一种1,2,3-三氮唑衍生物及其制备方法与应用
本公开属于有机合成化学技术领域,具体提供一种1,2,3-三氮唑衍生物及其制备方法与应用。
背景技术:
这里的陈述仅提供与本公开有关的背景信息,而不必然构成现有技术。
基叠氮化合物是一类非常重要的有机合成中间体。世界上第一例芳基叠氮化合物-苯基叠氮,是1864年德国化学家petergriess通过氨与过溴化重氮苯反应得到的。由于其独特的物理化学性质以及特殊的官能团,吸引了很多科学家的注意。然而,叠氮化合物一般都具有潜在的爆炸性,例如在热、光、摩擦或撞击条件下引入少量的外部能量后就会产生激烈的爆炸性分解。近年来,随着实验安全技术的提高,人们对叠氮化合物的认识也在不断加深,由于各方面的需要,该类化合物又重新获得了人们的高度重视。虽然叠氮化合物具有潜在的爆炸性,但它们却是一类高价值的有机合成中间体,通过它可以还原为氨基、参加点击反应、合成三氮唑类杂环等。近年来,叠氮化合物在化学、医学、药学、生物学、材料学等各个领域的广泛应用引起了人们更多的关注。
一般地,芳基叠氮化合物制备的方法有很多。例如,芳基叠氮化合物合成的经典方法是利用芳胺的重氮化:2009年,zarei等人报道了芳胺在硅载硫酸的作用下,与叠氮化钠在室温条件下发生重氮化反应合成相应的芳基叠氮化合物,该反应中产生的重氮盐中间体比较稳定,可以在室温条件下保存数日,且操作简便;其次,可以通过重氮转移反应:2002年,tilley课题组报道了邻位取代的芳基碘化物在正丁基锂的作用下先合成芳基锂,然后与对甲苯磺酰叠氮通过重氮转移反应得到目标反应;再次,可以通过有机硼酸催化偶联反应合成:2007年,tao课题组在室温条件下,用芳基硼酸与叠氮化钠在硫酸铜的催化下生成了相应的叠氮化合物;还可以通过卤代烃的亲核取代反应合成相应的叠氮化合物:2006年,varma课题组将卤代烃在微波条件下与叠氮化钠发生叠氮化反应合成相应的叠氮化合物。
芳基叠氮化合物是一种非常重要的有机合成中间体,因其含有叠氮基高能活性官能团。点击化学与叠氮化合物的应用密不可分,其中叠氮化合物与端炔的环加成反应是点击化学中最普遍的一类反应。近年来,叠氮化合物与炔烃的环加成反应成为了合成三氮唑类化合物的一类非常重要的反应。2002年,sharpless等人报道了铜催化的叠氮化合物与端炔在叔丁醇与水的混合溶剂中发生cuaac反应,该反应使用五水硫酸铜与抗坏血酸钠在室温下原位生成的一价铜作为催化剂,高效的选择性合成了1,2,3-三氮唑化合物;2014年,ramachary课题组报道了dbu催化的叠氮化合物与醛的环加成反应,并得到产率高的三氮唑化合物;2015年,jia课题组报道了碘化亚铜催化的(e)-1-溴-2-(2-硝基乙烯基)苯、叠氮化钠与醛的三组分一锅法高效地合成相应的目标产物。
然而,在本公开发明人在研究过程中发现,这些实现结构对称式的1,2,3-三氮唑衍生物方法中存在以下问题:1.反应条件复杂,难以轻易控制;2.反应需要多步进行,影响产率,产率不高;3.反应所需催化剂价格昂贵,反应成本高;4.合成的1,2,3-三氮唑类化合物结构简单,不能充分发挥其药物优势;5.反应副产物较多,影响提纯。
技术实现要素:
针对现有技术中1,2,3-三氮唑衍生物在制备过程中存在反应条件复杂,需要多步进行,合成的1,2,3-三氮唑类化合物结构简单,不能充分发挥其药物优势,.反应副产物较多,影响提纯的问题。
本公开的目的是提供一种由双金属催化芳基叠氮化合物合成结构对称式的1,2,3-三氮唑衍生物方法,能够直接利用芳基叠氮化合物与端炔通过一锅法偶联成1,2,3-三氮唑衍生物,形成一种结构对称式的1,2,3-三氮唑化合物结构,极大地拓展了三氮唑类化合物的药用价值。
