间位双取代化合物及其制备和在防治蚊虫、植物病菌病中的应用
1.本发明涉及间位双取代化合物及其制备和在防治蚊虫、植物病菌病中的应用,属于农业防护技术领域。
背景技术:
2.合理药物设计中的重大突破就是计算机辅助药物设计(computer aided drug design,cadd),生物信息学和计算机技术的飞速发展极大的促进了cadd的发展。cadd可通过计算机预测和筛选大分子数据库,选择最有前景的分子进行结构优化,确保尽可能好的先导化合物进行体外筛选,来降低与药物发现相关的成本,而且还可以缩短药物上市所需的时间。因此,计算机辅助药物设计受到了制药行业和学术界的广泛关注。基于计算机药物筛选原理的不同,cadd方法一般分为两类(beilstein j.org.chem.2016,12,2694-2718.):基于结构的药物设计方法(structure-based drug design,sbdd),基于配体的药物设计方法(ligand-based drug design,lbdd)。
3.sbdd是通过研究药物靶点结构的特征,及药物分子和受体之间的相互作用来进行药物设计,如果已知疾病相关药物靶点的三维结构,最常用的cadd技术是基于结构设计,sbdd主要包括分子对接方法和从头设计两种方法。
4.当药物靶点的蛋白质结构没有通过实验确定或者无法用计算方法预测结构时,通常使用lbdd方法。其主要依赖于了解已知的与受体结合的配体结构来进行药物设计。药效团模型和定量构效关系(quantitative structure-activity relationship,qsar)模型是目前比较流行的lbdd方法。
5.γ-氨基丁酸(gaba)受体是一种重要的药物分子靶标,异噁唑啉类及间双酰胺类化合物为21世纪发现的作用于gaba受体新位点的新化合物。
技术实现要素:
6.以gaba受体为研究模型,结合文献调研并结合前两部分衍生物的结构特征,以间双酰胺结构为基础,通过计算机辅助的手段设计并合成了十八个新颖骨架的间位双取代化合物,
7.本发明提供间位双取代化合物及其制备方法和在防治蚊虫、植物病菌病中的应用。本专利的间位双取代化合物具有很好的杀蚊幼虫及抗植物病菌活性。
8.本发明的间位双取代化合物为下述i-1~i-18所示的化合物(结构式一)。
[0009][0010]
结构式一
[0011]
上述化学结构式i-1~i-18的合成方法如下:
[0012]
间位双取代化合物i-1的合成:按照方程式一所示的方法制备,首先以无水二氯甲烷做溶剂,三乙胺做缚酸剂,在0℃下,将2-氯乙烷磺酰氯(i-1-b)与n-甲基苄胺(i-1-a)反应生成中间体i-1-c。1-二甲基哌啶-4-甲酸(i-1-d)和n,o-二甲基羟胺盐酸盐(i-1-e)在二氯甲烷经n,n
′‑
羰基二咪唑及三乙胺催化反应生成中间体i-1-f。随后在无水四氢呋喃溶液中,1,3-二溴苯(i-1-g)在正丁基锂催化下与中间体i-1-f反应得到中间体i-1-h,再和中间体i-1-c经钯催化偶连生成中间体i-1-i,最后经钯/碳氢气还原生成i-1。
[0013][0014]
方程式一
[0015]
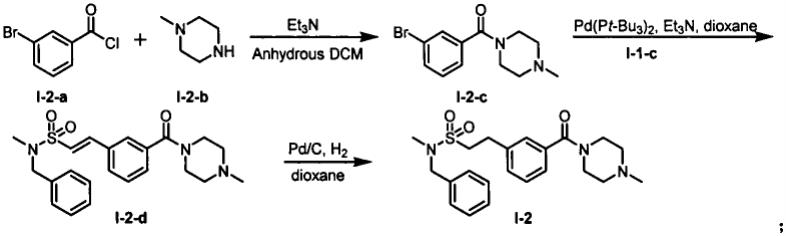
间位双取代化合物i-2的合成:按照方程式二所示的方法制备,首先以无水二氯甲烷做溶剂,三乙胺做缚酸剂,3-溴苯甲酰氯(i-2-a)和n-甲基哌嗪(i-2-b)反应生成中间体
i-2-c,随后与中间体i-1-c经钯催化偶连生成中间体i-2-d,最后经钯/碳氢气还原生成i-2。
[0016][0017]
方程式二
[0018]
间位双取代化合物i-3的合成:按照方程式三所示的方法制备,首先以无水二氯甲烷做溶剂,三乙胺做缚酸剂,3-溴苯甲酰氯(i-2-a)和2-甲氧基乙胺(i-3-a)反应生成中间体i-3-b,随后与中间体i-1-c经钯催化偶连生成中间体i-3-c,后经钯/碳氢气还原生成i-3-d,最后在硝基甲烷中,经过氧单磺酸钾、溴化钾催化脱苄基生成i-3。
[0019][0020]
方程式三
[0021]
间位双取代化合物i-4的合成:按照方程式四所示的方法制备,首先以无水二氯甲烷做溶剂,三乙胺做缚酸剂,6-溴吲哚(i-4-a)和甲氧基乙酰氯(i-4-b)反应生成中间体i-4-c,随后与中间体i-1-c经钯催化偶连生成中间体i-4-d,后在常压氢气下经钯/碳还原生成i-4-e,最后在高压氢气下经钯/碳、氢氧化钯/碳催化脱苄基生成i-4。
[0022][0023]
方程式四
[0024]
间位双取代化合物i-5的合成:按照方程式五所示的方法制备,首先在无水四氢呋喃溶液中,2,6-二溴吡啶(i-5-a)在正丁基锂催化下与中间体i-1-f反应得到中间体i-5-b,随后与中间体i-1-c经钯催化偶连生成中间体i-5-c,最后在常压氢气下经钯/碳还原生成i-5。
[0025][0026]
方程式五
[0027]
间位双取代化合物i-6的合成:按照方程式六所示的方法制备,首先先以n,n-二甲基甲酰胺做溶剂,三乙胺做缚酸剂,6-溴-2-吡啶羧酸(i-6-a)和n-甲基哌嗪(i-2-b)反应生成中间体i-6-b,随后与中间体i-1-c经钯催化偶连生成中间体i-6-c,最后在常压氢气下经钯/碳还原生成i-6。
[0028][0029]
方程式六
[0030]
间位双取代化合物i-7的合成:按照方程式七所示的方法制备,首先在氩气保护,-78℃下,(1-甲基吡咯烷-2-基)甲醇(i-7-f)与草酰氯、二甲基亚砜反应生成中间体i-7-g,后立即与甲苯磺酰肼(i-7-h)反应生成中间体i-7-i。2-(3-溴苯基)-1,3-二氧烷(i-7-a)与苯基乙烯基砜(i-7-b)经钯催化偶连生成中间体i-7-c,后在常压氢气下经钯/碳还原生成i-7-d,再经稀盐酸脱保护生成中间体i-7-e,最后与中间体i-7-i在碳酸铯催化下生成i-7。
[0031][0032]
