一种达泊西汀及盐酸达泊西汀的合成方法与流程
1.本发明涉及药物有机合成技术领域,具体涉及一种达泊西汀及盐酸达泊西汀的合成方法。
背景技术:
2.盐酸达泊西汀化学名为:(s)
‑
(+)
‑
n,n
‑
二甲基
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙胺盐酸盐,是一种选择性5
‑
羟胺再摄取抑制剂,具有半衰期短、不良反应较小等优点,由美国礼来制药公司研制并于2009年在欧洲上市,用于治疗男性早泄。
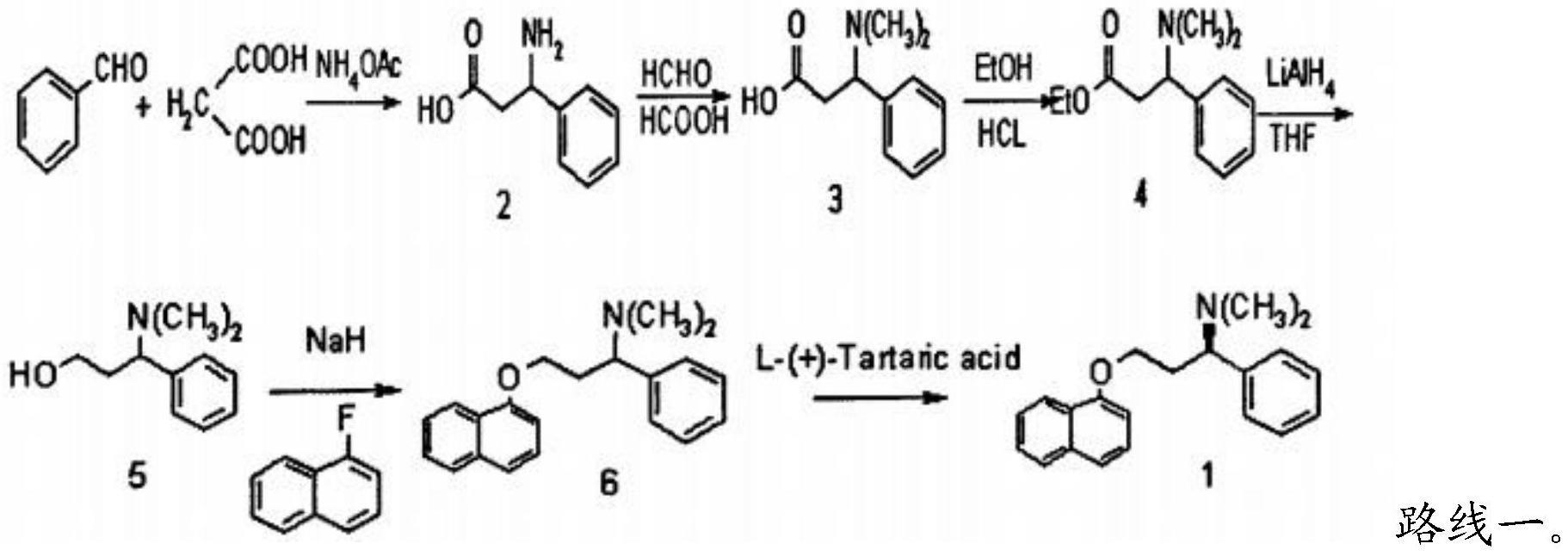
3.按制备原料的不同,盐酸达泊西汀的合成方法分为以下几类:以苯甲醛和丙二酸为起始,按照路线一反应得到达泊西汀(参见j.label.compd.radionpharm,1993,(31):305
‑
315、欧洲专利ep0288188和《中国新药杂志》,2008,17(24):2119
‑
2121),然而该类方法合成路线长,需要用到四氢铝锂还原,工艺易爆危险,并且最后酒石酸拆分,至少废弃50%以上的异构体,生产成本高。
[0004][0005]
以1
‑
萘酚和3
‑
苯基
‑1‑
溴丙烷为起始原料,按照路线二反应合成达泊西汀(参见欧洲专利ep0288188和张飞皇,陈瞍.达泊西汀的合成进展,《中国新药杂志》,2007,16(2):111
‑
113),该方法反应条件容易控制,路线也较简单,然而3
‑
苯基丙基溴价格偏高,而且同样需要酒石酸拆分,至少废弃50%以上的异构体,生产成高。
[0006][0007]
以手性化合物n
‑
boc
‑
(r)
‑
苯基甘氨酸为原料,按照路线三反应合成达泊西汀(参
见《中国新药杂志》,2008,17(24):2119
‑
2121);该方法的优点在于避免了用l
‑
(+)
‑
酒石酸拆分,提高了产率,然而手性化合物n
‑
boc
‑
(r)
‑
苯基甘氨酸不易得到并且价格昂高,另外用到了不易得的硼烷和极毒物nacn,反应路线也较繁琐。
[0008][0009]
以s
‑1‑
苯基
‑3‑
氯
‑1‑
丙醇和l
‑
萘酚为起始原料,按照路线四反应合成达泊西汀(参见张飞皇,陈瞍.达泊西汀的合成进展,《中国新药杂志》,2007,16(2):111
‑
113和nuclmedboil,1994,2l(4):649
‑
675);该方法路线简单,反应条件容易控制,避免了用l
‑
(+)
‑
酒石酸拆分,提高产率,但原料s
‑1‑
苯基
‑3‑
氯
‑1‑
丙醇价格比较贵,合成成本高。
[0010][0011]
以3
‑
苯基丙烯醛和n
‑
苄氧羰基羟胺为起始原料,按照路线五反应,合成达泊西汀盐酸盐(参见中国专利cn103304434);然而该方法反应路线繁琐,且使用了价格昂贵的手性催化剂和钯等贵重金属,生产成本高。
[0012]
技术实现要素:
[0013]
鉴于此,本发明的目的在于提供一种达泊西汀及盐酸达泊西汀的合成方法,本发明提供的合成方法,合成路线短、生产成本低且对环境污染小。
[0014]
为了实现上述发明目的,本发明提供以下技术方案:
[0015]
本发明提供了一种达泊西汀的合成方法,包括以下步骤:
[0016]
(1)将1
‑
萘酚、多聚甲醛、溴化氢和第一有机溶剂混合,进行溴甲基化反应,得到1
‑
萘酚溴甲醚;
[0017]
(2)在保护气氛下,将所述1
‑
萘酚溴甲醚、镁、引发剂和第二有机溶剂混合,进行取代反应,得到1
‑
萘酚溴甲醚格氏试剂溶液;
[0018]
(3)将所述1
‑
萘酚溴甲醚格氏试剂溶液、r
‑
环氧苯乙烷和第三有机溶剂混合,进行格氏反应,得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇;所述r
‑
环氧苯乙烷中的r包括甲苯磺酰基、甲磺酰基或间硝基苯磺酰基;
[0019]
(4)将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、溴化试剂和第四有机溶剂混合,进行溴化反应,得到s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;
[0020]
或,将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、酰氯试剂、碱性试剂和第五有机溶剂混合,进行磺酰化反应,得到s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;
[0021]
(5)将所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷和s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷中一种、二甲胺和第六有机溶剂混合,进行氨解取代反应,得到达泊西汀。
[0022]
优选的,步骤(1)中,所述多聚甲醛的摩尔量以甲醛计,所述1
‑
萘酚和多聚甲醛的摩尔比为1:(1~3)。
[0023]
优选的,步骤(1)中,所述溴甲基化反应的温度为0~50℃。
[0024]
优选的,步骤(2)中,所述1
‑
萘酚溴甲醚和镁的摩尔比为1:(1~2)。
[0025]
优选的,步骤(3)中,所述r
‑
环氧苯乙烷和1
‑
萘酚溴甲醚格氏试剂溶液中的1
‑
萘酚溴甲醚格氏试剂的摩尔比为1:(0.5~3);
[0026]
所述第二有机溶剂和第三有机溶剂独立地包括四氢呋喃、甲基叔丁基醚或乙醚。
[0027]
优选的,步骤(3)中,所述格氏反应的温度为
‑
78℃~
‑
10℃。
[0028]
优选的,步骤(4)中,所述溴化试剂包括n
‑
溴代琥珀酰亚胺和/或液溴;
[0029]
所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇和溴化试剂的摩尔比为1:(1~3)。
