一种贾斯帕霉素的合成方法与流程
本发明涉及核苷类化合物合成领域,特别涉及到贾斯帕霉素(英文名:jaspamycin,cas号:22242-96-2)的合成方法。
背景技术:
核苷类化合物由于它们的生物学活性,广泛的应用于抗病毒药物研究。贾斯帕霉素是一种有效的pka激活剂,最近科学家发现其是强大的细胞渗透性诱导剂(natcommun.2019mar29;10(1):1421),可在医学上被忽视的重要病原体中探索独立于camp的pka信号传导,为一些疾病的治疗带来新思路。而贾斯帕霉素目前报道的合成方法是通过四氰基乙烯(~3000元/100克)通过8步反应制得(tetrahedronletters,2013,vol.54,#40,p.5484–5488;bioorganicchemistry,2007,vol.35,#1,p.25-34),多步需要柱层析纯化,总收率低,生产成本非常高,还要用到氢化,重氮化等危险反应。
技术实现要素:
本发明的目的是提供一种贾斯帕霉素的合成方法,主要解决目前合成方法存在的反应步骤长,成本高的技术问题。
本发明技术方案如下:一种贾斯帕霉素的新合成方法,包括以下步骤:
(1)4-氯-5-碘-7h-吡咯并[2,3-d]嘧啶和1-乙酰基-2,3,5-三苯甲酰氧基-1-beta-d-呋喃核糖在n,o-双(三甲基甲硅烷基)乙酰胺和三氟甲磺酸三甲基硅酯的作用下生成化合物1,产物打浆提纯;
(2)化合物1与甲醇钠在甲醇里反应,得到化合物2,产物无需提纯;
(3)化合物2在氢氧化钠水溶液里加热反应生成化合物3,产物打浆提纯;
(4)化合物3与氰化亚铜在吡啶里加热反应,所得粗品打浆,重结晶得到目标产物。本发明的反应路线如下:
上述反应中,步骤1,n,o-双(三甲基甲硅烷基)乙酰胺和三氟甲磺酸三甲基硅酯在室温下加,加完后室温搅拌10分钟再升温反应,反应温度为75℃~85℃,优选反应温度为80℃,反应12-24小时,优选反应时间为18小时。步骤2,反应温度为20℃~30℃,优选反应温度为25℃,反应12-24小时,优选反应时间为18小时。步骤3,反应温度为回流,反应1-2小时,优选反应时间为1.5小时。步骤4,反应温度为100℃-115℃,优选反应温度为115℃回流,反应18-36小时,优选反应时间为24小时。
本发明的有益效果是:本发明只需4步,路线步骤短,所用试剂便宜,所用起始原料(4-氯-5-碘-7h-吡咯并[2,3-d]嘧啶,~1000元/100克),总收率达到36.5%。反应条件简单,避免了氢化,重氮化等危险反应,所有中间体和最终产品都无需柱层析提纯,只需打浆,结晶就可得到纯产品,操作简单,非常适于大量生产。
附图说明
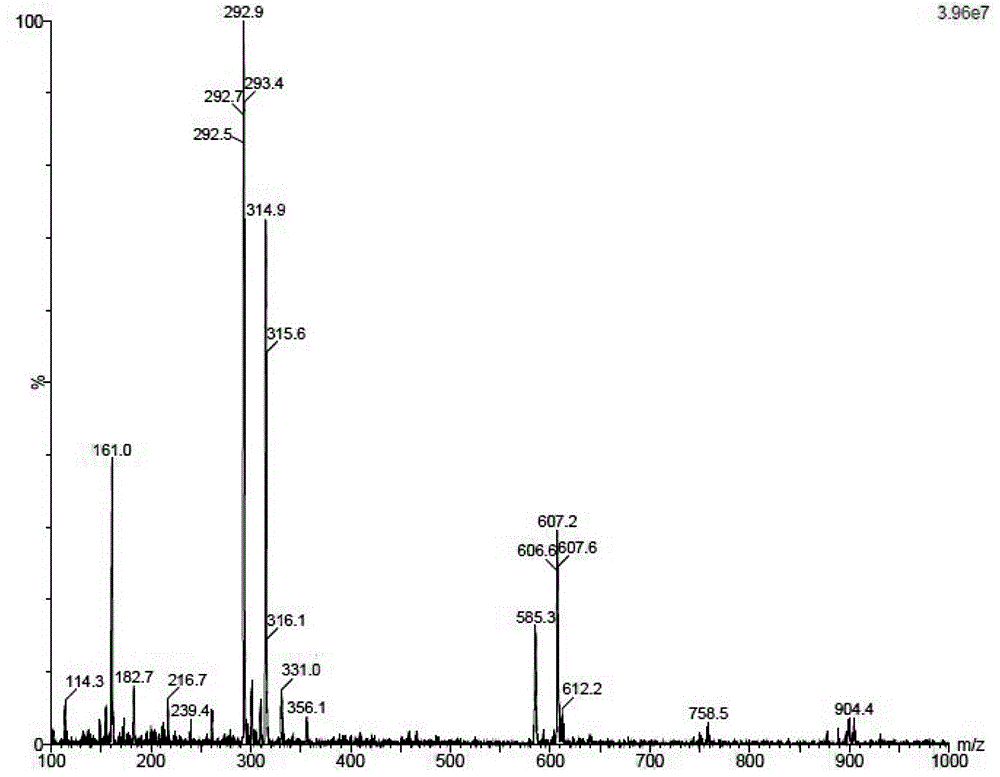
图1为本发明产物的质谱图。
图2为本发明产物的核磁图谱。
具体实施方式
实施例1:贾斯帕霉素。
步骤1:
搅拌下将n,o-双(三甲基甲硅烷基)乙酰胺(34毫升,134豪摩尔)加入4-氯-5-碘-7h-吡咯并[2,3-d]嘧啶(34克,120毫摩尔)在乙腈(1升)的悬浮液中。10分种后向其中加入1-乙酰基-2,3,5-三苯甲酰氧基-1-beta-d-呋喃核糖(72克,140毫摩尔)和三氟甲磺酸三甲基硅酯(25.5毫升,134毫摩尔)。反应液室温搅拌15分钟后,升温至80℃反应18小时。反应液冷却到室温,乙酸乙酯稀释倒入饱和碳酸氢钠水溶液中分液。有机相用饱和食盐水洗,干燥,浓缩。所得残余物在甲醇中打浆,过滤得白色固体化合物1(79克,90%收率)。
步骤2:
化合物1(79克,109毫摩尔)分散于甲醇(2升)中,加入甲醇钠(60克,109毫摩尔),反应液在25℃下搅拌18小时。反应液减压浓缩干。残余物化合物2直接用于下一步无需提纯。
步骤3:
上步所得化合物2(44克,109毫摩尔)加入2n氢氧化钠水溶液(2升)中,加热回流1.5小时。反应液冷却至室温,用浓盐酸调至中性,有白色固体析出。搅拌半小时,过滤得化合物3(25克,58%收率)。
步骤4:
化合物3(25克,63.6毫摩尔)和氰化亚铜(29.5克,329毫摩尔)加入吡啶(230毫升)中,氮气保护下,115℃回流24小时。反应液冷却至室温,减压浓缩干,残余物用氨水打浆过滤。所得固体用甲醇和乙酸乙酯结晶得到白色固体产品贾斯帕霉素(13克,70%收率)。ms(esi)m/z:292.9[m+h+]。1hnmr(400mhz,dmso-d6):δ12.56(s,1h),8.36(s,1h),8.10(d,j=4.0hz,1h),6.03(d,j=5.6hz,2h),5.52-5.19(br,3h),4.31(t,j=5.2hz,1h),4.10(t,j=4.4hz,1h),3.93(q,j=3.8hz,1h),3.66(dd,j=12.0,3.9hz,1h),3.56(dd,j=12.0,3.8hz,1h)。质谱图和核磁图谱参照图1、2。
实施例2,步骤1反应温度为75℃,反应时间24小时;步骤2反应温度为30℃,反应时间为12小时;步骤3回流1小时,步骤4反应温度为110℃,反应时间为30小时;其余同实施例1。
实施例3,步骤1反应温度为85℃,反应时间12小时;步骤2反应温度为20℃,反应时间24小时;步骤3回流2小时;步骤4反应温度为100℃,反应时间为36小时。其余同实施例1。
技术特征:
1.一种贾斯帕霉素的合成方法,其特征是:包括以下步骤:
(1)4-氯-5-碘-7h-吡咯并[2,3-d]嘧啶和1-乙酰基-2,3,5-三苯甲酰氧基-1-beta-d-呋喃核糖在n,o-双(三甲基甲硅烷基)乙酰胺和三氟甲磺酸三甲基硅酯的作用下生成化合物1,产物打浆提纯;
(2)化合物1与甲醇钠在甲醇里反应,得到化合物2,产物无需提纯;
(3)化合物2在氢氧化钠水溶液里加热反应生成化合物3,产物打浆提纯;
(4)化合物3与氰化亚铜在吡啶里加热反应,所得粗品打浆,重结晶得到目标产物;合成线路如下:
2.根据权利要求1所述的一种贾斯帕霉素的合成方法,其特征是:所述第一步,n,o-双(三甲基甲硅烷基)乙酰胺和三氟甲磺酸三甲基硅酯在室温下加,加完后室温搅拌10分钟再升温反应,反应温度为75℃~85℃,反应12-24小时。
3.根据权利要求2所述的一种贾斯帕霉素的合成方法,其特征是:反应温度为80℃,反应18小时。
4.根据权利要求1所述的一种贾斯帕霉素的合成方法,其特征是:所述第二步,反应温度为20℃~30℃,反应12-24小时。
5.根据权利要求4所述的一种贾斯帕霉素的合成方法,其特征是:所述第二步,反应温度为25℃,反应18小时。
6.根据权利要求1所述的一种贾斯帕霉素的合成方法,其特征是:所述第三步,为回流反应1-2小时。
7.根据权利要求1所述的一种贾斯帕霉素的合成方法,其特征是:所述第四步,反应温度为100℃-115℃,反应18-36小时。
8.根据权利要求7所述的一种贾斯帕霉素的合成方法,其特征是:所述第四步,反应温度为115℃,反应24小时。
技术总结
本发明涉及一种贾斯帕霉素的合成方法。主要解决目前合成方法步骤长,成本高的技术问题。本发明合成步骤:4‑氯‑5‑碘‑7H‑吡咯并[2,3‑d]嘧啶和1‑乙酰基‑2,3,5‑三苯甲酰氧基‑1‑beta‑D‑呋喃核糖在N,O‑双(三甲基甲硅烷基)乙酰胺和三氟甲磺酸三甲基硅酯的作用下生成化合物1,产物打浆提纯;化合物1与甲醇钠在甲醇里反应,得到化合物2,无需提纯;化合物2在氢氧化钠水溶液里加热反应生成化合物3,产物打浆提纯;化合物3与氰化亚铜在吡啶里加热反应,所得粗品打浆,重结晶得到目标产物。
技术研发人员:徐红岩;王鹏涛
受保护的技术使用者:康化(上海)新药研发有限公司
技术研发日:2021.03.10
技术公布日:2021.05.28
- 还没有人留言评论。精彩留言会获得点赞!