方程式七
[0033]
间位双取代化合物i-8的合成:按照方程式八所示的方法制备,首先3-溴苯甲酸甲酯(i-8-a)与苯基乙烯基砜(i-7-b)经钯催化偶连生成中间体i-8-b,后在常压氢气下经钯/碳还原生成i-8-c,再经稀盐酸水解生成中间体i-8-d,最后与n-甲基哌嗪(i-2-b)缩合生成i-8。
[0034][0035]
方程式八
[0036]
间位双取代化合物i-9的合成:按照方程式九所示的方法制备,首先6-溴-2-吡啶羧酸(i-9-b)与4-氟苯胺(i-9-a)缩合生成中间体i-9-c,最后与4-氨基-1-甲基哌啶(i-9-d)经钯催化偶连生成i-9。
[0037][0038]
方程式九
[0039]
间位双取代化合物i-10的合成:按照方程式十所示的方法制备,首先n-甲基-4-哌啶酮(i-10-d)与苯磺酰肼(i-7-h)反应生成中间体i-10-e。0℃下,3-溴-dl-苯丙氨酸(i-10-a)经硼氢化钠还原、甲醇稀释、氢氧化钠水溶液催化,然后再与三光气反应生成中间体i-10-b,后与甲酸在钯催化下生成溴被甲酰基取代中间体i-10-c,最后与中间体i-10-e在碳酸铯催化下生成i-10。
[0040][0041]
方程式十
[0042]
间位双取代化合物i-11的合成:按照方程式十一所示的方法制备,首先中间体i-10-b与乙烯基正丁醚(i-11-a)在钯催化下生成溴被乙酰基取代中间体i-11-b,最后在稀盐酸催化下与甲胺、多聚甲醛生成i-11。
[0043][0044]
方程式十一
[0045]
间位双取代化合物i-12的合成:按照方程式十二所示的方法制备,首先2,4-二溴苯酚(i-12-a)与4-氟苄脒盐酸盐(i-12-b)经1,3-偶极环加成得到中间体i-12-c,最后与4-氨基-1-甲基哌啶(i-9-d)经钯催化偶连生成i-12。
[0046][0047]
方程式十二
[0048]
间位双取代化合物i-13的合成:按照方程式十三所示的方法制备,首先4-溴苯甲酰氯(i-13-a)与6-溴-2-氨基吡啶(i-13-b)经缩合得到中间体i-13-c,最后与4-二甲氨基哌啶(i-13-d)经钯催化偶连生成i-13。
[0049][0050]
方程式十三
[0051]
间位双取代化合物i-14的合成:按照方程式十四所示的方法制备,首先间苯二甲酸单甲酯(i-14-a)与4-氟苯胺(i-9-a)缩合生成中间体i-14-b,然后经氢氧化钠水解得到中间体i-14-c,最后与n-甲基哌嗪(i-2-b)酰胺缩合生成i-14。
[0052][0053]
方程式十四
[0054]
间位双取代化合物i-15的合成:按照方程式十五所示的方法制备,邻二苯甲酸酐(i-15-a)与6-溴-2-氨基吡啶(i-13-b)在乙酸加热回流下缩合得到中间体i-15-b,后与苯基乙烯基砜(i-7-b)经钯催化偶连生成中间体i-15-c,后在常压氢气下经钯/碳还原、水合肼水解生成i-15-c,最后与1-甲基吡咯烷-3-甲酸(i-15-e)在三氯氧磷催化下缩合生成i-15。
[0055][0056]
方程式十五
[0057]
间位双取代化合物i-16的合成:按照方程式十六所示的方法制备,对氟苯硼酸(i-16-a)与6-溴-2-氨基吡啶(i-13-b)在经钯催化偶连生成中间体i-16-b,后与4-环己酮羧酸(i-16-c)在三氯氧磷催化下缩合生成中间体i-16-d,最后与二甲胺经三乙酰氧基硼氢化钠
还原胺化得到i-16。
[0058][0059]
方程式十六
[0060]
间位双取代化合物i-17的合成:按照方程式十七所示的方法制备,首先中间体i-13-c与1,4-二氧杂-螺[4,5]癸-7-烯-8-硼酸频哪醇酯(i-17-a)经钯催化偶连生成中间体i-17-b,在常压氢气下经钯碳还原生成中间体i-17-c,后经稀盐酸水解得到中间体i-17-d,最后与二甲胺经三乙酰氧基硼氢化钠还原胺化得到i-17。
[0061][0062]
方程式十七
[0063]
间位双取代化合物i-18的合成:按照方程式十八所示的方法制备,首先对氟苯甲醛(i-18-a)与4-溴邻苯二胺(i-18-b)在偏亚硫酸氢钠催化下加热回流得到环化中间体i-18-c,后与1,4-二氧杂-螺[4,5]癸-7-烯-8-硼酸频哪醇酯(i-17-a)经钯催化偶连生成中间体i-18-d,在常压氢气下经钯碳还原生成中间体i-18-e,后经稀盐酸水解得到中间体i-18-f,最后与二甲胺经三乙酰氧基硼氢化钠还原胺化得到i-18。
[0064][0065]
方程式十八
[0066]
本发明的间位双取代化合物i-1~i-18表现出较好的杀蚊幼虫和抗植物病菌活性,能较好的灭杀蚊幼虫及抑制黄瓜枯萎,花生褐斑,苹果轮纹,小麦纹枯,玉米小斑,西瓜炭疽,水稻恶苗,番茄早疫,小麦赤霉,水稻稻瘟,辣椒疫霉,油菜菌核,黄瓜灰霉,水稻纹枯14种植物病菌。
具体实施方式
[0067]
下述的实施例和生测试验结果可用来进一步说明本发明,但不意味着限制本发明。
[0068]
实施例1:间位双取代化合物i-1的合成。
[0069]
第一步,中间体i-1-c的合成。在0℃下,将2-氯乙烷磺酰氯(i-1-b,1.00ml,10.0mmol),滴加到n-甲基苄胺(i-1-a,1.29ml,10.0mmol)和三乙胺(4.86ml,35.0mmol)的无水二氯甲烷中。滴加完毕后,0℃下继续搅拌2h。然后将混合物用二氯甲烷稀释,盐水洗涤,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=6∶1)柱层析得淡黄色油状物1.43g,收率68%。1h nmr(400mhz,cdcl3)δ7.38-7.28(m,5h),6.45(dd,j=16.4,10.0hz,1h),6.25(d,j=16.4hz,1h),6.01(d,j=10.0hz,1h),4.24(s,2h),2.68(s,3h).