[0030]
优选的,步骤(4)中,所述酰氯试剂包括甲磺酰氯、对甲苯磺酰氯、间硝基苯磺酰氯和对硝基苯磺酰氯中的一种或几种;
[0031]
所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇和酰氯试剂的摩尔比为1:(0.9~3)。
[0032]
优选的,步骤(5)中,所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷与二甲胺的摩尔比为1:(1~5)。
[0033]
本发明还提供了一种盐酸达泊西汀的合成方法,包括以下步骤:利用氯化氢将上述技术方案所述合成方法得到的达泊西汀进行酸化,得到盐酸达泊西汀。
[0034]
本发明提供了一种达泊西汀的合成方法,包括以下步骤:(1)将1
‑
萘酚、多聚甲醛、溴化氢和第一有机溶剂混合,进行溴甲基化反应,得到1
‑
萘酚溴甲醚;(2)在保护气氛下,将所述1
‑
萘酚溴甲醚、镁、引发剂和第二有机溶剂混合,进行取代反应,得到1
‑
萘酚溴甲醚格氏试剂溶液;(3)将所述1
‑
萘酚溴甲醚格氏试剂溶液、r
‑
环氧苯乙烷和第三有机溶剂混合,进行格氏反应,得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇;所述r
‑
环氧苯乙烷中的r包括甲苯磺酰基、甲磺酰基或间硝基苯磺酰基;(4)将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、溴化试剂和第四有机溶剂混合,进行溴化反应,得到s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;或,将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、酰氯试剂、碱性试剂和第五有机溶剂混合,进行磺酰化反应,得到s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;(5)将所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷和
s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷中一种、二甲胺和第六有机溶剂混合,进行氨解取代反应,得到达泊西汀。
[0035]
进一步的,利用氯化氢将上述技术方案所述合成方法得到的达泊西汀进行酸化,得到盐酸达泊西汀。
[0036]
本发明提供的合成方法,合成路线短,产物收率高,产品质量好,原材料易得、成本低,对环境污染小,适宜工业化生产。
具体实施方式
[0037]
本发明提供了一种达泊西汀的合成方法,包括以下步骤:
[0038]
(1)将1
‑
萘酚、多聚甲醛、溴化氢和第一有机溶剂混合,进行溴甲基化反应,得到1
‑
萘酚溴甲醚;
[0039]
(2)在保护气氛下,将所述1
‑
萘酚溴甲醚、镁、引发剂和第二有机溶剂混合,进行取代反应,得到1
‑
萘酚溴甲醚格氏试剂溶液;
[0040]
(3)将所述1
‑
萘酚溴甲醚格氏试剂溶液、r
‑
环氧苯乙烷和第三有机溶剂混合,进行格氏反应,得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇;所述r
‑
环氧苯乙烷中的r包括甲苯磺酰基、甲磺酰基或间硝基苯磺酰基;
[0041]
(4)将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、溴化试剂和第四有机溶剂混合,进行溴化反应,得到s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;
[0042]
或,将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、酰氯试剂、碱性试剂和第五有机溶剂混合,进行磺酰化反应,得到s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;
[0043]
(5)将所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷和s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷中一种、二甲胺和第六有机溶剂混合,进行氨解取代反应,得到达泊西汀;
[0044]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0045]
本发明将1
‑
萘酚、多聚甲醛、溴化氢和第一有机溶剂混合,进行溴甲基化反应,得到1
‑
萘酚溴甲醚。
[0046]
在本发明中,所述多聚甲醛的摩尔量以甲醛计,所述1
‑
萘酚和多聚甲醛的摩尔比优选为1:(1~3),进一步优选为1:(1.5~2.5),更优选为1:2。本发明对于所述多聚甲醛的分子量没有特殊限定,采用本领域技术人员熟知的分子量即可。在本发明中,所述溴化氢优选为干燥的溴化氢气体。在本发明中,所述第一有机溶剂优选为二氯甲烷;本发明对于所述第一有机溶剂的用量没有特殊限定,采用本领域技术人员熟知的用量即可;在本发明的实施例中,所述1
‑
萘酚的物质的量和第一有机溶剂的质量之比优选为1mol:400g。
[0047]
在本发明中,所述混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述混合的顺序优选为将1
‑
萘酚、多聚甲醛和第一有机溶剂混合后通入溴化氢。
[0048]
在本发明中,所述溴甲基化反应的温度优选为0~50℃,进一步优选为10~40℃,更优选为20~30℃。在本发明中,所述溴甲基化反应过程中发生的反应如式(1)所示:
[0049][0050]
