一种烷基/烯基取代含氮杂环化合物的制备方法
1.本发明属于有机合成技术领域,具体涉及烷基/烯基取代含氮杂环化合物的制备方法。
背景技术:
2.烷基/烯基取代含氮杂环化合物是一类用途比较广泛的有机合成中间体,其在香精香料、医药生产、功能材料以及有机合成中具有重要的应用价值,因此,发展高效简洁的合成烷基/烯基取代含氮杂环化合物的方法具有重要意义。文献调查发现,近年来这两类化合物的合成方法大都是以2
‑
甲基含氮杂环化合物和伯醇为原料,经金属比如ru、ir、pt、mn、fe、ni或co催化(j.org.chem.,2020,85,2277
‑
2290;j.org.chem.,2017,82,4113
‑
4120;eur.j.org.chem.,2020,4942
‑
4949;org.lett.,2019,21,7514
‑
7518;chem.lett.,2019,48,1192
‑
1195;angew.chem.int.ed.,2018,57,9126
‑
9130;angew.chem.int.ed.,2018,57,9131
‑
9135;chem.commun.,2018,54,12369
‑
12372;chem.commun.,2020,56,249
‑
252;chem.commun.,2019,55,7530
‑
7533;j.org.chem.,2019,84,9819
‑
9825;),甚至是需要金属和配体的螯合物参与才能进行反应,由此给工业生产后处理和环境保护带来了麻烦。
3.综上所述,虽然关于以甲基含氮杂环化合物和伯醇为原料制备烷基/烯基取代含氮杂环化合物的合成方法有大量报道,但无法兼顾合成方法绿色和工业化生产成本低廉。基于此,本发明提供了一种无金属和无配体参与的碱促进含氮杂环化合物2
‑
甲基c
‑
烷基化或烯基化的反应方法,该方法可以绿色高效的合成系列烷基/烯基取代含氮杂环化合物。
技术实现要素:
4.针对上述问题,本发明提供了一种烷基/烯基取代含氮杂环化合物的制备新方法,以解决现有合成方法应用受限的问题。
5.为解决上述技术问题,本发明所采用的技术方案是:
6.一种烷基/烯基取代含氮杂环化合物的制备方法,包括下列步骤:
7.(1)将2
‑
甲基含氮杂环化合物、伯醇、碱和添加剂加入有机溶剂中在一定温度下反应24h,然后冷却至室温,得到反应液;
8.(2)将步骤(1)所得反应液进行浓缩,分离纯化,即得烷基/烯基取代含氮杂环化合物。
9.进一步,所述2
‑
甲基含氮杂环化合物为2
‑
甲基吡嗪、2
‑
甲基吡啶氮氧化物、2
‑
甲基喹啉衍生物、2
‑
甲基喹喔啉衍生物和2
‑
甲基苯并恶唑衍生物中的至少一种;2
‑
甲基吡嗪、2
‑
甲基吡啶氮氧化物、2
‑
甲基喹啉衍生物、2
‑
甲基喹喔啉衍生物和2
‑
甲基苯并恶唑衍生物的结构式分别为:
10.11.r1选自h、ome、f、cl、br。
12.进一步,所述伯醇为芳基甲醇或烷基伯醇,其中芳基甲醇包括苄醇、2
‑
甲基苄醇、3
‑
甲基苄醇、3
‑
氟苄醇、3
‑
氯苄醇、3
‑
溴苄醇、4
‑
甲基苄醇、4
‑
氟苄醇、4
‑
氯苄醇、4
‑
溴苄醇、4
‑
乙基苄醇、4
‑
异丙基苄醇、4
‑
联苯甲醇、2
‑
萘甲醇、2
‑
吡啶甲醇、2
‑
噻吩甲醇和2
‑
呋喃甲醇中的至少一种,烷基伯醇包括c4
‑
c10直链末端醇、环丙甲醇、环己甲醇中的至少一种。
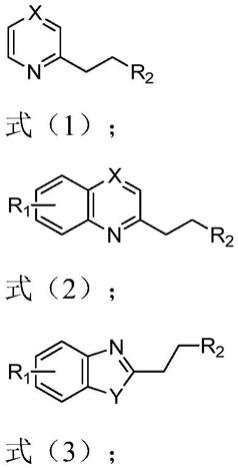
13.进一步,所述烷基取代含氮杂环化合物具有如式(1)、式(2)或式(3)所示的结构:
[0014][0015]
式(1)中,r2选自c3
‑
c9直链烷基、环丙甲基、环己甲基、ph、chph、meph、etph、i
‑
prph、omeph、fph、clph、brph、cf3ph、4
‑
biphenyl、1
‑
naphthyl、2
‑
pyridyl、2
‑
thiophyl、2
‑
furanyl;x为ch或n;
[0016]
式(2)中,r1选自h、ome、f、cl、br;r2选自c3
‑
c9直链烷基、环丙甲基、环己甲基、ph、chph、meph、etph、i
‑
prph、omeph、fph、clph、brph、cf3ph、4
‑
biphenyl、1
‑
naphthyl、2
‑
pyridyl、2
‑
thiophyl、2