13
c nmr(100mhz,cdcl3)δ135.7,133.2,128.7,128.4,128.0,127.9,53.8,34.1.
[0070]
第二步,中间体i-1-f的合成。室温下,向1-二甲基哌啶-4-甲酸(i-1-d,2.86g,20.0mmol)的二氯甲烷(60ml)溶液中加入n,n
′‑
羰基二咪唑(3.24g,20.0mmol),室温搅拌1h,然后加入n,o-二甲基羟胺盐酸盐(i-1-e,2.54g,26.0mmol),三乙胺(4.2ml,60.0mmol)。反应搅拌过夜,后分别用饱和碳酸钠水溶液、水洗涤,无水硫酸钠干燥,并在减压下浓缩。经(v(二氯甲烷)∶v(甲醇)=80/1
→
30∶1)柱层析得白色油状物2.31g,收率62%。1h nmr(400mhz,cdcl3)δ3.69(s,3h),3.17(s,3h),2.9-2.86(m,2h),2.65-2.54(m,1h),2.26(s,3h),1.99-1.93(m,2h),1.88-1.70(m,4h).
13
c nmr(100mhz,cdcl3)δ176.1,61.3,55.1,46.3,37.5,32.0,28.2.mass spectrometry:hrms-esi(m/z):calcd for c9h
19
n2o2[m+h]
+
187.1447,found,187.1443.
[0071]
第三步,中间体i-1-h的合成。将1,3-二溴苯(i-1-g,849.3mg,3.6mmol)的thf(5ml)溶液冷却至-78℃,滴加正丁基锂(1.5ml,3.6mmol,2.4m)。在-78℃下搅拌30min后,将i-1-f(558.5mg,3.0mmol)的thf(3ml)溶液缓慢滴加到反应液中,使混合物在1小时内升至0℃,随后用饱和氯化铵溶液淬灭。乙酸乙酯萃取,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
50∶1)柱层析得棕色固体549.3mg,收率64%,熔点72-74℃。1h nmr(400mhz,cdcl3)δ7.95(s,1h),7.76(d,j=7.2hz,1h),7.59(d,j=7.2hz,1h),7.26(t,j=7.2hz,1h),3.17-3.03(m,1h),2.92-2.79(m,2h),2.24(s,3h),2.12-1.99(m,2h),1.85-1.70(m,4h).
13
c nmr(100mhz,cdcl3)δ201.1,137.8,135.7,131.2,130.3,126.7,123.0,55.0,46.3,42.9,28.4.mass spectrometry:hrms-esi(m/z):calcd for c
13h17
brno[m+h]
+
282.0494,found,282.0488.
[0072]
第四步,中间体i-1-i的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六环,后加入化合物i-1-h(1.12g,4.0mmol),i-1-c(844.3mg,4.0mmol),三乙胺(1.1ml,8.0mmol)和pd(pt-bu3)2(204.4mg,0.4mmol,10mol%),氩气置换气三次。混合物于70℃加热反应24h。冷却至室温,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
50∶1)柱层析得白色固体1.52g,收率92%,熔点134-136℃。1h nmr(400mhz,cdcl3)δ8.02(s,1h),7.95(d,j=7.6hz,1h),7.64(d,j=7.6hz,1h),7.56-7.49(m,2h),7.40-7.29(m,5h),6.75(d,j=15.6hz,1h),4.32(s,2h),3.24-3.15(m,1h),2.98-2.89(m,2h),2.75(s,3h),2.32(s,3h),2.15-2.05(m,2h),1.91-1.82(m,4h).
13
c nmr(100mhz,cdcl3)δ202.1,141.4137.0,135.7,133.5,132.3,130.3,129.6,128.9,128.6,128.2,127.9,124.0,55.3,54.0,46.6,43.4,
34.4,28.8.mass spectrometry:hrms-esi(m/z):calcd for c
23h29
n2o3s[m+h]
+
413.1899,found,413.1894.
[0073]
第五步,i-1的合成。向100ml圆底烧瓶中加入化合物i-1-i(1.24g,3.0mmol),10%钯碳(638.5mg,0.6mmol),后加入30ml 1,4-二氧六环。氢气置换气5次,室温搅拌72h。经硅藻土垫过滤,二氯甲烷洗涤,减压浓缩,得到白色固体1.01g,产率81%,熔点93-94℃。1h nmr(400mhz,cdcl3)δ7.83-7.77(m,2h),7.46-7.40(m,2h),7.40-7.30(m,5h),4.35(s,2h),3.28-3.15(m,5h),2.94(d,j=11.6hz,2h),2.79(s,3h),2.31(s,3h),2.14-2.05(m,2h),1.90-1.81(m,4h).
13
c nmr(100mhz,cdcl3)δ202.6,139.0,136.9,135.8,133.1,129.3,128.9,128.4,128.2,128.2,127.0,55.4,54.0,51.7,46.6,43.4,34.5,29.6,28.9.mass spectrometry:hrms-esi(m/z):calcd for c
23h31
n2o3s[m+h]
+
415.2055,found,415.2053.
[0074]
实施例2:间位双取代化合物i-2的合成。
[0075]
第一步,中间体i-2-c的合成。将3-溴苯甲酰氯(i-2-a,1.45ml,11.0mmol)缓慢滴加入冰浴下的n-甲基哌嗪(i-2-b,1.1ml,10.0mmol)和三乙胺(2.8ml,20mmol)的二氯甲烷(50ml)溶液中。反应升至室温搅拌1h。加水淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
40∶1)柱层析得黄色固体2.51g,收率89%,熔点65-67℃。1h nmr(400mhz,cdcl3)δ7.57-7.52(m,2h),7.35-7.25(m,2h),3.89-3.62(m,2h),3.57-3.30(m,2h),2.59-2.43(m,2h),2.41-2.24(m,5h).
13
c nmr(100mhz,cdcl3)δ168.4,137.7,132.62(s),130.0,129.9,125.5,122.5,55.1,54.5,47.5,46.0,42.0.mass spectrometry:hrms-esi(m/z):calcd for c
12h16
brn2o[m+h]
+
283.0446,found,283.0441.