所述溴甲基化反应后,本发明优选还包括将所述溴甲基化反应的反应液置于分液漏斗中进行分层,将所得有机相进行干燥剂干燥后过滤,将所得滤液进行浓缩,得到1
‑
萘酚溴甲醚。在本发明中,所述干燥剂优选为无水硫酸钠。在本发明中,所述过滤的目的是除去干燥剂。在本发明中,所述浓缩的方式优选为减压蒸馏,本发明对于所述浓缩的温度和时间没有特殊限定,浓缩至恒重即可。
[0051]
得到1
‑
萘酚溴甲醚后,本发明在保护气氛下,将所述1
‑
萘酚溴甲醚、镁、引发剂和第二有机溶剂混合,进行取代反应,得到1
‑
萘酚溴甲醚格氏试剂溶液。
[0052]
在本发明中,所述1
‑
萘酚溴甲醚和镁的摩尔比优选为1:(1~2),进一步优选为1:(1.2~1.8),更优选为1:(1.5~1.6)。在本发明中,所述引发剂优选包括碘或溴乙烷;本发明对于所述引发剂的用量没有特殊限定,采用本领域技术人员熟知的引发剂的用量即可。在本发明中,所述保护气氛优选为氮气或氩气。
[0053]
在本发明中,所述1
‑
萘酚溴甲醚、镁、引发剂和第二有机溶剂混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述混合的顺序优选为将1
‑
萘酚溴甲醚溶解于部分第二有机溶剂中,得到1
‑
萘酚溴甲醚溶液;将镁、引发剂和剩余第二有机溶剂混合,得到悬浮液;将所述1
‑
萘酚溴甲醚溶液滴加到所述悬浮液中。在本发明中,所述1
‑
萘酚溴甲醚溶液优选分两次滴加到所述混合悬浮液中,具体的,将部分1
‑
萘酚溴甲醚溶液滴加到所述悬浮液中,加热回流至反应液颜色褪去,然后在滴加剩余1
‑
萘酚溴甲醚溶液。在本发明中,所述部分1
‑
萘酚溴甲醚溶液优选占所述1
‑
萘酚溴甲醚溶液的5~10%,更优选为6.5~7%;本发明对于所述滴加的速度没有特殊限定,采用本领域技术人员熟知的滴加速度即可。在本发明中,所述第二有机溶剂优选包括四氢呋喃、甲基叔丁基醚或乙醚。本发明对于所述第二有机溶剂的用量没有特殊限定,采用本领域技术人员熟知的用量即可,在本发明的实施例中,所述1
‑
萘酚溴甲醚的物质的量和部分第二有机溶剂的体积之比优选为1mol:600ml,所述镁的物质的量和剩余第二有机溶剂的体积之比优选为1mol:250ml。
[0054]
在本发明中,所述取代反应的温度优选为55~75℃,进一步优选为60~70℃,所述取代反应的的时间优选为2~3h,进一步优选为2.5h。在本发明中,所述取代反应过程中发生的反应如式(2)所示:
[0055][0056]
所述取代反应后,本发明优选还包括将所述取代反应的体系冷却至室温,得到1
‑
萘酚溴甲醚格氏试剂溶液。本发明对于所述冷却的方式没有特殊限定,采用本领域技术人
员熟知的冷却方式即可。
[0057]
得到1
‑
萘酚溴甲醚格氏试剂溶液后,本发明将所述1
‑
萘酚溴甲醚格氏试剂溶液、r
‑
环氧苯乙烷和第三有机溶剂混合,进行格氏反应,得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇;所述r
‑
环氧苯乙烷中的r包括甲苯磺酰基、甲磺酰基或间硝基苯磺酰基。
[0058]
在本发明中,所述r
‑
环氧苯乙烷和1
‑
萘酚溴甲醚格氏试剂溶液中的1
‑
萘酚溴甲醚格氏试剂的摩尔比优选为1:(0.5~3),进一步优选为1:(1~2.5),更优选为1:2。在本发明中,所述第三有机溶剂优选包括四氢呋喃、甲基叔丁基醚或乙醚。本发明对于所述第二有机溶剂的用量没有特殊限定,采用本领域技术人员熟知的用量即可,在本发明的实施例中,所述r
‑
环氧苯乙烷的物质的量和第三有机溶剂的体积之比优选为1mol:400ml。
[0059]
在本发明中,所述1
‑
萘酚溴甲醚格氏试剂溶液、r
‑
环氧苯乙烷和第三有机溶剂混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述混合的顺序优选为将r
‑
环氧苯乙烷和第三有机溶剂混合后滴加1
‑
萘酚溴甲醚格氏试剂溶液。本发明对于所述滴加的速度没有特殊限定,采用本领域技术人员熟知的滴加速度,保证反应体系的温度不高于
‑
10℃即可。
[0060]
在本发明中,所述格氏反应的温度为优选为
‑
78℃~
‑
10℃,进一步优选为
‑
65~
‑
25℃,更优选为
‑
50℃~
‑
40℃;所述格氏反应的时间优选为1~4h,进一步优选为1.5~3.5h,更优选为2~3h,所述格氏反应的时间以滴加完所述1
‑
萘酚溴甲醚格氏试剂溶液开始计时。在本发明中,所述格氏反应过程中发生的反应如式(3)所示:
[0061][0062]
所述格氏反应后,本发明优选还包括将所述格氏反应的体系的温度升温至室温,加入饱和氯化铵水溶液搅拌后静置分层,将所得水相用有机溶剂萃取,将所得有机相进行无水硫酸钠干燥后过滤,将所得滤液浓缩至恒重,得到浓缩液,将所得浓缩液进行重结晶,得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇。本发明对于所述饱和氯化铵水溶液的用量没有特殊限定,采用本领域技术人员熟知的用量即可;在本发明的实施例中,所述r
‑
环氧苯乙烷的物质的量与饱和氯化铵水溶液的质量之比优选为1mol:450~600g,进一步优选为1mol:500~550g;所述搅拌的温度优选为室温,所述搅拌的时间优选为1h。在本发明中,所述萃取用有机溶剂优选包括二氯甲烷、四氢呋喃、甲基叔丁基醚或乙醚;所述萃取的次数优选为2~3次;所述r
‑
环氧苯乙烷的物质的量与单次萃取用有机溶剂的体积之比优选为1mol:400~700ml,进一步优选为1mol:450~600ml。在本发明中,所述浓缩的方式优选为减压蒸馏。在本发明中,所述重结晶的操作优选包括在所述浓缩液中加入乙酸乙酯加热回流,冷却至室温后加入石油醚,在搅拌条件下冷却至0~5℃,固体析出量不再增加后,过滤,将所得滤饼进行干燥,得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇。在本发明中,所述r