‑
furanyl;x为ch或n;
[0017]
式(3)中,r1选自h、ome、f、cl、br;r2选自c3
‑
c9直链烷基、环丙甲基、环己甲基、ph、chph、meph、etph、i
‑
prph、omeph、fph、clph、brph、4
‑
biphenyl、1
‑
naphthyl、2
‑
pyridyl、2
‑
thiophyl、2
‑
furanyl;y为o或s;
[0018]
进一步,所述烯基取代含氮杂环化合物具有如式(4)、式(5)或式(6)所示的结构:
[0019]
[0020][0021]
式(4)中,ar选自ph、meph、etph、i
‑
prph、omeph、fph、clph、brph、cf3ph、4
‑
biphenyl、1
‑
naphthyl、2
‑
pyridyl、2
‑
thiophyl、2
‑
furanyl;x为ch或n;
[0022]
式(5)中,r1选自h、ome、f、cl、br;ar选自ph、meph、etph、i
‑
prph、omeph、fph、clph、brph、cf3ph、4
‑
biphenyl、1
‑
naphthyl、2
‑
pyridyl、2
‑
thiophyl、2
‑
furanyl;x为ch或n;
[0023]
式(6)中,r1选自h、ome、f、cl、br;ar选自ph、meph、etph、i
‑
prph、omeph、fph、clph、brph、4
‑
biphenyl、1
‑
naphthyl、2
‑
pyridyl、2
‑
thiophyl、2
‑
furanyl。
[0024]
进一步,所述碱为叔丁醇锂、叔丁醇钠、叔丁醇钾、碳酸铯、碳酸钠、氢氧化钾、醋酸钾、甲醇钠和三乙胺中的至少一种,具体可为叔丁醇钾。
[0025]
进一步,所述添加剂为三苯基膦、三环己基膦、三(2
‑
甲氧基苯基)膦、1,10
‑
菲啰啉、2,2'
‑
联吡啶、喹啉、四甲基乙二胺和18
‑
冠醚
‑
6中的至少一种,具体可为18
‑
冠醚
‑
6。
[0026]
进一步,所述有机溶剂为二甲基亚砜、四氢呋喃、乙腈、1,4
‑
二氧六环、甲苯、叔丁醇、1,2
‑
二氯乙烷中的至少一种,具体可为甲苯。
[0027]
进一步,所述伯醇采用芳基甲醇或烷基伯醇时,按下述反应条件制得烷基取代含氮杂环化合物:所述2
‑
甲基含氮杂环化合物与伯醇的物质的量之比为1:2~2:1,具体可为1:1.5;步骤(1)中的碱的用量为0.5~2.0当量,具体可为1.0当量;步骤(1)中的添加剂的用量为0.5当量;步骤(1)中的反应温度为120~160℃,具体可为150℃。
[0028]
进一步,所述伯醇采用芳基甲醇时,按下述反应条件制得烯基取代含氮杂环化合物:所述2
‑
甲基含氮杂环化合物与伯醇的物质的量之比为1:2~2:1,具体可为1:1.5;步骤(1)中的碱的用量为0.5~2.0当量,具体可为1.5当量;步骤(1)中的添加剂的用量为0.5当量;所述步骤(1)中的反应温度为40~110℃,具体可为100℃。
[0029]
进一步,所述步骤(2)中的分离纯化为:将反应液浓缩后产生的浓缩物以正己烷、乙酸乙酯组成的混合物为展开剂,进行薄层色谱分离,正己烷和乙酸乙酯的体积比为(3~5):1,具体可为体积比4:1。
[0030]
本发明的有益效果:本发明方法能够避免使用贵金属,不需任何配体参与,不需要惰性气体保护,仅使用了廉价无污染的叔丁醇钾和18
‑
冠醚
‑
6,在一定温度和溶剂条件下即可高效合成烷基/烯基取代含氮杂环化合物,收率高达95%,操作方便,适合推广应用,预期在香精香料、医药生产、功能材料以及有机合成领域得到广泛应用。
附图说明
[0031]
图1为实施例1所得产物2
‑
苯乙基吡嗪的核磁氢谱。
[0032]
图2为实施例1所得产物2
‑
苯乙基吡嗪的核磁碳谱。
[0033]
图3为实施例5所得产物2
‑
苯乙烯基吡嗪的核磁氢谱。
[0034]
图4为实施例5所得产物2
‑
苯乙烯基吡嗪的核磁碳谱。
具体实施方式
[0035]
下面结合具体实施例对本发明做进一步的说明,但本发明的保护范围并不局限于此。
[0036]
实施例1
[0037]
本实施例的烷基取代含氮杂环化合物为2
‑
苯乙基吡嗪,结构式为:
[0038][0039]
本实施例的烷基取代含氮杂环化合物的制备方法,反应路线如下所示:
[0040][0041]
具体采用以下步骤:
[0042]
1)取2
‑
甲基吡嗪0.2mmol、苄醇0.3mmol、叔丁醇钾0.2mmol和18
‑
冠醚
‑
6 0.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于150℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0043]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得33mg目标产物。
[0044]
本实施例的目标产品收率为90%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.51(s,1h),8.39(d,j=2.4hz,1h),8.35(s,1h),7.27(t,j=7.12hz,2h),7.18(dd,j=10.1,7.3hz,3h),3.15
‑
3.10(m,2h),3.09
‑
3.05(m,2h);
13
c nmr(100mhz,cdcl3)ppm:156.8,144.7,144.1,142.4,140.8,128.5,128.4,126.2,37.2,35.4。
[0045]
实施例2
[0046]
本实施例的烷基取代含氮杂环化合物为2
‑
(3
‑
甲基苯乙基)吡嗪,结构式为:
[0047][0048]
本实施例的烷基取代含氮杂环化合物的制备路线可参考实施例1,具体采用以下步骤:
[0049]
1)取2
‑
甲基吡嗪0.2mmol、3
‑
甲基苄醇0.3mmol、叔丁醇钾0.2mmol和18
‑
冠醚
‑
6 0.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于150℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0050]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得34mg目标产物。
[0051]
本实施例的目标产品收率为85%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.51(dd,j=2.3,1.8hz,1h),8.40(d,j=2.5hz,1h),8.36(d,j=
1.2hz,1h),7.16(t,j=7.8hz,1h),7.01(d,j=6.4hz,2h),6.97(d,j=7.8hz,1h),3.10(td,j=8.2,2.0hz,2h),3.03(td,j=6.3,1.8hz,2h),2.31(s,3h);
13
c nmr(100mhz,cdcl3)ppm:156.9,144.7,144.1,142.3,140.7,138.1,129.2,128.4,127.0,125.4,37.3,35.4,21.4。
[0052]
实施例3
[0053]
本实施例的烷基取代含氮杂环化合物为2
‑
(2
‑
呋喃乙基)吡嗪,结构式为:
[0054][0055]
本实施例的烷基取代含氮杂环化合物的制备路线可参考实施例1,具体采用以下步骤:
[0056]
1)取2
‑
甲基吡嗪0.2mmol、2
‑
呋喃甲醇0.3mmol、叔丁醇钾0.2mmol和18
‑
冠醚
‑
6 0.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于150℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0057]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得28mg目标产物。
[0058]
本实施例的目标产品收率为81%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.53(s,1h),8.41(d,j=2.4hz,1h),8.40(s,1h),7.12
‑
7.10(m,1h),6.89(dd,j=4.9,3.5hz,1h),6.76(d,j=3.0hz,1h),3.32(t,j=7.2hz,2h),3.18(t,j=8.0hz,2h);
13
