl2,其中dmf:ch2cl2的体积比为4:1。第一次偶联的时间为40min,之后以dmf洗涤;第二次偶联的时间为40min。其中,dic与hobt的摩尔比为1:1.25;hatu、hobt与diea的摩尔比为1:1:2。
[0061]
本发明中,eq为摩尔当量,是一个在化学或生物科学中使用的单位,用于表示物质的量。在一般的偶联步骤中,氨基酸的添加量为3eq,但是在偶联第32位和第31位氨基酸的步骤中,由于缩合反应不容易发生,如果氨基酸的投料量不足,则会导致合成效率的严重下降。本发明研究表明,当提高氨基酸投料量后,缩合反应效率显著提高。因此,在本发明中,偶联第32位和第31位氨基酸的步骤中,氨基酸的投料量为5eq。除偶联第32位和第31位氨基酸外,其他偶联步骤中,氨基酸的投料量为3eq。
[0062]
此外,对于第32位和第31位氨基酸的偶联效果还与偶联剂的选择密切相关,在这两位氨基酸的偶联中,每个氨基酸的偶联都依次以dic/hobt和hatu/hobt/diea为偶联试剂,经过两次偶联反应;第一次偶联的溶剂为dmf,第二次偶联的溶剂为dmf和ch2cl2,其中dmf:ch2cl2的体积比为4:1。第一次偶联的时间为40min,之后以dmf洗涤;第二次偶联的时间为40min。其中,dic与hobt的摩尔比为1:1.25;hatu、hobt与diea的摩尔比为1:1:2。
[0063]
本发明中,除第41位、32位、31位、25
‑
26位氨基酸外,其他偶联步骤的偶联剂为dic/hobt、hatu/hobt/diea、hbtu/hobt/diea或pybop/hobt/diea中的任意一种或多种。一些实施例中,其他偶联步骤的偶联剂为dic/hobt,在这些偶联步骤中,各反应物的物质的量之比为n氨基酸:hobt:dic=1:1.25:1。所述偶联的溶剂选自dmf、ch2cl2、nmp。一些实施例中,所述偶联的溶剂为dmf。
[0064]
具体的,在本发明中,26
‑
氧酰异aβ1
‑
42
‑
ctc树脂的制备包括:
[0065]
①
制备fmoc
‑
ala
–
ctc树脂,以20%pip/dmf脱除fmoc保护基团,脱除时间为25min,然后与fmoc
‑
ile
‑
oh偶联,偶联剂为diea/hatu(摩尔比3:1)或者为pybop/diea,溶剂为dmf,氨基酸的投料量为3eq;
[0066]
②
依次偶联ile、val、val、gly、gly、val、met、leu、gly;各步偶联以20%pip/dmf反应25min脱除fmoc保护,偶联剂为dic/hobt(摩尔比8.6:6.9)或者偶联剂为pybop/hobt(摩尔比为1:1),溶剂为dmf,氨基酸的投料量为3eq;
[0067]
③
偶联第31位氨基酸和第32位氨基酸,以各步偶联以20%pip/dmf反应25min脱除
fmoc保护,fmoc
‑
ile
31
‑
oh和fmoc
‑
ile
32
‑
oh投料量均为5eq,偶联过程中先以dic/hobt(摩尔比1:1.25)为偶联剂,以dmf为溶剂,偶联40min后,以dmf洗涤后再以hatu/hobt/diea(摩尔比1:1:2)为偶联剂,以dmf为溶剂,偶联40min;
[0068]
④
依次偶联ala、gly、lys、asn,各步偶联以20%pip/dmf反应25min脱除fmoc保护,偶联剂为dic/hobt(摩尔比8.6:6.9)或者偶联剂为pybop/hobt(摩尔比为1:1),溶剂为dmf,各氨基酸的投料量为3eq;
[0069]
⑤
偶联gly
‑
ser,以20%pip/dmf反应25min脱除fmoc保护,boc
‑
ser(fmoc
‑
gly)
‑
oh的投料量为3eq,偶联过程中先以dic/hobt(摩尔比1:1.25)为偶联剂,以dmf为溶剂,偶联40min后,以dmf洗涤后再以hatu/hobt/diea(摩尔比1:1:2)为偶联剂,以dmf/dcm为溶剂(体积比4:1),偶联40min;
[0070]
⑥