‑
环氧苯乙烷的物质的量与乙酸乙酯的体积之比优选为1mol:400~600ml,进一步优选为1mol:450~500ml;所述r
‑
环氧苯乙烷的物质的量与石油醚的体积之比优选为1mol:700~1000ml,进一步优选为1mol:800~900ml。本发明对于所述搅拌的速度没有特殊限定,采用本领技术人员熟知的搅拌速度即可。在本发明中,所述干燥的方式优选为烘干,本发明对于所述干燥的温度和时间没有特殊限定,采用本领域技术人员熟知的干燥温度和时间即可。
[0063]
得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇后,本发明将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、溴化试剂和第四有机溶剂混合,进行溴化反应,得到s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇中的r包括甲苯磺酰基、甲磺酰基或间硝基苯磺酰基。
[0064]
在本发明中,所述溴化试剂优选包括n
‑
溴代琥珀酰亚胺(nbs)和/或液溴。在本发明中,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇和溴化试剂的摩尔比优选为1:(1~3),进一步优选为1:(1.5~2.5),更优选为1:2。本发明对于所述第四有机溶剂的种类没有特殊限定,能够溶解r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇即可;在本发明的实施例中,所述第四有机溶剂优选包括二氯甲烷或氯仿;本发明对于所述第四有机溶的用量没有特殊限定,采用本领域技术人员熟知的用量即可;在本发明的实施例中,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和第四有机溶剂的质量之比优选为1mol:3000g。
[0065]
在本发明中,所述混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。
[0066]
本发明对于所述溴化反应的温度没有特殊限定,能够回流即可;在本发明的实施例中,当采用二氯甲烷为第四有机溶剂时,所述溴化反应的温度的温度为39.8℃,当采用氯仿为第四有机溶剂时,所述溴化反应的温度的温度为61.2℃;所述溴化反应的时间优选为6~10h,更优选为8h;所述溴化反应优选使用tlc监测至溴化反应完全。在本发明中,所述溴化反应过程中发生的反应如式(4)所示:
[0067][0068]
所述溴化反应后,本发明优选还包括将所述溴化反应的体系冷却至室温,加入水搅拌,静置分层,得到水相和有机相,萃取所得水相,将萃取所得有机相和分层所得有机相合并后依次进行水洗、无水硫酸钠干燥和过滤,将所得滤液浓缩至恒重,得到浓缩物,将所得浓缩物进行重结晶,得到s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷。本发明对于所述水的用量没有特殊限定,采用本领域技术人员熟知的用量即可;在本发明的实施例中,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和水的体积之比优选为1mol:1500ml。在本发明中,所述搅拌的时间优选为1h。在本发明中,所述浓缩的方式优选为减压蒸馏。在本发明中,所述重结晶用有机溶剂优选为乙酸乙酯和石油醚,所述乙酸乙酯和石油醚的体积比优选为1:(1~5),更优选为1:(2~4),最优选为1:3;本发明对于所述重结晶用有机溶剂的用量没有特殊限定,采用本领域技术人员熟知的有机溶剂用量即可。
[0069]
得到r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇后,本发明将所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、酰氯试剂、碱性试剂和第五有机溶剂混合,进行磺酰化反应,得到s
‑1‑
磺酰氧基
‑1‑
苯
基
‑3‑
(1
‑
萘氧基)丙烷。
[0070]
在本发明中,所述酰氯试剂优选包括甲磺酰氯、对甲苯磺酰氯、间硝基苯磺酰氯和对硝基苯磺酰氯中的一种或几种;所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇和酰氯试剂的摩尔比优选为1:(0.9~3),进一步优选为1:(1~2.5),更优选为1:(1.5~2)。在本发明中,所述碱性试剂优选包括吡啶、三乙胺和4
‑
二甲氨基吡啶(dmap)中的一种或几种。在本发明中,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇和碱性试剂的摩尔比优选为1:(1.2~38),进一步优选为1:(5~20)。本发明对于所述第五有机溶剂的种类没有特殊限定,能够溶解r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇即可;在本发明的实施例中,所述第五有机溶剂优选包括二氯甲烷或氯仿。
[0071]
在本发明中,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、酰氯试剂、碱性试剂和第五有机溶剂混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、酰氯试剂、碱性试剂和第五有机溶剂混合的顺序优选为将r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇、碱性试剂和部分第五有机溶剂混合,得到混合溶液;将酰氯试剂和剩余第五有机溶剂混合,得到酰氯试剂溶液;将所述混合溶液的温度冷却至
‑