c nmr(100mhz,cdcl3)ppm:156.1,144.8,144.1,143.3,142.5,126.8,124.7,123.5,37.3,29.2。
[0059]
实施例4
[0060]
本实施例的烷基取代含氮杂环化合物为2
‑
戊基喹啉,结构式为:
[0061][0062]
本实施例的烷基取代含氮杂环化合物的制备路线可参考实施例1,具体采用以下步骤:
[0063]
1)取2
‑
甲基喹啉0.2mmol、正丁醇0.3mmol、叔丁醇钾0.2mmol和18
‑
冠醚
‑
6 0.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于150℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0064]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得32mg目标产物。
[0065]
本实施例的目标产品收率为80%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.04(dd,j=8.4,2.5hz,2h),7.75(d,j=8.0hz,1h),7.60
‑
7.70(m,1h),7.49
‑
7.42(m,1h),7.28(d,j=8.4hz,1h),2.95(dd,j=8.6,7.2hz,2h),1.86
‑
1.73(m,2h),1.39
‑
1.30(m,4h),0.80(t,j=7.1hz,3h);
13
c nmr(100mhz,cdcl3)ppm:163.2,148.0,136.2,129.3,129.0,127.5,126.7,125.6,121.4,39.4,31.8,29.8,22.6,14.0。
[0066]
实施例5
[0067]
本实施例的烯基取代含氮杂环化合物为(e)
‑2‑
苯乙烯基吡嗪,结构式为:
[0068][0069]
本实施例的烯基取代含氮杂环化合物的制备方法,反应路线如下所示:
[0070][0071]
具体采用以下步骤:
[0072]
1)取2
‑
甲基吡嗪0.2mmol、苄醇0.3mmol、叔丁醇钾0.3mmol和18
‑
冠醚
‑
60.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于100℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0073]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得35mg目标产物。
[0074]
本实施例的目标产品收率为95%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.64(d,j=1.2hz,1h),8.54(t,j=2.1hz,1h),8.40(d,j=2.4hz,1h),7.75(d,j=16.1hz,1h),7.59(d,j=7.3hz,2h),7.40(t,j=7.0hz,2h),7.35
‑
7.32(m,1h),7.16(d,j=16.1hz,1h);
13
c nmr(100mhz,cdcl3)ppm:151.3,144.3,143.8,142.8,136.1,135.2,129.0,128.9,127.3,124.0。
[0075]
实施例6
[0076]
本实施例的烯基取代含氮杂环化合物为(e)
‑2‑
(3
‑
甲基苯乙烯基)吡嗪,结构式为:
[0077][0078]
本实施例的烯基取代含氮杂环化合物的制备路线可参考实施例5,具体采用以下步骤:
[0079]
1)取2
‑
甲基吡嗪0.2mmol、3
‑
甲基苄醇0.3mmol、叔丁醇钾0.3mmol和18
‑
冠醚
‑
6 0.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于100℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0080]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得31mg目标产物。
[0081]