依次偶联val、asp、glu、ala、phe、phe、val、leu、lys、gln、his、his、val、glu、tyr、gly、ser、asp、his、arg、phe、glu、ala和asp,各步偶联以10%pip/dmf反应5min,重复2次脱除fmoc保护,偶联剂为dic/hobt(摩尔比8.6:6.9)或者偶联剂为pybop/hobt(摩尔比为1:1),溶剂为dmf,各氨基酸的投料量为3eq。
[0071]
本发明中,所述固相合成后,切割树脂的裂解剂为苯甲硫醚、苯酚、edt、碘化铵和水中的至少一种与tfa的混合液。所述裂解剂中tfa的浓度不小于80%。在切割试剂中加入碘化铵,能够避免了氧化杂质met(o)的产生。一些具体实施例中,所述裂解剂由tfa、苯甲硫醚、苯酚、edt、水和碘化胺组成。其中tfa:苯甲硫醚:苯酚:edt:水=87.5:2.5:2.5:5:2.5;碘化胺的浓度为1mg/ml。所述裂解的步骤为:26
‑
氧酰异aβ1
‑
42
‑
树脂复合物中加入裂解剂进行反应,反应温度为0℃~40℃,反应时间为1小时~4小时。
[0072]
切割树脂后,获得26
‑
氧酰异aβ1
‑
42粗品,以高效液相色谱纯化,获得26
‑
氧酰异aβ1
‑
42纯品。纯化的色谱条件为,色谱柱:30mm的聚合物(聚苯乙烯,粒径10um 300a,色谱柱长度25cm,直径是30mm)或c18柱,流动相a:0.25%甲酸水溶液,流动相b:0.1%tfa/乙腈溶液;b相在0
‑
5min由5%变到22%;在5
‑
50min内,b相由22%变到42%,运行45min,流动相温度为60℃。
[0073]
本发明中,所述酰基迁移为将26
‑
氧酰异aβ1
‑
42溶解于乙腈水溶液中,以氨水调节ph值为7.4,25℃反应4小时,制得aβ1
‑
42。
[0074]
在具体实施例中,本发明制备方法进一步优选包括如下步骤:
[0075]
(1)制备fmoc
‑
ala
‑
树脂复合物。
[0076]
向带有过滤装置的杯状反应容器中加入树脂。向所述容器中加入溶剂dcm溶胀树脂30min,用dcm溶解fmoc
‑
ala
‑
oh和酯化试剂后,加入到树脂中,在25℃反应70min。反应结束后,加入树脂体积三分之一倍的甲醇/diea混合溶液(体积比为1:1),室温反应30min后过滤。然后加入溶剂dmf,洗涤树脂五次,抽滤。其中所述的树脂为2
‑
氯三苯甲基氯树脂,所述的酯化试剂为diea,各物质间的比例为n(fmoc
‑
ala
‑
oh):n(diea):n(2
‑
氯三苯甲基氯树脂)=0.3:1:1。
[0077]
(2)制备保护的26
‑
氧酰异aβ1
‑
42树脂复合物。
[0078]
在上述的fmoc
‑
ala
‑
树脂复合物中加入体积比为20%pip/dmf溶液,进行脱保护反应,25℃下反应25min。反应完毕后,抽滤。然后加入dmf洗涤树脂5次。
[0079]
向反应器中加入c端第二个氨基酸fmoc
‑
ile
‑
oh和缩合试剂,25℃下反应70min。反
应结束后加入dmf洗涤树脂,过滤。所述的缩合试剂优选为hatu/diea,pybop/diea。其中所述的反应物物质的量之比为:nfmoc
‑
ile
‑
oh:hatu:diea=1:1:3。
[0080]
按照26
‑
氧酰异aβ1
‑
42从c端到n端的顺序,依次偶联氨基酸,循环脱保护反应和缩合反应的操作,缩合试剂可为dic/hobt、hatu/hobt/diea、hbtu/hobt/diea和pybop/hobt/diea中的一种或多种,溶剂为dmf、ch2cl2、nmp中的一种或两种的混合物,合成26
‑
氧酰异aβ1
‑
42
‑
树脂复合物,其中优选的反应物物质的量之比为:n氨基酸:hobt:dic=1:1.25:1。
[0081]
其中gly25
‑
ser26用boc
‑
ser(fmoc
‑
gly)
‑
oh为原料的偶联优选方法为用dic/hobt,溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf:ch2cl2为4:1。
[0082]1‑
25位氨基酸的fmoc的脱除优选10%哌啶/dmf溶液,时间为2次5min的方式。
[0083]
(3)制备26
‑
氧酰异aβ1
‑
42粗品。
[0084]
向保护的26
‑
氧酰异aβ1
‑