5~0℃后滴加酰氯试剂溶液。在本发明中,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和部分第五有机溶剂的质量之比优选为1mol:0~1500g;所述酰氯试剂的物质的量和剩余第五有机溶剂的质量之比优选为1mol:450~1000g。本发明对于所述酰氯试剂溶液的滴加的速度没有特殊限定,能够保证反应体系的温度≤5℃即可。
[0072]
在本发明中,所述磺酰化反应的温度优选为室温,所述磺酰化反应的时间优选为1~2h,进一步优选为1.5h。在本发明中,所述磺酰化反应过程中发生的反应如式(5)所示:
[0073][0074]
所述磺酰化反应后,本发明优选还包括将所述磺酰化反应得到的反应液进行后处理,所述后处理的方式优选包括第一后处理或第二后处理。在本发明中,所述第一后处理包括以下步骤:依次采用稀盐酸水溶液、碳酸氢钠水溶液和水洗涤所述反应液,然后用无水硫酸钠干燥后过滤,将所得滤液浓缩至恒重,得到浓缩物,将所述浓缩物进行重结晶,得到s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;所述稀盐酸水溶液的浓度优选为5wt%,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和稀盐酸水溶液的体积之比优选为1mol:1500ml;所述碳酸氢钠水溶液的浓度优选为5wt%,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和碳酸氢钠水溶液的体积之比优选为1mol:1500ml;所述水优选为蒸馏水,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和水的体积之比优选为1mol:1500ml;所述浓缩的方式优选为常压蒸馏;所述重结晶用有机溶剂优选包括乙酸乙酯和石油醚,所述乙酸乙酯和石油醚的体积比优选为1:(1~5),更优选为1:(2~4),最优选为1:3;本发明对于所述重结晶用有机溶剂的用量没有特殊限定,采用本领域技术人员熟知的有机溶剂用量即可。在本发明中,所述第二后处理包括以下步骤:将所述反应液倒入冰水中,加入有机溶剂进行萃取,将所得有机相
用无水硫酸钠干燥后过滤,将所得滤液浓缩至恒重,得到浓缩物,将所述浓缩物进行重结晶,得到s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷;所述冰水的温度优选为0℃,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和冰水的体积之比优选为1mol:5000ml;所述萃取的次数优选为3~4次,所述萃取用有机溶剂优选包括二氯甲烷,所述r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇的物质的量和单次萃取用有机溶剂的质量之比优选为1mol:2000ml;所述浓缩的方式优选为常压蒸馏;所述重结晶用有机溶剂优选包括乙酸乙酯和石油醚,所述乙酸乙酯和石油醚的体积比优选为1:(1~5),更优选为1:(2~4),最优选为1:3,本发明对于所述重结晶用有机溶剂的用量没有特殊限定,采用本领域技术人员熟知的有机溶剂用量即可。
[0075]
得到s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷后,本发明将所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷和s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷中一种、二甲胺和第六有机溶剂混合,进行氨解取代反应,得到达泊西汀。
[0076]
在本发明中,所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷与二甲胺的摩尔比优选为1:(1~5),进一步优选为1:(2~4),更优选为1:(2.5~3)。在本发明中,所述二甲胺优选以二乙胺溶液形式使用,所述二甲胺溶液优选为二乙胺水溶液或二乙胺四氢呋喃溶液,所述二乙胺水溶液的浓度优选为40wt%,所述二乙胺四氢呋喃溶液的浓度优选为2mol/l。在本发明中,所述第六有机溶剂优选包括四氢呋喃或乙腈;所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷的物质的量与第六有机溶剂的体积之比优选为1mol:2500ml。
[0077]
在本发明中,所述混合过程中优选还加入三乙胺,所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷与三乙胺的摩尔比优选为1:0.5。
[0078]
在本发明中,所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷、二甲胺和第六有机溶剂混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述混合的顺序优选为将s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷溶解于第六有机溶剂后加入二甲胺混合。在本发明中,当所述制备原料还包括三乙胺时,所述三乙胺的假如说时机与所述二乙胺相同。
[0079]
在本发明中,所述氨解取代反应的温度优选为室温,所述氨解取代反应的时间优选为20~30h,进一步优选为25~26h;所述氨解取代反应优选使用tlc监测至氨解取代反应完全。在本发明中,所述氨解取代反应过程中发生的反应如式(6)或式(7)所示:
[0080][0081]