本公开一个或一些实施方式中,提供一种1,2,3-三氮唑衍生物,其结构如式(1)所示,
式(1)
其中,r选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c4-c8直链或支链的烷基中的一种。
本公开一个或一些实施方式中,提供式(1)所示化合物的制备方法,其合成路径如下所示,
其中,r选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c4-c8直链或支链的烷基中的一种。
本公开一个或一些实施方式中,提供式(1)所示化合物的制备方法,包括如下步骤:以芳基叠氮化合物与端炔作为原料,在一价铜盐和二价钯盐的催化作用下,加入添加剂、溶剂,通过一锅法偶联成结构对称式的1,2,3-三氮唑衍生物,
其中,端炔中端部选自给电子或缺电子的芳基、杂芳基、烷基和烷氧基。
本公开一个或一些实施方式中,提供上述化合物或上述合成式(1)所示化合物的方法在制备抗炎药物中的应用。
上述技术方案中的一个或一些技术方案具有如下优点或有益效果:
1)本公开提供了一种双金属催化芳基叠氮基化合物合成结构对称式的1,2,3-三氮唑衍生物新方法,极大地发挥1,2,3-三氮唑化合物本身的结构优势,具有很高的生物药用价值。
2)本公开通过一锅法合成结构对称式1,2,3-三氮唑衍生物,该方法简便、高效,所用到的原料简单易得且无毒、步骤少、条件温和、成本低,操作简单,产物纯度较高,条件易于控制,适合工业大规模生产。
附图说明
构成本公开一部分的说明书附图用来提供对本公开的进一步理解,本公开的示意性实施例及其说明用于解释本公开,并不构成对本公开的不当限定。
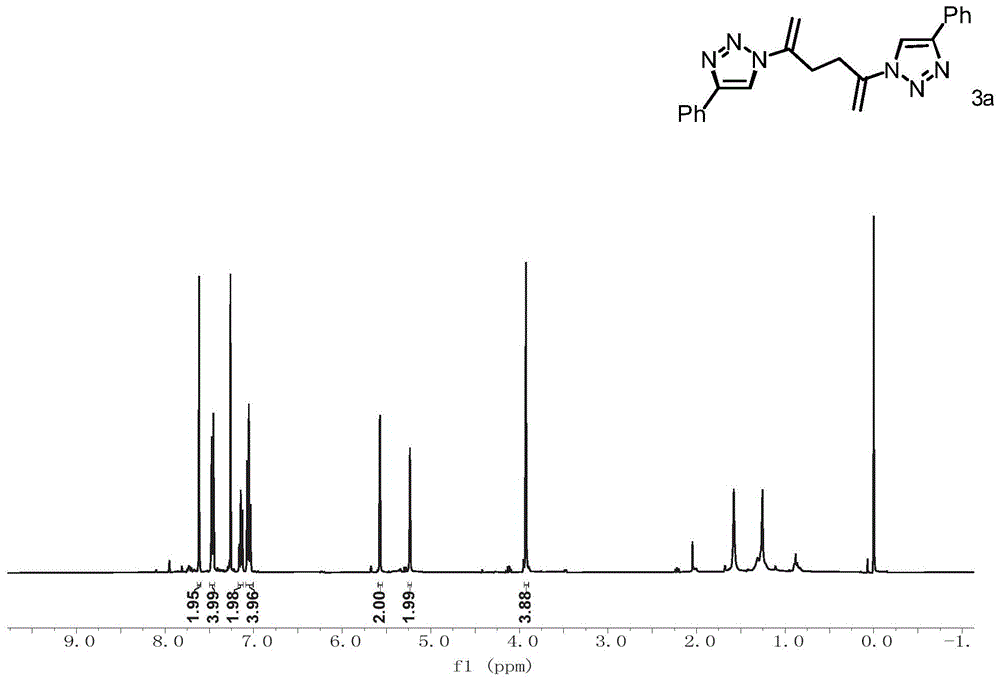
图1为本公开实施例3制备的化合物3a的1h-nmr的核磁共振谱图;
图2为本公开实施例3制备的化合物3a的13c-nmr的核磁共振谱图;
图3为本公开实施例9制备的化合物3b的1h-nmr的核磁共振谱图;
图4为本公开实施例9制备的化合物3b的13c-nmr的核磁共振谱图;
图5为本公开实施例10制备的化合物3c的1h-nmr的核磁共振谱图;
图6为本公开实施例10制备的化合物3c的13c-nmr的核磁共振谱图;
图7为本公开实施例11制备的化合物3d的1h-nmr的核磁共振谱图;
图8为本公开实施例11制备的化合物3d的13c-nmr的核磁共振谱图。
具体实施方式
下面将对本公开实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本公开的一部分实施例,而不是全部实施例。基于本公开的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本公开保护的范围。
针对现有技术中1,2,3-三氮唑衍生物在制备过程中存在反应条件复杂,需要多步进行,合成的1,2,3-三氮唑类化合物结构简单,不能充分发挥其药物优势,.反应副产物较多,影响提纯的问题,
本公开的目的是提供一种由双金属催化芳基叠氮化合物合成结构对称式的1,2,3-三氮唑衍生物方法,能够直接利用芳基叠氮化合物与端炔通过一锅法偶联成1,2,3-三氮唑衍生物,形成一种结构对称式的1,2,3-三氮唑化合物结构,极大地拓展了三氮唑类化合物的药用价值。
本公开一个或一些实施方式中,提供一种1,2,3-三氮唑衍生物,其结构如式(1)所示,
式(1)
其中,r选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c4-c8直链或支链的烷基中的一种。