42树脂复合物中加入裂解试剂,裂解试剂是tfa、苯甲硫醚、苯酚、edt、碘化铵和水中的至少一种与tfa的混合液,所述的混合液中tfa的浓度不小于80%;向保护的26
‑
氧酰异aβ1
‑
42
‑
树脂复合物中加入切割试剂进行反应的温度为0℃~40℃,反应时间为1小时~4小时。优选比例为tfa:苯甲硫醚:苯酚:edt:水=87.5:2.5:2.5:5:2.5,加入碘化铵(1mg/ml),裂解试剂在0
‑
10度下预先冷却;于冰水浴中将裂解试剂加入到26
‑
氧酰异aβ1
‑
42树脂肽中,反应30min之后,逐渐升至25℃,继续裂解2h。裂解液中加入预冷的乙醚离心沉淀后得到26
‑
氧酰异aβ1
‑
42粗品;
[0085]
(4)制备aβ1
‑
42纯品。
[0086]
将26
‑
氧酰异aβ1
‑
42粗品先冻干后。冻干固体用体积比为5%的甲醇或乙腈水溶液溶解,采用制备型高效液相色谱纯化,得到26
‑
氧酰异aβ1
‑
42精品。纯化的色谱条件为,色谱柱为30mm的聚合物(聚苯乙烯,粒径10um 300a,色谱柱长度25cm,直径是30mm),流动相a相为0.25%甲酸水溶液,b相为0.1%tfa/乙腈溶液;b相在0
‑
5min由5%变到22%;在5
‑
50min内,b相由22%变到42%,运行45min,流动相温度为60℃。
[0087]
另外,本发明中一些常用的缩写具有如下含义:
[0088]
本发明涉及到的缩合试剂英文缩写含义如下所示:2
‑
ctc树脂:2
‑
氯三苯基氯树脂diea:n,n
‑
二异丙基乙胺wang树脂:对烷氧基苯甲醇树脂hbtu:苯并三氮唑
‑
n,n,n’,n
’‑
四甲基脲六氟磷酸酯hatu:2
‑
(7
‑
氮杂苯并三氮唑)
‑
四甲基脲六氟磷酸盐pybop:六氟磷酸苯并三氮唑
‑1‑
基
‑
氧基三吡咯烷dic:n,n
‑
二异丙基碳二亚胺tfa:三氟乙酸hobt:1
‑
羟基苯并三唑dcm:二氯甲烷nmp:n
‑
甲基吡咯烷酮pip:哌啶fmoc:芴甲氧羰酰基
dbu:1,8
‑
二氮杂二环十一碳
‑7‑
烯edt:1,2
‑
乙二硫醇dmf:n,n
‑
二甲基甲酰胺obzl:n
‑
叔丁氧羰基
‑
l
‑
丝氨酸苄酯edc
·
hcl:1
‑
乙基
‑
(3
‑
二甲基氨基丙基)碳酰二亚胺盐酸盐dmap:4
‑
二甲氨基吡啶ar:氩气pe:ea石油醚:乙酸乙酯
[0089]
下面结合实施例,进一步阐述本发明:
[0090]
实施例1:boc
‑
ser(fmoc
‑
gly)
‑
oh的合成
[0091]
在1l单口瓶中加入化合物fmoc
‑
gly
‑
oh(2.97g,10mmol),化合物boc
‑
ser
‑
obzl(2.95g,88.4mmol),edc.hcl(2.3g,12mmol),加500ml dcm溶解完全,加入dmap(0.122g,0.2mmol),ar保护,反应约5h,取样hplc检测反应完全;反应液用1mol/l hcl水溶液洗2次,饱和食盐水洗2次,无水硫酸镁干燥,蒸干,得白色固体5.1g;将白色固体溶于100ml meoh,加10%pd/c,h2置换后反应2.5h,取样点板检测,化合物反应完全;滤去pd/c,蒸干,过硅胶柱;展开剂:pe:ea=3:1加入1滴乙酸;洗脱剂:pe:ea=5:1;得到纯品2.84g。
[0092]
实施例2:制备aβ1
‑
42
[0093]
(1)fmoc
‑
ala
‑
ctc树脂的合成。
[0094]
称取取代度为0.98mmol/g的2
‑
ctc树脂10g,加入到杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;称取3mmol fmoc
‑
ala
‑
oh,加入dcm和diea于反应杯中,氮气鼓泡反应90min之后;加入3ml甲醇和diea封端反应30min,dmf洗涤4次,dcm洗涤1次之后用甲醇抽干,可得到fmoc
‑
gly
‑
ctc树脂;取少量树脂用紫外分光光度计测得取代度为0.23mmol/g(三次平均值)。
[0095]
(2)2
‑
ctc树脂ile
41
的偶联。
[0096]
将实施例1制备的取代度为0.23mmol/g的fmoc
‑
ala
‑