所述氨解取代反应后,本发明优选还包括将所述氨解取代反应得到的反应液倒入水中,然后加入有机溶剂进行萃取,将所得有机相用无水硫酸钠干燥后过滤,将所得滤液浓缩至恒重,得到达泊西汀;所述水优选为蒸馏水,所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷的物质的量和水的质量之比优选为1mol:3500~5000g。所述萃取前本发明优选还包括将加入水的反应液的ph值调节至12.5~13.5,进一步优选为13;所述ph值调节采用的碱优选为naoh水溶液,所述naoh水溶液的浓度优选为5mol/l;本发明对于所述碱的用量没有特殊限定,能够将ph值调节至12.5~13.5即可。在本发明中,所述萃取的次数优选为2~3次,所述萃取用有机溶剂优选包括二氯甲烷,所述s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷或s
‑1‑
磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷的物质的量和单次萃取用有机溶剂的质量之比优选为1mol:2500~3000ml。在本发明中,所述浓缩的方式优选为常压蒸馏。
[0082]
本发明还提供了一种盐酸达泊西汀的合成方法,包括以下步骤:利用氯化氢将上述技术方案所述合成方法得到的达泊西汀进行酸化,得到盐酸达泊西汀。
[0083]
在本发明中,所述盐酸达泊西汀的合成方法优选包括以下步骤:将所述达泊西汀溶剂于第七有机溶剂中,利用氯化氢将所得泊西汀溶液的ph值调节至2~3,进行酸化,得到盐酸达泊西汀。
[0084]
在本发明中,所述氯化氢优选为干燥的氯化氢气体。在本发明中,所述第七有机溶剂优选包括甲醇;所述达泊西汀的物质的量和第七有机溶剂的体积之比优选为1mol:1500~2000ml。
[0085]
在本发明中,所述ph值进一步优选为2.5。在本发明中,所述酸化的温度优选为0~5℃,本发明对于所述酸化的时间没有特殊限定,酸化至析出的固体的质量不再增加为准。
[0086]
所述酸化后,本发明优选还包括将所述酸化的体系过滤,将所得滤饼进行干燥,得到盐酸达泊西汀。在本发明中,所述干燥的方式优选为真空干燥,本发明对于所述干燥的温度和时间没有特殊限定,采用本领域技术人员熟知的干燥条件干燥至恒重即可。
[0087]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0088]
实施例1
[0089]
(1)1
‑
萘酚溴甲醚
[0090]
在1000ml反应瓶中加入400g二氯甲烷、144.2g 1
‑
萘酚(1mol)和36g多聚甲醛(1.2mol),搅拌成悬浮液,通入干燥的溴化氢气体,控制体系温度不超过30℃,直到固体全溶,将反应液转入分液漏斗中,得到的下层有机相中加入无水硫酸钠干燥,过滤,将所得滤液减压蒸馏至恒重,得到1
‑
萘酚溴甲醚(淡黄色油状物液体)。
[0091]
(2)r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇
[0092]
将0.5mol 1
‑
萘酚溴甲醚与300ml四氢呋喃混合均匀,得到1
‑
萘酚溴甲醚溶液;在氮气保护下,将加入150ml四氢呋喃、14.6g镁条(0.6mol)和0.1g碘混合均匀,滴加20ml 1
‑
萘酚溴甲醚溶液,缓慢加热至回流,当反应夜色变浅褪去,滴加剩余的1
‑
萘酚溴甲醚溶液,滴加完毕后继续保温回流反应2h,冷却至室温,过滤除去多余的镁,得到1
‑
萘氧基乙基溴化镁格式试剂溶液,氮气保护备用。
[0093]
将60g(r)
‑
环氧苯乙烷(0.5mol)与200ml无水四氢呋喃搅拌混合,冷却至
‑
50℃,滴加上述制备的1
‑
萘氧基乙基溴化镁格式试剂溶液,控制滴加速度使反应温度不高于
‑
40℃,滴加完毕后保温反应2h,关闭冷冻,自然升温至室温,然后滴加250g饱和氯化铵水溶液,在室温条件下搅拌1h,静置分层,得到水相和有机相,将所得水相用二氯甲烷萃取,单次萃取用二氯甲烷体积为300ml,萃取3次,合并有机相后加入无水硫酸钠干燥,过滤,将所得滤液浓缩至恒重,在得到的暗红色浓缩液中加入200ml乙酸乙酯,加热至回流,冷却至升温,加入500ml石油醚,在搅拌条件下冷却至0~5℃,有固体析出,过滤,将所得滤饼烘干至恒重,得r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇(白色固体,92.33g,两步反应总收率为66.34%,hplc检测纯度为98.62%)。
[0094]
(3)s
‑1‑
甲磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷
[0095]
将25.2g甲磺酰氯(0.22mol)溶解于100g二氯甲烷中,得到甲磺酰氯溶液;在1000ml反应瓶中加入300g二氯甲烷、55.7g r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇(0.2mol)、24.3g三乙胺(0.24mol)和1g dmap,搅拌溶解均匀,冷却至
‑
5~0℃后滴加甲磺酰氯溶液,控制滴加速度使反应液温度不高于5℃,滴加完毕,自然升温至室温后搅拌1h,依次用300ml浓度为5wt%的稀盐酸、300ml浓度为5wt%的碳酸氢钠溶液和300ml纯净水洗涤,然后用无水硫酸钠干燥,过滤,将所得滤液常压蒸馏至恒重,将所得浓缩物用乙酸乙酯和石油醚的混合溶剂(乙酸乙酯和石油醚体积比=1:3)重结晶,得到s
‑1‑
甲磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷(白色固体,68.25g,收率为95.74%,hplc检测得到纯度为97.69%)。
[0096]
(4)盐酸达泊西汀
[0097]
将71.3g s
‑1‑
甲磺酰氧基