优选的,所述芳基选自苯基或取代苯基;
或,所述芳基选自苯基和被卤素、烷基或烷氧基所取代的苯基;
或,所述卤素选自f、cl、br;
或,所述烷基选自c1-c6直链或支链烷基;
或,所述c1-c6直链烷基选自甲基、乙基、正丙基,正丁基;
或,所述c1-c6支链烷基选自叔丁基、正戊基;
或,所述烷氧基选自c1-c2直链或支链烷氧基;
或,所述c1-c2直链或支链烷氧基选自甲氧基、乙氧基;
或,所述杂芳基含有一个或多个杂原子,所述杂原子选自n、o和s。
本公开一个或一些实施方式中,提供式(1)所示化合物的制备方法,其合成路径如下所示,
其中,r选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c4-c8直链或支链的烷基中的一种。
本公开一个或一些实施方式中,提供式(1)所示化合物的制备方法,包括如下步骤:以芳基叠氮化合物与端炔作为原料,在一价铜盐和二价钯盐的催化作用下,加入添加剂、溶剂,通过一锅法偶联成结构对称式的1,2,3-三氮唑衍生物,
其中,端炔中端部选自给电子或缺电子的芳基、杂芳基、烷基和烷氧基。
优选的,一价铜盐为含有一价铜的化合物,如碘化亚铜、溴化亚铜、氯化亚铜、噻吩-2-甲酸亚铜、四氟硼酸四乙腈铜、硫化亚铜、溴化亚铜二甲硫醚。该实施方式的一种或多种实施例中,一价铜盐为硫化亚铜、碘化亚铜和噻吩-2-甲酸亚铜。该催化剂能够提高原料的转化率和产物的产率。当一价铜盐为硫化亚铜时,能够进一步提高该1,2,3-三氮唑衍生物的产率。二价钯盐为二价钯的化合物,如pd(pph3)4、pd(pph3)2cl2、醋酸钯。该实施方式的一种或多种实施例中,当一价铜盐为硫化亚铜时,二价钯盐为醋酸钯时,能够进一步提高该1,2,3-三氮唑衍生物的产率。
优选的,添加剂为三乙胺、dbu、pmdeta、naoh、na2co3等。当添加剂为dbu时,能够提高原料的转化率和产物的产率。
优选的,反应温度为60~100℃。该温度能够提高原料的转化率,同时提高产物的产率。当反应的温度为80±8℃时,能够进一步提高原料的转化率和产物的产率。
优选的,为了使端炔与酚取代的烯基叠氮化合物混合均匀,还包括如下步骤,将原料添加至溶剂中溶解,在加入添加剂及催化剂,加热进行反应。
溶剂选自甲醇、甲苯、n,n-二甲基甲酰胺(dmf)、乙腈(ch3cn)、二甲基亚砜(dmso)、1,2-二氯乙烷(dce)中的一种或多种。优选的,溶剂为二甲亚砜(dmso)和乙腈时,该溶剂提高原料的转化率,同时提高产物的产率。进一步优选的,当溶剂为二甲亚砜(dmso)时,原料的转化率、产物的产率更高。
优选的,端炔与酚取代的烯基叠氮化合物的摩尔比为1~5:1~8。进一步优选的,端炔与酚取代的烯基叠氮化合物的摩尔比为2:5。
优选的,一价铜盐的添加量为原料总质量的10%~50%。
进一步优选的,一价铜盐的添加量为原料总质量的20%。
优选的,二价钯盐的添加量为原料总质量的1%~5%。
进一步优选的,二价钯盐的添加量为原料总质量的2%。
优选的,反应时间为0~6h,反应时间不为0。
进一步优选的,反应时间为4±0.5h。
为了提高该1,2,3-三氮唑衍生物的纯度,该实施方式的一种或多种实施例中,将反应后溶液加入萃取溶剂进行萃取获得有机相,将有机相中的溶剂去除,进行硅胶柱层析,获得结构对称式的1,2,3-三氮唑衍生物。
优选的,萃取采用的萃取溶剂为1,2-二氯乙烷、甲苯、硝基甲烷、乙酸乙酯、乙醚、正己烷、环己烷、石油醚或二氯甲烷中的一种或多种。