ctc树脂于杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;加入20%pip/dmf脱除fmoc保护基团,脱除时间为25min,再加入dmf洗涤5次,dcm洗1次,dmf洗涤1次,茚三酮检测树脂显色,同时需要保证哌啶洗涤干净无残留。称取2.4g fmoc
‑
ile
‑
oh(6.9mmol),2.6g hatu(6.9mmol),溶解于50ml dmf中,加入3.5ml diea(21mmol)反应2min之后,倒入杯状反应柱中,氮气鼓泡反应70min。
[0097]
(3)制备保护的26
‑
氧酰异aβ1
‑
42树脂复合物。
[0098]
向反应器中加入100ml 20%pip/dmf溶液室温反应25min,再加入dmf洗涤6次,dcm洗1次,茚三酮检测树脂显色。6.9mmol fmoc
‑
val
‑
oh,8.6mmol hobt及6.9mmol dic,溶于50ml dmf中,将所得溶液加入到反应器中,25℃下反应70min。反应结束后,过滤。然后加入80ml dmf,洗涤树脂六遍。
[0099]
按照26
‑
氧酰异aβ1
‑
42c端到n端的顺序,依次偶联氨基酸,循环脱保护反应和缩合反应。其中fmoc
‑
ile
31
‑
oh,fmoc
‑
ile
32
‑
oh的偶联方法为氨基酸投料5eq,反应浓度为0.3mmol/ml,dic/hobt为偶联试剂(氨基酸:dic:hobt=1:1:1.25),溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf。
[0100]
boc
‑
ser(fmoc
‑
gly)
‑
oh的偶联方法为氨基酸投料3eq,反应浓度为0.3mmol/ml,
dic/hobt为偶联试剂,溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf:dcm=4:1。
[0101]1‑
25位氨基酸的fmoc的脱除采用10%哌啶/dmf溶液,时间为2次5min的方式。
[0102]
其他氨基酸fmoc的脱除采用20%哌啶/dmf溶液,时间为25min,脱除一次。
[0103]
(4)制备保护的26
‑
氧酰异aβ1
‑
42粗品。
[0104]
将上述26
‑
氧酰异aβ1
‑
42树脂复合物1g放置于冰水浴中,将裂解试剂(质量比为tfa:苯甲硫醚:苯酚:edt:水=87.5:2.5:2.5:5:2.5,加入碘化铵(1mg/ml))在0~10℃下预先冷却,加入到26
‑
氧酰异aβ1
‑
42树脂肽中;于冰水浴中先反应30min之后,逐渐升至25℃,继续裂解2h。裂解液中加入预冷的15倍体积的乙醚离心沉淀后得到26
‑
氧酰异aβ1
‑
42粗品270mg,粗品分析图见图1;
[0105]
(5)制备aβ1
‑
42纯品。
[0106]
将270mg 26
‑
氧酰异aβ1
‑
42粗品溶于体积比为5%的甲醇水溶液中,经高效液相色谱纯化得到26
‑
氧酰异aβ1
‑
42精品,纯化条件包括:高效液相色谱,色谱柱为30mm的聚合物(聚苯乙烯,粒径10um 300a,色谱柱长度25cm,直径是30mm),流动相a相为0.25%甲酸水溶液,b相为0.1%tfa/乙腈溶液;b相在0
‑
5min由5%变到22%;在5
‑
50min内,b相由22%变到42%,运行45min,流动相温度为60℃。纯化后,经冷冻干燥后得64.8mg纯品,纯度为98.798%,总产率12.49%。
[0107]
将纯品溶解于乙腈/水(体积比为1:1)中至纯品浓度为1mg/ml,加入千分之一氨水调ph=7.4,25℃下反应4小时得到aβ1
‑
42,冷冻干燥后获得固体58.3mg,纯度为98.298%。高效液相色谱图见图2,质谱图见图3。
[0108]
实施例3:制备aβ1
‑
42
[0109]
(1)fmoc
‑
ala
‑
ctc树脂的合成。
[0110]
称取取代度为0.98mmol/g的2
‑
ctc树脂10g,加入到杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;称取1mmol fmoc
‑
ala
‑