[0076]
第二步,中间体i-2-d的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六环,后加入化合物i-2-c(1.13g,4.0mmol),i-1-c(844.3mg,4.0mmol),三乙胺(1.1ml,8.0mmol)和pd(pt-bu3)2(204.4mg,0.4mmol,10mol%),氩气置换气三次。混合物于70℃加热反应24h。冷却至室温,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
50∶1)柱层析得无色油状物1.43g,收率87%。1h nmr(400mhz,cdcl3)δ7.54-7.40(m,5h),7.37-7.28(m,5h),6.71(d,j=15.6hz,1h),4.29(s,2h),3.81(s,2h),3.43(s,2h),2.73(s,3h),2.49(s,2h),2.40-2.29(m,5h).
13
c nmr(100mhz,cdcl3)δ169.4,141.4,137.0,135.7,133.4,129.6,129.4,129.1,128.9,128.6,128.2,126.7,123.8,55.4,55.4,54.8,47.9,46.2,42.3,34.4.mass spectrometry:hrms-esi(m/z):calcd for c
22h28
n3o3s[m+h]
+
414.1851,found,414.1846.
[0077]
第三步,i-2的合成。向100ml圆底烧瓶中加入化合物i-2-d(1.24g,3.0mmol),10%钯碳(478.9mg,0.45mmol),后加入30ml 1,4-二氧六环。氢气置换气5次,室温搅拌72h。经硅藻土垫过滤,二氯甲烷洗涤,减压浓缩,得到无色液体1.16g,收率91%。1h nmr(400mhz,cdcl3)δ7.39-7.29(m,6h),7.28-7.24(m,3h),4.35-4.31(m,2h),3.80(s,2h),3.43(s,2h),3.25-3.13(m,4h),2.78(s,3h),2.49(s,2h),2.33(s,5h).
13
c nmr(100mhz,cdcl3)δ170.1,138.8,136.6,135.8,129.8,129.1,128.9,128.4,128.2,127.2,125.6,55.5,54.9,53.9,51.5,47.8,46.1,42.2,34.4,29.5.mass spectrometry:hrms-esi(m/z):calcd for c
22h30
n3o3s[m+h]
+
416.2008,found,416.2003.
[0078]
实施例3:间位双取代化合物i-3的合成。
[0079]
第一步,中间体i-3-b的合成。将3-溴苯甲酰氯(i-2-a,1.46ml,11.0mmol)缓慢滴
nmr(100mhz,cdcl3)δ167.3,143.7,129.9,126.3,125.5,120.3,119.3,71.6,59.1,46.6,27.6.mass spectrometry:hrms-esi(m/z):calcd for c
11h13
brno2[m+h]
+
270.0130,found,270.0126.
[0085]
第二步,中间体i-4-d的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六环,后加入化合物i-4-c(807.0g,3.0mmol),i-1-c(633.2mg,3.0mmol),三乙胺(0.84ml,6.0mmol)和pd(pt-bu3)2(153.3mg,0.3mmol,10mol%),氩气置换气三次。混合物于70℃加热反应24h。冷却至室温,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=1∶1)柱层析得白色固体1.04g,收率89%,熔点108-109℃。1h nmr(400mhz,cdcl3)δ8.45(s,1h),7.45(d,j=15.2hz,1h),7.36-7.33(m,4h),7.33-7.26(m,1h),7.22(d,j=7.8hz,1.2h),7.12(dd,j=7.8,1.0hz,1.2h),6.72(d,j=15.2hz,1h),4.27(s,2h),4.17(s,2h),4.08(t,j=8.4hz,2h),3.52(s,3h),3.23(t,j=8.4hz,2h),2.70(s,3h).
13
c nmr(100mhz,cdcl3)δ167.9,143.8,142.7,135.9,134.4,132.4,128.8,128.5,128.0,125.6,125.2,121.9,115.3,72.4,59.5,54.0,47.0,34.3,28.4.mass spectrometry:hrms-esi(m/z):calcd for c
21h25
n2o4s[m+h]
+
401.1535,found,401.1529.
[0086]
第三步,中间体i-4-e的合成。向100ml圆底烧瓶中加入化合物i-4-d(800.3mg,2.0mmol),10%钯碳(425.7mg,0.4mmol),后加入20ml 1,4-二氧六环。氢气置换气5次,室温搅拌72h。经硅藻土垫过滤,二氯甲烷洗涤,减压浓缩,得到白色固体直接进行下一步。
[0087]
第四步,i-4的合成。将中间体i-4-e(201.1mg,0.5mmol),乙醇(2ml),10%p/c(80.0mg,0.075mmol),20%pd(oh)2/c(52.7mg,0.075mmol)加入高压反应釜,置于4mpa氢气氛围下反应14h。反应结束后,将混合物用硅藻土过滤,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=1∶1)柱层析得得白色固体43.6mg,收率28%,熔点158-160℃。1h nmr(400mhz,cdcl3)δ8.13(s,1h),7.13(d,j=7.6hz,1h),6.90(d,j=7.6hz,1h),4.29-4.21(m,1h),4.15(s,2h),4.04(t,j=8.4hz,2h),3.51(s,3h),3.31-3.25(m,2h),3.18(t,j=8.4hz,2h),3.12-3.05(m,2h),2.75(d,j=5.3hz,3h).
13
c nmr(100mhz,cdcl3)δ167.6,143.4,137.6,129.7,124.9,124.3,116.8,72.3,59.4,52.3,47.0,30.1,29.4,28.0.mass spectrometry:hrms-esi(m/z):calcd for c
14h20
n2o4s[m+h]
+
313.1222,found,313.1215.
[0088]
实施例5:间位双取代化合物i-5的合成。
[0089]
第一步,中间体i-5-b的合成。将2,6-二溴吡啶(i-5-a,852.8mg,3.6mmol)的thf(5ml)溶液冷却至-78℃,滴加正丁基锂(1.5ml,3.6mmol,2.4m)。在-78℃下搅拌30min后,将i-1-f(558.5mg,3.0mmol)的thf(3ml)溶液缓慢滴加到反应液中,使混合物在1小时内升至0℃,随后用饱和氯化铵溶液淬灭。乙酸乙酯萃取,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
40∶1)柱层析得深棕色油状物473.8mg,收率56%。1h nmr(400mhz,cdcl3)δ7.96(dd,j=7.6,0.8hz,1h),7.68(t,j=7.6hz,1h),7.64(dd,j=7.6,0.8hz,1h),3.71(tt,j=11.6,3.7hz,1h),2.93-2.87(m,2h),2.29(s,3h),2.12(td,j=11.6,2.4hz,2h),1.94-1.86(m,2h),1.81-1.70(m,2h).
13
c nmr(100mhz,cdcl3)δ202.4,153.6,141.3,139.4,131.7,121.4,55.2,46.6,41.8,28.3.mass spectrometry:hrms-esi(m/z):calcd for c
12h16
brn2o[m+h]
+
283.0446,found,283.0442.