优选的,萃取采用的萃取溶剂为1,2-二氯乙烷、甲苯、硝基甲烷、乙酸乙酯、乙醚、正己烷、环己烷、石油醚或二氯甲烷中的一种或多种;
进一步优选的,萃取采用的萃取溶剂为二氯甲烷;
优选的,所述萃取进行1~3次,每次使用5~20ml萃取溶剂;
优选的,获得有机相采用无水硫酸镁进行干燥,再去除有机溶剂;
优选的,硅胶柱层析的洗脱液为石油醚和乙酸乙酯;
优选的,石油醚和乙酸乙酯的体积比为1~30:1~6;
进一步优选的,石油醚和乙酸乙酯的体积比为10:3。采用该洗脱液能够获得纯度更高的1,2,3-三氮唑衍生物。
为了使得本领域技术人员能够更加清楚地了解本公开的技术方案,以下将结合具体的实施例详细说明本公开的技术方案。
实施例1
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加碘化亚铜(0.0095g,0.050mmol)、醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为76%。
实施例2
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入cutc(0.0095g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为45%。
实施例3
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为91%。
实施例4
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),四(三苯基膦)钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为80%。
实施例5
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的乙腈中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为66%。
实施例6
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为53%
实施例7
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、氢氧化钠(0.0660ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为35%
实施例8
将化合物1a即苯乙炔(0.0270ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、pmdeta(0.0520ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3a的产率为28%
实施例1~8的反应如下式所示:
化合物3a:
1hnmr(400mhz,cdcl3),如图1所示,δ7.62(s,2h),7.50–7.44(m,4h),7.18–7.12(m,2h),7.09–7.00(m,4h),5.57(d,j=1.3hz,2h),5.23(d,j=1.3hz,2h),3.93(s,4h).13cnmr(100mhz,cdcl3),如图2所示,δ146.94,139.81,130.88,128.56,127.92,125.73,119.47,110.94,56.73.hrms(esi)m/zcalculatedforc22h20n6[m+na]+:563.2398,found:563.2378.