oh,加入dcm和diea于反应杯中,氮气鼓泡反应90min之后;加入3ml甲醇和diea封端反应30min,dmf洗涤4次,dcm洗涤1次之后用甲醇抽干,可得到fmoc
‑
gly
‑
ctc树脂;取少量树脂用紫外分光光度计测得取代度为0.07mmol/g(三次平均值)。
[0111]
(2)2
‑
ctc树脂ile
41
的偶联。
[0112]
将实施例1制备的取代度为0.07mmol/g的fmoc
‑
ala
‑
ctc树脂于杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;加入20%pip/dmf脱除fmoc保护基团,脱除时间为25min,再加入dmf洗涤5次,dcm洗1次,国药dmf洗涤1次,茚三酮检测树脂显色,同时需要保证哌啶洗涤干净无残留。称取0.74g fmoc
‑
ile
‑
oh(2.1mmol),0.78g hatu(2.1mmol),溶解于15ml dmf中,加入1.05ml diea(6.3mmol)反应2min之后,倒入杯状反应柱中,氮气鼓泡反应70min。
[0113]
(3)制备保护的26
‑
氧酰异aβ1
‑
42树脂复合物。
[0114]
向反应器中加入100ml 20%pip/dmf溶液室温反应25min,再加入dmf洗涤6次,dcm洗1次,茚三酮检测树脂显色。取2.1mmol fmoc
‑
val
‑
oh,2.1mmol hatu,2.1mmol hobt及6.3mmol diea,溶于15ml dmf中,将所得溶液加入到反应器中,25℃下反应70min。反应结束后,过滤。然后加入25ml dmf,洗涤树脂六遍。
[0115]
按照26
‑
氧酰异aβ1
‑
42c端到n端的顺序,依次偶联氨基酸,循环脱保护反应和缩合反应。当肽链长度为16个氨基酸的时候,取少量树脂做质谱检测,质谱图见图4,aβ27
‑
42目标分子量为1511.9,检测分子量是1511.6(二价为756.8),1397.8(二价为699.9)是缺失ile的分子量。boc
‑
ser(fmoc
‑
gly)
‑
oh的偶联方法为氨基酸投料3eq,反应浓度为0.3mmol/ml,dic/hobt为偶联试剂,溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf:dcm=4:1。
[0116]1‑
25位氨基酸的fmoc的脱除采用10%哌啶/dmf溶液,时间为2次5min的方式。其他氨基酸fmoc的脱除采用20%哌啶/dmf溶液,时间为25min,脱除一次。
[0117]
(4)制备保护的26
‑
氧酰异aβ1
‑
42粗品。
[0118]
将上述26
‑
氧酰异aβ1
‑
42树脂复合物1g放置于冰水浴中,将裂解试剂加入到26
‑
氧酰异aβ1
‑
42树脂肽中,质量比为tfa:苯甲硫醚:苯酚:edt:水=87.5:2.5:2.5:5:2.5,加入碘化铵(1mg/ml),裂解试剂在0~10℃下预先冷却;于冰水浴中先反应30min之后,逐渐升至25℃,继续裂解2h。裂解液中加入预冷的15倍体积的乙醚离心沉淀后得到26
‑
氧酰异aβ1
‑
42粗品粗品112mg;
[0119]
(5)制备aβ1
‑
42纯品。
[0120]
将26
‑
氧酰异aβ1
‑
42粗品溶于体积比为5%的甲醇水溶液中,经高效液相色谱纯化得到26
‑
氧酰异aβ1
‑
42精品,纯化条件为:高效液相色谱,色谱柱为30mm的聚合物(聚苯乙烯,粒径10um 300a,色谱柱长度25cm,直径是30mm),流动相a相为0.25%甲酸水溶液,b相为0.1%tfa/乙腈溶液;b相在0
‑
5min由5%变到22%;在5
‑
50min内,b相由22%变到42%,运行45min,流动相温度为60℃。纯化后,经冷冻干燥后得11.8mg纯品,纯度为98.703%,总产率6.72%。将纯品溶解于乙腈/水(体积比为1:1)中,浓度为1mg/ml,加入千分之一氨水调ph=7.4,25℃下反应4小时得到aβ1
‑
42,冷冻干燥后获得固体10.3mg,纯度为97.651%。高效液相色谱图见图4,质谱图见图5。
[0121]
实施例4:制备aβ1
‑
42
[0122]
(1)fmoc
‑
ala
‑
ctc树脂的合成。