[0090]
第二步,中间体i-5-c的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六环,后加入化合物i-5-b(846.1g,3.0mmol),i-1-c(633.2mg,3.0mmol),三乙胺(0.84ml,
6.0mmol)和pd(pt-bu3)2(153.3mg,0.3mmol,10mol%),氩气置换气三次。混合物于70℃加热反应24h。冷却至室温,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
40∶1)柱层析得黄色固体921.2g,收率74%,熔点108-109℃。1h nmr(400mhz,cdcl3)δ8.03(dd,j=7.6,0.8hz,1h),7.90(t,j=7.6hz,1h),7.56-7.51(m,2h),7.38-7.28(m,6h),4.36(s,2h),3.79(tt,j=11.6,3.6hz,1h),2.98-2.91(m,2h),2.79(s,3h),2.33(s,3h),2.13(td,j=11.6,2.4hz,2h),1.98-1.90(m,2h),1.87-1.76(m,2h).
13
c nmr(100mhz,cdcl3)δ203.3,153.1,150.5,140.3,138.4,135.7,128.9,128.5,128.2,127.8,127.8,123.4,55.4,54.0,46.7,42.1,34.3,28.5.mass spectrometry:hrms-esi(m/z):calcd for c
22h28
n3o3s[m+h]
+
414.1851,found,414.1846.
[0091]
第三步,i-5的合成。向100ml圆底烧瓶中加入化合物i-5-c(1.24g,3.0mmol),10%钯碳(638.5mg,0.6mmol),后加入30ml 1,4-二氧六环。氢气置换气5次,室温搅拌72h。经硅藻土垫过滤,二氯甲烷洗涤,减压浓缩,得到浅黄色油状物1.01g,收率81%。1h nmr(400mhz,cdcl3)δ7.88(d,j=7.8hz,1h),7.77(t,j=7.8hz,1h),7.39-7.28(m,6h),4.35(s,2h),3.80-3.70(m,1h),3.59-3.52(m,2h),3.42-3.34(m,2h),2.91(d,j=11.2hz,2h),2.79(s,3h),2.28(s,3h),2.07(t,j=10.8hz,2h),1.90(d,j=11.8hz,2h),1.84-1.71(m,2h).
13
c nmr(100mhz,cdcl3)δ203.6,157.0,152.5,137.8,135.8,128.9,128.3,128.2,126.7,120.7,55.5,53.9,48.9,46.6,42.1,34.3,31.3,28.4.mass spectrometry:hrms-esi(m/z):calcd for c
22h30
n3o3s[m+h]
+
416.2008,found,416.2005.
[0092]
实施例6:间位双取代化合物i-6的合成。
[0093]
第一步,中间体i-6-b的合成。室温下,向6-溴-2-吡啶羧酸(2.02g,10.0mmol)和n-甲基哌嗪(i-2-b,1.22ml,11.0mmol)的dmf(40ml)溶液,加入edci盐酸盐(2.88g,15.0mmol),hobt(2.03g,15.0mmol),三乙胺(2.1ml,15.0mmol)。混合物室温搅拌16h。减压除去挥发物,用二氯甲烷萃取,合并有机相用饱和碳酸钠水溶液洗涤,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
20∶1)柱层析得黄色固体2.03g,收率72%,熔点65-67℃。1h nmr(400mhz,cdcl3)δ7.68(t,j=7.6hz,1h),7.61(d,j=7.6hz,1h),7.55(d,j=7.6hz,1h),3.81(t,j=4.8hz,2h),3.61(t,j=4.8hz,2h),2.51(t,j=4.8hz,2h),2.43(t,j=4.8hz,2h),2.33(s,3h).
13
c nmr(100mhz,cdcl3)δ165.5,154.7,140.5,139.4,129.0,122.8,55.1,54.5,47.1,46.0,42.3.mass spectrometry:hrms-esi(m/z):calcd for c
12h16
brn2o[m+h]
+
283.0446,found,283.0442.
[0094]
第二步,中间体i-6-c的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六环,后加入化合物i-6-b(566.1m g,2.0mmol),i-1-c(422.1mg,2.0mmol),三乙胺(0.84ml,6.0mmol)和pd(pt-bu3)2(102.2mg,0.2mmol,10mol%),氩气置换气三次。混合物于70℃加热反应48h。冷却至室温,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
40∶1)柱层析得淡黄色油606.4g,收率73%。1h nmr(400mhz,cdcl3)δ7.86(t,j=7.6hz,1h),7.63(d,j=7.6hz,1h),7.48(d,j=14.8hz,1h),7.41(d,j=7.6hz,1h),7.38-7.34(m,4h),7.34-7.26(m,2h),4.32(s,2h),3.86(t,j=4.8hz,2h),3.59(t,j=4.8hz,2h),2.75(s,3h),2.54(t,j=4.8hz,2h),2.42(t,j=4.8hz,2h),2.34(s,3h).
13
c nmr(100mhz,cdcl3)δ166.0,154.7,150.0,140.1,138.4,135.7,128.9,128.5,128.2,127.8,125.6,124.8,55.4,54.8,54.0,47.3,46.2,42.4,34.3.mass spectrometry:hrms-esi(m/z):calcd for c
21h27
n4o3s[m+h]
+
415.1804,found,415.1797.
[0095]
第三步,i-6的合成。向100ml圆底烧瓶中加入化合物i-6-c(1.24g,3.0mmol),10%钯碳(638.5mg,0.6mmol),后加入30ml 1,4-二氧六环。氢气置换气5次,室温搅拌72h。经硅藻土垫过滤,二氯甲烷洗涤,减压浓缩,得到浅黄色油状物1.09g,收率87%。1h nmr(400mhz,cdcl3)δ7.75(s,1h),7.47(d,j=7.6hz,1h),7.39-7.27(m,6h),4.32(s,2h),3.88-3.79(m,2h),3.58-3.47(m,4h),3.38-3.30(m,2h),2.76(s,3h),2.55-2.49(m,2h),2.43-2.36(m,2h),2.32(s,3h).
13
c nmr(100mhz,cdcl3)δ167.5,156.7,153.9137.9,135.8,128.8,128.3,128.1,124.1,121.8,55.4,54.7,53.8,48.8,47.2,46.1,42.2,34.3,31.4.mass spectrometry:hrms-esi(m/z):calcd for c
21h29
n4o3s[m+h]
+
417.1960,found,417.1956.