实施例9
将化合物1b即3-甲基苯乙炔(0.0320ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3b的产率为83%。
化合物3b:
1hnmr(400mhz,cdcl3),如图3所示,δ7.67(s,2h),7.46–7.42(m,2h),7.25(q,j=2.6,1.9hz,2h),7.02–6.92(m,4h),5.56(d,j=1.3hz,2h),5.21(d,j=1.3hz,2h),3.93(s,4h),2.24(s,6h).13cnmr(100mhz,cdcl3),如图4所示,δ148.27,140.40,138.11,130.38,128.78,128.46,126.23,122.94,119.18,109.66,54.22,24.02.hrms(esi)m/zcalculatedforc33h36n6[m+na]+:539.2878,found:539.2868.
实施例10
将化合物1c即4-乙基苯乙炔(0.0350ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3c的产率为86%。
化合物3c:
1hnmr(400mhz,cdcl3),如图5所示,δ7.55(s,2h),7.42–7.36(m,4h),6.90–6.84(m,4h),5.59(d,j=1.3hz,2h),5.21(d,j=1.3hz,2h),3.92(s,4h),2.57(q,j=7.6hz,4h),1.21(t,j=7.6hz,6h).13cnmr(100mhz,cdcl3),图6所示,δ147.58,144.55,140.17,128.01,126.52,126.23,120.41,108.82,57.75,28.58,15.90.hrms(esi)m/zcalculatedforc35h40n6[m+na]+:575.2698,found:575.2675.
实施例11
将化合物1d即4-叔丁基苯乙炔(0.0450ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.1024g,0.25mmol)、dbu(0.0450ml,2.5mmol),加入到1ml的dmso中,在80℃的条件下溶解,随后向体系中加入硫化亚铜(0.0080g,0.05mmol),醋酸钯(0.0100g,0.005mmol),在氮气的保护下,继续加热搅拌四小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:3)得到化合物3d的产率为88%。
化合物3d:
1hnmr(400mhz,cdcl3),如图7所示,δ7.71(s,2h),7.57–7.50(m,4h),7.24–7.20(m,4h),5.64(d,j=1.3hz,2h),5.16(d,j=1.3hz,2h),3.95(s,4h),1.28(s,18h).13cnmr(100mhz,cdcl3),图8所示,δ153.57,148.37,138.95,127.92,125.04,119.49,104.88,55.50,34.60,30.84.hrms(esi)m/zcalculatedforc39h48n6[m+na]+:575.2698,found:575.2689.
实施例12
本实施例提供化合物3a、3b、3c、3d抑制宫颈癌hela细胞活性效果测试,包括如下步骤:
1.取宫颈癌hela细胞以6×104/ml的细胞密度接种于培养板中,每孔100μl,置37℃、5%co2及饱和湿度的条件下培养过夜。
2.待其贴壁后,分别加入化合物3a、3b、3c、3d使其终浓度达到1μg/ml、2μg/ml、3μg/ml、4μg/ml、5μg/ml,每组设5个复孔,每孔终体积为200μl。对照组加入等量的dmem培养液。
3.培养24h和48h后,每孔加入mtt(5mg/ml))30μl,继续培养6h。
4.离心去上清,每孔加入dmso150μl溶解结晶颗粒。
5.酶标仪于580nm波长测定吸光度(d),计算不同时间、不同浓度的阿霉素对宫颈癌hela细胞的增殖抑制率,实验重复3次。
6.计算增殖抑制率=[(对照组d570—实验组d570)/对照组d570]×100%。
ic50是指在用药后存活的细胞数量减少一半时所需的药物浓度。在mtt法中,就是以对照组吸光度od值减少一半所需的药物浓度为ic50。除此以外,半数抑制浓度的含义还相当于药物对培养细胞的最小致死剂量的平均值,作为反映药效的定量指标,在各种药物的筛选中广泛应用。
具体的,根据公式:抑制率=1-加药组od值/对照组od值,计算化合物的ic50值
经测试,所有化合物的ic50值均低于8.0μg/kg,
通过上述测试结果可以看出,本公开实施例中所有化合物均有良好抑制肿瘤细胞活性的效果。
以上所揭露的仅为本公开的优选实施例而已,当然不能以此来限定本公开之权利范围,因此依本公开申请专利范围所作的等同变化,仍属本公开所涵盖的范围。
- 还没有人留言评论。精彩留言会获得点赞!