[0123]
称取取代度为0.98mmol/g的2
‑
ctc树脂10g,加入到杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;称取5mmol fmoc
‑
ala
‑
oh,加入dcm和diea于反应杯中,氮气鼓泡反应90min之后;加入3ml甲醇和diea封端反应30min,dmf洗涤4次,dcm洗涤1次之后用甲醇抽干,可得到fmoc
‑
gly
‑
ctc树脂;取少量树脂用紫外分光光度计测得取代度为0.33mmol/g(三次平均值)。
[0124]
(2)2
‑
ctc树脂ile
41
的偶联。
[0125]
将实施例1制备的取代度为0.33mmol/g的fmoc
‑
ala
‑
ctc树脂于杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;加入20%pip/dmf脱除fmoc保护基团,脱除时间为25min,再加入dmf洗涤5次,dcm洗1次,国药dmf洗涤1次,茚三酮检测树脂显色,同时需要保证哌啶洗涤干净无残留。称取3.5g fmoc
‑
ile
‑
oh(10mmol),5.2g pybop(10mmol),溶解于70ml dmf中,加入5ml diea(30mmol)反应2min之后,倒入杯状反应柱中,氮气鼓泡反应70min。
[0126]
(3)制备保护的26
‑
氧酰异aβ1
‑
42树脂复合物。
[0127]
向反应器中加入100ml 20%pip/dmf溶液室温反应25min,再加入dmf洗涤6次,dcm
洗1次,茚三酮检测树脂显色。取10mmol fmoc
‑
val
‑
oh,10mmol pybop,10mmol hobt及30mmol diea,溶于70ml dmf中,将所得溶液加入到反应器中,25℃下反应70min。反应结束后,过滤。然后加入100ml dmf,洗涤树脂六遍。
[0128]
按照26
‑
氧酰异aβ1
‑
42c端到n端的顺序,依次偶联氨基酸,循环脱保护反应和缩合反应。其中fmoc
‑
ile
31
‑
oh,fmoc
‑
ile
32
‑
oh的偶联方法为氨基酸投料5eq,反应浓度为0.3mmol/ml,dic/hobt为偶联试剂(氨基酸:dic:hobt=1:1:1.25),溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf。
[0129]
boc
‑
ser(fmoc
‑
gly)
‑
oh的偶联方法为氨基酸投料3eq,反应浓度为0.3mmol/ml,dic/hobt为偶联试剂,溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf:dcm=4:1。
[0130]1‑
25位氨基酸的fmoc的脱除采用10%哌啶/dmf溶液,时间为2次5min的方式。其他氨基酸fmoc的脱除采用20%哌啶/dmf溶液,时间为25min,脱除一次。
[0131]
(4)制备保护的26
‑
氧酰异aβ1
‑
42粗品。
[0132]
将上述26
‑
氧酰异aβ1
‑
42树脂复合物放置于冰水浴中,将裂解试剂加入到26
‑
氧酰异aβ1
‑
42树脂肽中,质量为tfa:苯甲硫醚:苯酚:edt:水=87.5:2.5:2.5:5:2.5,加入碘化铵(1mg/ml),裂解试剂在0~10℃下预先冷却;于冰水浴中先反应30min之后,逐渐升至25℃,继续裂解2h。裂解液中加入预冷的15倍体积的乙醚离心沉淀后得到26
‑
氧酰异aβ1
‑
42粗品320mg;
[0133]
(5)制备aβ1
‑
42纯品。
[0134]
将26
‑
氧酰异aβ1
‑
42粗品溶于体积比为5%的甲醇水溶液中,经高效液相色谱纯化得到26
‑
氧酰异aβ1
‑
42精品,冷冻干燥后得72.6mg,纯度为97.361%,总产率12.20%。纯化条件为:30mm的聚合物(聚苯乙烯,粒径10um 300a,色谱柱长度25cm,直径是30mm),流动相a相为0.25%甲酸水溶液,b相为0.1%tfa/乙腈溶液;b相在0
‑
5min由5%变到22%;在5
‑