[0096]
实施例7:间位双取代化合物i-7的合成。
[0097]
第一步,中间体i-7-c的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六环,后加入化合物2-(3-溴苯基)-1,3-二氧烷(i-7-a,0.30ml,2.0mmol),苯基乙烯基砜(i-7-b,336.4mg,2.0mmol),三乙胺(0.56ml,4.0mmol)和pd(pt-bu3)2(102.2mg,0.2mmol,10mol%),氩气置换气三次。混合物于70℃加热反应48h。冷却至室温,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=5∶1)柱层析得白色固体483.5mg,收率76%,熔点78-80℃。1h nmr(400mhz,cdcl3)δ7.98-7.92(m,2h),7.69(d,j=15.6hz,1h),7.65-7.60(m,2h),7.54(dd,j=15.2,8.0hz,3h),7.48(d,j=7.8hz,1h),7.41(t,j=7.6hz,1h),6.89(d,j=15.6hz,1h),5.79(s,1h),4.16-4.00(m,4h).
13
c nmr(100mhz,cdcl3)δ142.2,140.7,139.2,133.6,132.7,129.6,129.5,129.3,127.9,127.8,126.3,103.1,65.5.mass spectrometry:hrms-esi(m/z):calcd for c
17h17
o4s[m+h]
+
317.0848,found,317.0842.
[0098]
第二步,中间体i-7-d的合成。向100ml圆底烧瓶中加入化合物i-7-c(776.0mg,2.4mmol),10%钯碳(383.2mg,0.36mmol),后加入30ml 1,4-二氧六环。氢气置换气5次,室温搅拌24h。经硅藻土垫过滤,减压浓缩,得到白色固体738.6mg,产率97%。
[0099]
第三步,中间体i-7-e的合成。将中间体i-7-d(632.2mg,2.0mmol)加入1n盐酸(12ml)及丙酮(22ml)的溶液中,室温下反应4h。用饱和碳酸氢钠水溶液中和,减压旋出部分挥发物
[185]
。残余物用乙酸乙酯和水萃取,合并有机相用盐水洗涤,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=3∶1)柱层析得白色油506.1mg,收率92%。1h nmr(400mhz,cdcl3)δ9.94(s,1h),7.95-7.90(m,2h),7.71(dt,j=6.8,1.6hz,1h),7.69-7.64(tt,j=7.6,2.0hz,1h),7.61(s,1h),7.57(t,j=7.6hz,2h),7.44(m,2h),3.42-3.36(m,2h),3.17-3.10(m,2h).
13
c nmr(100mhz,cdcl3)δ192.1,138.9,138.7,136.9,134.6,134.1,129.7,129.5,129.0,128.9,128.2,57.1,28.5.mass spectrometry:hrms-esi(m/z):calcd for c
15h15
o3s[m+h]
+
275.0742,found,275.0733.
[0100]
第四步,中间体i-7-g的合成。氩气保护,-78℃搅拌下,将草酰氯(0.179ml,2.05mmol)滴加到dmso(0.150ml,2.11mmol)的dcm(3.0ml,0.70m)溶液中。在该温度下搅拌20min后,将溶液升至-50℃,并将(1-甲基吡咯烷-2-基)甲醇(i-7-f,230mg,2.0mmol)的dcm(1.0ml,2.0m)溶液经反应瓶壁以缓慢的速度加入(以进行充分的预冷却)。-50℃下搅拌30min后,反应物变混浊。冷却至-78℃,将三乙胺(0.307ml,2.20mmol)滴加至反应瓶中。在-60℃下搅拌30min后,形成浑浊的沉淀。然后将正戊烷(5.0ml)加入混合物中搅拌10min。然
后经硅藻土过滤,并用正戊烷多次冲洗,减压下除去挥发物,得到无色油。该物质始终保持在氮气气氛下,无需进一步纯化直接进行下一步(由于其在23℃以上的溶液中会缓慢分解,该化合物必须立即使用)
[0101]
第五步,中间体i-7-i的合成。向甲苯磺酰肼(i-7-h,931.2mg,2.0mmol)的甲醇(8ml)溶液中加入化合物i-7-g,室温搅拌过夜。减压浓缩,得浅黄色固体172.6mg,两步收率31%,熔点109-111℃。1h nmr(400mhz,cdcl3)δ7.78(m,3h),7.27(d,j=8.0hz,2h),6.82(d,j=7.2hz,1h),3.22(t,j=7.2hz,1h),2.84(q,j=8.0hz,1h),2.40(s,3h),2.26(q,j=8.8hz,1h),2.13(s,3h),2.03-1.92(m,1h),1.90-1.72(m,2h),1.69-1.56(m,1h).
13
c nmr(100mhz,cdcl3)δ150.6,143.7,136.0,129.5,127.8,67.4,56.4,40.0,29.3,22.8,21.6.mass spectrometry:hrms-esi(m/z):calcd for c
13h20
n3o2s[m+h]
+
282.1276,found,282.1269.
[0102]
第六步,i-7的合成。将化合物i-7-i(267.1mg,1.0mmol),碳酸铯(488.7mg,1.5mmol)和i-7-e(274.1mg,1.0mmol)置于圆底烧瓶。氩气置换气三次,然后加入1,4-dioxane(2ml),加热至110℃反应18h。将混合物冷却至室温,用饱和氯化铵水溶液淬灭,并用二氯甲烷萃取。合并有机相用无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
30∶1)柱层析得黄色油状物92.3mg,收率25%。1h nmr(400mhz,cdcl3)δ7.92(d,j=7.6hz,2h),7.78(d,j=6.4hz,1h),7.70-7.62(m,2h),7.56(t,j=7.6hz,2h),7.39-7.32(m,2h),3.40-3.26(m,3h),3.09(dd,j=10.0.,6.4hz,3h),2.94(dd,j=16.4,8.8hz,1h),2.81-2.72(m,1h),2.36(s,3h),2.25(dd,j=17.6,9.2hz,1h),2.13(dt,j=13.6,8.4hz,1h),1.84-1.67(m,2h),1.50-1.39(m,1h).
13
c nmr(100mhz,cdcl3)δ199.2,138.9,138.2,137.7,134.0,133.2,129.5,129.3,128.2,127.8,126.9,62.2,57.3,56.9,43.7,40.9,31.6,28.7,22.3.mass spectrometry:hrms-esi(m/z):calcd for c
21h26
no3s[m+h]
+
372.1633,found,372.1625.