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷(0.2mol)加入到反应瓶中,加入500ml四氢呋喃搅拌溶解,在室温条件下加入200ml浓度为2.0mol/l的二甲胺四氢呋喃溶液和10.1g三乙胺(0.1mol)混合均匀,在室温条件下反应20h,tlc监测反应完全后,将所得反应液倒入1000ml水中,加入150ml浓度为5mol/l的naoh溶液,调ph值至13,用二氯甲烷萃取2次,单次二氯甲烷用量为500ml,将所得有机相用无水硫酸钠干燥,然后过滤,将所得滤液浓缩至恒重,得到达泊西汀;
[0098]
在所述达泊西汀中加入200ml甲醇,搅拌至全部溶解,在室温下通入干燥的氯化氢气体至ph值为2~3,冷却至0~5℃,有颗粒状固体析出,过滤,将所得滤饼真空干燥至恒重,
得到盐酸达泊西汀(51.73g,收率为75.7%,hplc检测纯度为99.68%)。
[0099]
盐酸达泊西汀的h
‑
nmr表征数据:1h
‑
nmr[(cd3cl),400mhz]:δ8.09(d,1h,j=8.00hz,ar
‑
h),7.81(d,1h,j=7.96hz,ar
‑
h),7.67
‑
7.69(t,2h,ar
‑
h),7.41
‑
7.48(m,6h,ar
‑
h),7.27(t,1h,j=7.95hz,ar
‑
h),6.68(d,1h,j=7.47hz,ar
‑
h),4.74(d,1h,j=6.76hz,ar
‑
h),4.09(m,1h,och2),3.67(m,1h,och2),2.97(m,1h,2位ch2),2.74(m,1h,2位ch2),2.55(d,3h,j=4.16hz,n
‑
ch3),2.84(d,3h,j=3.93hz,n
‑
ch3)。
[0100]
实施例2
[0101]
(1)1
‑
萘酚溴甲醚
[0102]
在1000ml反应瓶中加入400g二氯甲烷、144.2g 1
‑
萘酚(1mol)和36g多聚甲醛(1.2mol),搅拌成悬浮液,通入干燥的溴化氢气体,控制体系温度不超过30℃,直到固体全溶,将反应液转入分液漏斗中,得到的下层有机相中加入无水硫酸钠干燥,过滤,将所得滤液减压蒸馏至恒重,得到1
‑
萘酚溴甲醚(淡黄色油状物液体)。
[0103]
(2)r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇
[0104]
将0.5mol 1
‑
萘酚溴甲醚与300ml甲基叔丁基醚混合均匀,得到1
‑
萘酚溴甲醚溶液;在氮气保护下,将加入150ml甲基叔丁基醚、18.2g镁条(0.75mol)和1ml溴乙烷混合均匀,滴加15ml 1
‑
萘酚溴甲醚溶液,缓慢加热至回流,当反应夜色变浅褪去,滴加剩余的1
‑
萘酚溴甲醚溶液,滴加完毕后继续保温回流反应1h,冷却至室温,过滤除去多余的镁,得到1
‑
萘氧基乙基溴化镁格式试剂溶液,氮气保护备用。
[0105]
将66g(r)
‑
环氧苯乙烷(0.55mol)与200ml无水甲基叔丁基醚搅拌混合,冷却至
‑
60℃,滴加上述制备的1
‑
萘氧基乙基溴化镁格式试剂溶液,控制滴加速度使反应温度不高于
‑
45℃,滴加完毕后保温反应1.5h,关闭冷冻,自然升温至室温,然后滴加250g饱和氯化铵水溶液,在室温条件下搅拌1h,静置分层,得到水相和有机相,将所得水相用甲基叔丁基醚萃取1次,甲基叔丁基醚用量为250ml,,合并有机相后用无水硫酸钠干燥,过滤,将所得滤液浓缩至恒重,在得到的暗红色浓缩液中加入250ml乙酸乙酯,加热至回流,冷却至升温,加入400ml石油醚,在搅拌条件下冷却至0~5℃,有固体析出,过滤,将所得滤饼烘干至恒重,得r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇(白色固体,89.65g,两步反应总收率为64.42%,hplc检测纯度为98.57%)。
[0106]
(3)s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷
[0107]
在1000ml反应瓶中加入600g氯仿、55.7g r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇(0.2mol)和37.4gnbs(0.21mol),搅拌混合均匀,加热回流8h,tlc监测反应完全后,冷却至室温,加入300ml水,在所得水相中加入150ml乙酸乙酯萃取,将所得有机相用150ml水洗涤后用无水硫酸钠干燥,过滤,将所得滤液常压蒸馏至恒重,将所得浓缩物用乙酸乙酯和石油醚的混合溶剂(乙酸乙酯和石油醚体积比=1:2.5)重结晶,得到s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷(白色固体,63.79g,收率为93.47%,hplc检测得到纯度为97.72%)。
[0108]
(4)盐酸达泊西汀
[0109]
将68.25g s
‑1‑
溴
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷(0.2mol)加入到反应瓶中,加入500ml四氢呋喃搅拌溶解,在室温条件下加入300ml浓度为2.0mol/l的二甲胺四氢呋喃溶液和10.1g三乙胺(0.1mol)混合均匀,在室温条件下反应20h,tlc监测反应完全后,将所得反应液倒入1000ml水中,加入150ml浓度为5mol/l的naoh溶液,调ph值至13,用二氯甲烷萃取2
次,二氯甲烷单次用量为500ml,将所得有机相用无水硫酸钠干燥,然后过滤,将所得滤液浓缩至恒重,得到达泊西汀;
[0110]
在所述达泊西汀中加入300ml甲醇,搅拌至全部溶解,在室温下通入干燥的氯化氢气体至ph值为2~3,冷却至0~5℃,有颗粒状固体析出,过滤,将所得滤饼真空干燥至恒重(53.17g,收率为77.8%,hplc检测纯度为99.62%)。
[0111]
盐酸达泊西汀的h
‑