50min内,b相由22%变到42%,运行45min,流动相温度为60℃。将多肽溶解于乙腈/水中,浓度为1mg/ml,加入千分之一氨水调ph=7.4,25℃下反应4小时得到aβ1
‑
42冷冻干燥后固体64.3mg,纯度为97.153%。高效液相色谱图见图6,质谱图见图7。
[0135]
实施例5:制备aβ1
‑
42
[0136]
(1)fmoc
‑
ala
‑
ctc树脂的合成。
[0137]
称取取代度为0.98mmol/g的2
‑
ctc树脂3g,加入到杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;称取1mmol fmoc
‑
ala
‑
oh,加入dcm和diea于反应杯中,氮气鼓泡反应90min之后;加入3ml甲醇和diea封端反应30min,dmf洗涤4次,dcm洗涤1次之后用甲醇抽干,可得到fmoc
‑
gly
‑
ctc树脂;取少量树脂用紫外分光光度计测得取代度为0.25mmol/g(三次平均值)。
[0138]
(2)2
‑
ctc树脂ile
41
的偶联。
[0139]
将实施例1制备的取代度为0.25mmol/g的fmoc
‑
ala
‑
ctc树脂于杯状反应柱中,用dcm溶胀树脂30分钟后,抽滤除去dcm;加入20%pip/dmf脱除fmoc保护基团,脱除时间为25min,再加入dmf洗涤5次,dcm洗1次,dmf洗涤1次,茚三酮检测树脂显色,同时需要保证哌啶洗涤干净无残留。称取2.4g fmoc
‑
ile
‑
oh(2.25mmol),2.6g hatu(2.25mmol),溶解于15ml dmf中,加入1.1ml diea(6.75mmol)反应2min之后,倒入杯状反应柱中,氮气鼓泡反应
70min。
[0140]
(3)制备保护的26
‑
氧酰异aβ1
‑
42树脂复合物。
[0141]
向反应器中加入40ml 20%pip/dmf溶液室温反应25min,再加入dmf洗涤6次,dcm洗1次,茚三酮检测树脂显色。取2.25mmol fmoc
‑
val
‑
oh,2.8mmol hobt及2.25mmol dic,溶于15ml dmf中,将所得溶液加入到反应器中,25℃下反应70min。反应结束后,过滤。然后加入50ml dmf,洗涤树脂六遍。
[0142]
按照26
‑
氧酰异aβ1
‑
42c端到n端的顺序,依次偶联氨基酸,循环脱保护反应(20%pip/dmf脱除fmoc保护基团,脱除时间为25min,脱除1次)和缩合反应至肽链合成结束。其中fmoc
‑
ile
31
‑
oh,fmoc
‑
ile
32
‑
oh的偶联方法为氨基酸投料5eq,反应浓度为0.3mmol/ml,dic/hobt为偶联试剂,溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf。
[0143]
boc
‑
ser(fmoc
‑
gly)
‑
oh的偶联方法为氨基酸投料3eq,反应浓度为0.3mmol/ml,dic/hobt为偶联试剂,溶剂为dmf偶联40min之后抽滤,dmf洗涤树脂1次之后再选用hatu/hobt/diea再次偶联40min,溶剂为dmf:dcm=4:1。
[0144]
所有氨基酸fmoc的脱除采用20%哌啶/dmf溶液,时间为25min,脱除一次。
[0145]
(4)制备保护的26
‑
氧酰异aβ1
‑
42粗品。
[0146]
将上述26
‑
氧酰异aβ1
‑
42树脂复合物1g放置于冰水浴中,将裂解试剂(比例为tfa:苯甲硫醚:苯酚:edt:水=87.5:2.5:2.5:5:2.5,加入碘化铵(1mg/ml))在0
‑
10℃下预先冷却,加入到26
‑
氧酰异aβ1
‑
42树脂肽中;于冰水浴中先反应30min之后,逐渐升至25℃,继续裂解2h。裂解液中加入预冷的15倍体积的乙醚,离心沉淀后得到26
‑
氧酰异aβ1
‑
42粗品130mg,粗品质谱图见图9,高效液相色谱图见图10;因为没有正确的目标分子量,所得质荷比为酯键断裂之后的残肽(分子量:1598,二价为799.70),所以难以制备出aβ1
‑
42纯品。
[0147]
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1