本实施例的目标产品收率为80%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.62(d,j=1.2hz,1h),8.52(t,j=2.1hz,1h),8.38(d,j=2.4hz,1h),7.71(d,j=16.1hz,1h),7.39(d,j=8.8hz,2h),7.28(t,j=7.6hz,1h),7.13(t,j=8.0hz,2h),2.38(s,3h);
13
c nmr(100mhz,cdcl3)ppm:151.4,144.3,143.7,142.7,138.4,
136.0,135.3,129.8,128.7,127.9,124.6,123.9,21.4。
[0082]
实施例7
[0083]
本实施例的烯基取代含氮杂环化合物为(e)
‑2‑
苯乙烯基吡啶,结构式为:
[0084][0085]
本实施例的烯基取代含氮杂环化合物的制备路线可参考实施例5,具体采用以下步骤:
[0086]
1)取2
‑
甲基吡啶氮氧化物0.2mmol、苄醇0.3mmol、叔丁醇钾0.3mmol和18
‑
冠醚
‑
6 0.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于100℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0087]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得28mg目标产物。
[0088]
本实施例的目标产品收率为79%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.61(d,j=4hz,1h),7.66(td,j=7.8,1.8hz,2h),7.59(t,j=9.1hz,2h),7.38(t,j=7.8hz,3h),7.30(t,j=7.3hz,1h),7.19
‑
7.13(m,2h);
13
c nmr(100mhz,cdcl3)ppm:155.5,149.6,136.6,136.5,132.7,128.8,128.4,128.0,127.1,122.1,122.0。
[0089]
实施例8
[0090]
本实施例的烯基取代含氮杂环化合物为(e)
‑2‑
(2
‑
([1,1'
‑
联二苯基]
‑4‑
基)
‑
乙烯基)
‑6‑
甲氧基喹啉(stb
‑
8),该化合物目前已经被广泛用作治疗阿尔茨海默病β淀粉样斑块的显像剂,结构式为:
[0091][0092]
本实施例的烯基取代含氮杂环化合物的制备路线可参考实施例5,具体采用以下步骤:
[0093]
1)取6
‑
甲氧基
‑2‑
甲基喹啉0.2mmol、4
‑
联苯甲醇0.3mmol、叔丁醇钾0.3mmol和18
‑
冠醚
‑
6 0.1mmol,加入1ml的甲苯中制成混合物,将该混合物置于5ml的schlenk管内,置于100℃的加热模块中,反应24h后,冷却至室温,得到反应液;
[0094]
2)将反应液进行浓缩得浓缩物,将浓缩物以正己烷、乙酸乙酯按体积比4:1组成的混合溶剂为展开剂,以硅胶为吸附剂,进行薄层色谱分离,得52mg目标产物。
[0095]
本实施例的目标产品收率为70%,对目标产品进行核磁表征,结果如下:1h nmr(400mhz,cdcl3)ppm:8.00(dd,j=16.4,8.9hz,2h),7.63(ddd,j=6.6,5.5,2.6hz,8h),7.48
‑
7.41(m,3h),7.35(ddd,j=7.6,5.3,3.9hz,2h),7.05(d,j=2.7hz,1h),3.92(s,3h);
13
c nmr(100mhz,cdcl3)ppm:156.6,152.7,143.3,140.1,139.6,134.8,134.1,131.7,129.7,128.1,127.8,127.3,126.6,126.5,126.4,126.0,121.3,118.6,104.3,54.6。
[0096]
本发明的烷基/烯基取代含氮杂环化合物的其他实施例中,步骤1)中,各原料的配比、具体反应条件可以在本发明限定的比例范围内进行适应性调整,可得到预期的目标产
物。在不脱离本发明宗旨的前提下,对上述实施例中的各个具体参数进行变更,形成多个具体的实施例,均为本发明的常见变化范围,在此不再一一详述。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1