[0103]
实施例8:间位双取代化合物i-8的合成。
[0104]
第一步,中间体i-8-b的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六环,后加入化合物3-溴苯甲酸甲酯(i-8-a,430.1mg,2.0mmol),苯基乙烯基砜(i-7-b,336.4mg,2.0mmol),三乙胺(0.56ml,4.0mmol)和pd(pt-bu3)2(102.2mg,0.2mmol,10mol%),氩气置换气三次。混合物于70℃加热反应48h。冷却至室温,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=6∶1)柱层析得黄色油状物413.9mg,收率69%。1h nmr(400mhz,cdcl3)δ8.14(s,1h),8.05(d,j=7.6hz,1h),7.97-7.91(m,2h),7.69(d,j=15.6hz,1h),7.63(dd,j=14.0,7.6hz,2h),7.54(t,j=7.6hz,2h),7.46(t,j=7.6hz,1h),6.96(d,j=15.6hz,1h),3.91(s,3h).
13
c nmr(100mhz,cdcl3)δ166.2,141.2,140.4,133.7,132.9,132.8,132.0,131.2,129.5,129.42,128.8,127.8,52.5.mass spectrometry:hrms-esi(m/z):calcd for c
16h15
o4s[m+h]
+
303.0691,found,303.0686.
[0105]
第二步,中间体i-8-c的合成。向100ml圆底烧瓶中加入化合物i-8-b(604.7mg,2.0mmol),10%钯碳(319.3mg,0.3mmol),后加入60ml甲苯。氢气置换气5次,室温搅拌24h。经硅藻土垫过滤,减压浓缩,得到白色固体605.8mg,产率99%,熔点135-136℃。1h nmr(400mhz,cdcl3)δ7.97-7.92(m,2h),7.91-7.86(m,1h),7.78(s,1h),7.68(t,j=7.6hz,1h),7.59(t,j=7.6hz,2h),7.37-7.33(m,2h),3.90(s,3h),3.42-3.34(m,2h),3.15-3.07
(m,2h).
13
c nmr(100mhz,cdcl3)δ166.9,139.1,137.9,134.0,133.1,130.8,129.6,129.5,129.1,128.4,128.2,57.4,52.4,28.7.mass spectrometry:hrms-esi(m/z):calcd for c
16h17
o4s[m+h]
+
305.0848,found,305.0839.
[0106]
第三步,中间体i-8-d的合成。将化合物i-8-c(604.1mg,2.0mmol)和氢氧化钠(160mg,4.0mmol)溶解在水(3.0ml),四氢呋喃(3.0ml)和甲醇(3.0ml)混合溶液中室温搅拌1h。反应结束后减压下除去挥发物,将混合物用甲基叔丁基醚洗涤两次,然后用1m盐酸将水层调节至ph=4-5,析出大量白色固体,过滤得4-(2-(苯基磺酰基)乙基)苯甲酸(i-8-d)白色粉末548.2mg,产率99%。未纯化直接进行下一步。
[0107]
第四步,i-8的合成。将化合物i-8-d(290.1mg,1.0mmol)和1-甲基哌嗪(i-2-b,0.12ml,1.1mmol)的dmf(5ml)混合物冷却至0℃,后加入hatu(456.3mg,1.1mmol),diea(0.33ml,2.0mmol),在0℃下继续搅拌1h。减压下除去挥发物,混合物用乙酸乙酯稀释,并用饱和柠檬酸水溶液洗涤。分离有机层,依次用饱和碳酸钠水溶液、盐水洗涤,无水硫酸钠干燥。减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
30∶1)柱层析得白色油273.7mg,产率74%。1h nmr(400mhz,cdcl3)δ7.88-7.81(m,2h),7.59(t,j=7.4hz,1h),7.49(t,j=7.6hz,2h),7.22(t,j=7.8hz,1h),7.16-7.08(m,3h),3.68(s,2h),3.37-3.25(m,4h),3.03-2.94(m,2h),2.38(s,2h),2.22(s,5h).
13
c nmr(100mhz,cdcl3)δ169.7,138.7,137.9,136.2,133.8,129.5,129.3,128.7,127.89,126.9,125.3,57.0,55.08,54.5,47.4,45.9,41.9,28.4.mass spectrometry:hrms-esi(m/z):calcd for c
20h25
n2o3s[m+h]
+
373.1586,found,373.1583.
[0108]
实施例9:间位双取代化合物i-9的合成。
[0109]
第一步,中间体i-9-c的合成。将化合物6-溴-2-吡啶羧酸(i-9-b,1.01g,5.0mmol)和4-氟苯胺(i-9-a,0.52ml,5.5mmol)的dmf(9ml)混合物冷却至0℃,后加入hatu(2,09g,5.5mmol),diea(1.65ml,10.0mmol),在0℃下继续搅拌1h。减压下除去挥发物,混合物用乙酸乙酯稀释,并用饱和柠檬酸水溶液洗涤。分离有机层,依次用饱和碳酸钠水溶液、盐水洗涤,无水硫酸钠干燥。减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=10∶1)柱层析得浅棕色固体978.1mg,产率85%,熔点89-91℃。1h nmr(400mhz,cdcl3)δ9.65(s,1h),8.26(d,j=7.6hz,1h),7.81-7.66(m,4h),7.12-7.06(m,2h).
13
c nmr(100mhz,cdcl3)δ160.6,159.7(d,j=242.0hz),150.9,140.6,140.1,133.5(d,j=2.7hz),131.2,121.8(d,j=8.0hz),121.6,115.9(d,j=22.6hz).mass spectrometry:hrms-esi(m/z):calcd for c
12
h9brfn2o[m+h]
+
294.9882,found,294.9879.
[0110]
第二步,i-9的合成。化合物i-9-c(293.0mg,1.0mmol),4-氨基-1-甲基哌啶(i-9-d,0.25ml,2.0mmol),pd2(dba)3(55.0mg,0.06mmol),rac-binap(112.1mg,0.18mmol),叔丁醇钾(480.5mg,5.0mmol)和甲苯(10ml)加入到100ml圆底烧瓶中,氩气置换气三次。加热至70℃反应24h后,将混合物冷却至室温并加入乙酸乙酯,过滤除去固体,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
20∶1)柱层析得淡黄色固体123.7mg,收率38%,熔点124-125℃。1h nmr(400mhz,cdcl3)δ9.85(s,1h),7.72-7.66(m,2h),7.61-7.53(m,2h),7.07(t,j=8.8hz,2h),6.56(dd,j=8.0,1.2hz,1h),4.56(d,j=7.6hz,1h),3.73-3.61(m,1h),2.87(d,j=11.2hz,2h),2.34(s,3h),2.22-2.09(m,4h),1.61(td,j=13.6,3.6hz,2h).
13
c nmr(100mhz,cdcl3)δ162.6,159.3(d,j=251.0hz),156.5,138.9,134.2(d,j=3.0hz),121.2
(d,j=8.0hz),115.9(d,j=22.4hz),111.8,111.1,54.7,48.5,46.5,32.5.mass spectrometry:hrms-esi(m/z):calcd for c
18h22
fn4o[m+h]
+
329.1778,found,329.1772.