nmr表征数据:1h
‑
nmr[(cd3cl),400mhz]:δ8.09(d,1h,j=8.00hz,ar
‑
h),7.81(d,1h,j=7.96hz,ar
‑
h),7.67
‑
7.69(t,2h,ar
‑
h),7.41
‑
7.48(m,6h,ar
‑
h),7.27(t,1h,j=7.95hz,ar
‑
h),6.68(d,1h,j=7.47hz,ar
‑
h),4.74(d,1h,j=6.76hz,ar
‑
h),4.09(m,1h,och2),3.67(m,1h,och2),2.97(m,1h,2位ch2),2.74(m,1h,2位ch2),2.55(d,3h,j=4.16hz,n
‑
ch3),2.84(d,3h,j=3.93hz,n
‑
ch3)。
[0112]
实施例3
[0113]
(1)1
‑
萘酚溴甲醚
[0114]
在1000ml反应瓶中加入400g二氯甲烷、144.2g 1
‑
萘酚(1mol)和36g多聚甲醛(1.2mol),搅拌成悬浮液,通入干燥的溴化氢气体,控制体系温度不超过30℃,直到固体全溶,将反应液转入分液漏斗中,得到的下层有机相中加入无水硫酸钠干燥,过滤,将所得滤液减压蒸馏至恒重,得到1
‑
萘酚溴甲醚(淡黄色油状物液体)。
[0115]
(2)r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇
[0116]
将0.5mol 1
‑
萘酚溴甲醚与300ml乙醚混合均匀,得到1
‑
萘酚溴甲醚溶液;在氮气保护下,将加入150ml乙醚、12.1g镁条(0.6mol)和3ml溴乙烷混合均匀,滴加15ml 1
‑
萘酚溴甲醚溶液,缓慢加热至回流,待乙醚沸腾后滴加剩余的1
‑
萘酚溴甲醚溶液,滴加时间为1h,滴加完毕后继续保温回流反应3h,冷却至室温,过滤除去多余的镁,得到1
‑
萘氧基乙基溴化镁格式试剂溶液,氮气保护备用。
[0117]
将72g(r)
‑
环氧苯乙烷(0.6mol)与500ml无水乙醚搅拌混合,冷却至
‑
70℃,滴加上述制备的1
‑
萘氧基乙基溴化镁格式试剂溶液,控制滴加速度使反应温度不高于
‑
50℃,滴加完毕后保温反应1h,关闭冷冻,自然升温至室温,然后滴加300g饱和氯化铵水溶液,在室温条件下搅拌1h,静置分层,得到水相和有机相,将所得水相用二氯甲烷萃取,单次萃取用二氯甲烷体积为300ml,萃取3次,合并有机相后加入无水硫酸钠干燥,过滤,将所得滤液浓缩至恒重,在得到的暗红色浓缩液中加入200ml乙酸乙酯,加热至回流,冷却至升温,加入600ml石油醚,在搅拌条件下冷却至0~5℃,有固体析出,过滤,将所得滤饼烘干至恒重,得r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇(白色固体,92.06g,两步反应总收率为66.15%,hplc检测纯度为98.33%)。
[0118]
(3)s
‑1‑
(4
‑
甲基苯磺酰氧基)
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷
[0119]
将41.8g对甲苯磺酰氯(0.22mol)溶解于200g二氯甲烷中,得到对甲苯磺酰氯溶液;在1000ml反应瓶中加入600g吡啶、55.7g r
‑3‑
(1
‑
萘氧基)
‑1‑
苯基丙醇(0.2mol)、24.3g三乙胺(0.24mol)和1g dmap,搅拌溶解均匀,冷却至
‑
5~0℃后滴加甲磺酰氯溶液,控制滴加速度使反应液温度不高于3℃,滴加完毕,自然升温至室温后搅拌1h,将反应液倒入1000ml冰水中,用二氯甲烷萃取3次,单次萃取用二氯甲烷体积为400g,合并有机相后用无水硫酸钠干燥,过滤,将所得滤液常压蒸馏至恒重,将所得浓缩物用乙酸乙酯和石油醚的混合溶剂(乙酸乙酯和石油醚体积比=1:3.5)重结晶,得到s
‑1‑
(4
‑
甲基苯磺酰氧基)
‑1‑
苯
基
‑3‑
(1
‑
萘氧基)丙烷(白色固体,76.27g,收率为88.17%,hplc检测得到纯度为98.21%)。
[0120]
(4)盐酸达泊西汀
[0121]
将43.3g s
‑1‑
(4
‑
甲基苯磺酰氧基)
‑1‑
苯基
‑3‑
(1
‑
萘氧基)丙烷(0.1mol)加入到反应瓶中,加入250ml乙腈搅拌溶解,在室温条件下加入100g浓度为40wt%的二甲胺水溶液混合均匀,在室温条件下反应30h,tlc监测反应完全后,将所得反应液倒入350ml水中,用二氯甲烷萃取2次,单次二氯甲烷用量为300ml,合并有机相后用无水硫酸钠干燥,然后过滤,将所得滤液浓缩至恒重,得到达泊西汀;
[0122]
在所述达泊西汀中加入200ml甲醇,搅拌至全部溶解,在室温下通入干燥的氯化氢气体至ph值为2~3,冷却至0~5℃,有颗粒状固体析出,过滤,将所得滤饼真空干燥至恒重(26.22g,收率为76.7%,hplc检测纯度为99.55%)。
[0123]
盐酸达泊西汀的h
‑
nmr表征数据:1h
‑
nmr[(cd3cl),400mhz]:δ8.09(d,1h,j=8.00hz,ar
‑
h),7.81(d,1h,j=7.96hz,ar
‑
h),7.67
‑
7.69(t,2h,ar
‑
h),7.41
‑
7.48(m,6h,ar
‑
h),7.27(t,1h,j=7.95hz,ar
‑
h),6.68(d,1h,j=7.47hz,ar
‑
h),4.74(d,1h,j=6.76hz,ar
‑
h),4.09(m,1h,och2),3.67(m,1h,och2),2.97(m,1h,2位ch2),2.74(m,1h,2位ch2),2.55(d,3h,j=4.16hz,n
‑
ch3),2.84(d,3h,j=3.93hz,n
‑
ch3)。
[0124]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1