[0111]
实施例10:间位双取代化合物i-10的合成。
[0112]
第一步,中间体i-10-b的合成。0℃下,将3-溴-dl-苯丙氨酸(i-10-a,4.88g,20.0mmol)加入硼氢化钠(1.82g,48.0mmol)的thf(60ml)溶液中,得到白色悬浮液。在0℃下搅拌5min后,然后加入碘(5.08g,20.0mmol)的thf(30ml)溶液,缓慢滴加1h,室温继续搅拌5h,然后加热至70℃搅拌18h。将得到的白色悬浮液冷却至室温,缓慢加入甲醇稀释混合物,直至溶液变澄清。减压浓缩,得到白色糊状物。后加入氢氧化钾水溶液(9g,20%m/m,45ml)稀释,于室温搅拌16h,加入二氯甲烷稀释。分离各相,水层用二氯甲烷萃取,合并有机相,无水硫酸钠干燥,在减压下除去所有挥发物得透明的油状物。未纯化直接进行下一步。
[0113]
氩气保护、冰浴下,缓慢将三光气(1.90g,6.4mmol)的无水二氯甲烷(6ml)溶液于45min内滴加到3-溴-dl-苯丙氨醇(3.66g,16.0mmol),三乙胺(4.9ml,35.2mmol)的无水二氯甲烷(45ml)悬浮液中。将混合物在0℃下再搅拌15min,后升至室温搅拌1h。然后加入二氯甲烷及饱和氯化铵水溶液,继续搅拌20min,将混合物转移到分液漏斗中。分离水层,用水洗涤有机层,合并的水层用二氯甲烷萃取,然后将合并的有机相用无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=2∶1)柱层析得白色粉末3.31g,两步收率81%,熔点96-98℃。1h nmr(400mhz,cdcl3)δ7.42(d,j=7.6hz,1h),7.34(s,1h),7.22(t,j=7.6hz,1h),7.11(d,j=7.6hz,1h),5.58(s,1h),4.51-4.42(m,1h),4.16-4.04(m,2h),2.85(d,j=6.8hz,2h).
13
c nmr(100mhz,cdcl3)δ159.6,138.3,132.2,130.6,130.5,127.8,123.1,69.5,53.6,41.1.mass spectrometry:hrms-esi(m/z):calcd for c
10h11
brno2[m+h]
+
255.9973,found,255.9969.
[0114]
第二步,中间体i-10-c的合成。将pd(oac)2(6.8mg,0.03mmol,6mol%),bupad2(22.0mg,0.06mmol,12mol%)和甲酸钠(170mg,2.5mmol)加入50ml圆底烧瓶并用氩气抽换气三次,后将i-10-b(127.5mg,0.5mmol)的dmf(2ml)溶液加入到反应瓶中。然后加入甲酸(37.8ul,1.0mmol)和乙酸酐(94.5ul,1.0mmol),混合物于30℃搅拌1h。加入三乙胺(0.14ml,1.0mmol),加热至110℃下搅拌16h。反应结束后,将混合物用乙酸乙酯稀释并用水洗涤三次。然后将合并的有机层减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=1∶1)柱层析得黄色油42.8mg,收率42%。1h nmr(400mhz,cdcl3)δ9.95(s,1h),7.75(d,j=7.2hz,1h),7.70(s,1h),7.53-7.42(m,2h),6.65(s,1h),4.42(t,j=7.2hz,1h),4.19-4.07(m,2h),3.01-2.88(m,2h).
13
c nmr(100mhz,cdcl3)δ192.3,159.8,137.2,136.8,135.4,129.9,129.7,129.1,69.38(s),53.5,40.9.mass spectrometry:hrms-esi(m/z):calcd for c
11h12
no3[m+h]
+
206.0817,found,206.0811.
[0115]
第三步,中间体i-10-e的合成。向甲苯磺酰肼(i-7-h,1.86g,10mmol)的甲醇(40ml)溶液中加入化合物i-10-d(1.13g,20mmol),室温搅拌过夜。减压浓缩,得棕黄色固体2.80g,当量收率,熔点128-130℃。1h nmr(400mhz,cdcl3)δ7.82(d,j=8.0hz,2h),7.30(d,j=8.0hz,2h),2.47(t,j=5.6hz,2h),2.45-2.40(m,5h),2.39-2.33(m,4h),2.27(s,3h).
13
c nmr(100mhz,cdcl3)δ158.9,144.1,135.4,129.7,128.2,55.7,54.2,45.8,34.5,26.9,21.7.mass spectrometry:hrms-esi(m/z):calcd for c
13h20
n3o2s[m+h]
+
282.1276,found,282.1271.
[0116]
第四步,i-10的合成。将化合物i-10-e(154.6mg,0.5mmol),碳酸铯(244.4mg,0.75mmol)和i-10-c(123.1mg,0.6mmol)置于圆底烧瓶。氩气置换气三次,然后加入1,4-dioxane(3ml),加热至110℃反应18h。将混合物冷却至室温,用饱和氯化铵水溶液淬灭,并用二氯甲烷萃取。合并有机相用无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=80/1
→
30∶1)柱层析得黄色油状物65.7mg,收率41%。1h nmr(400mhz,cdcl3)δ7.83(d,j=7.6hz,1h,arh),7.77(s,1h),7.45(t,j=7.6hz,1h),7.40(d,j=7.6hz,1h),6.24(s,1h),4.51-4.43(m,1h),4.15(dd,j=10.4,4.8hz,2h),3.26-3.16(m,1h),2.94(t,j=9.8hz,4h),2.31(s,3h),2.09(td,j=11.2,3.6hz,2h1.90-1.79(m,4h).
13
c nmr(100mhz,cdcl3)δ202.6,159.5,136.9,136.8,133.7,129.4,128.9,127.3,69.5,55.3,53.7,46.5,43.3,41.3,28.9,28.8.mass spectrometry:hrms-esi(m/z):calcd for c
17h23
n2o3[m+h]
+
303.1709,found,303.1704.
[0117]
实施例11:间位双取代化合物i-11的合成。
[0118]
第一步,中间体i-11-b的合成。向干燥的三颈瓶中加入化合物i-10-b(510.0mg,2.0mmol),pd(oac)2(22.6mg,0.10mmol,5mol%),1,3-双(二苯基膦)丙烷(82.6mg,0.20mmol,10mol%),氩气置换气三次,加入乙二醇(4ml),后加入乙烯基正丁醚(i-11-a,0.78ml,6.0mmol),三乙胺(0.70ml,5.0mmol)。加热至145℃搅拌24h。反应结束后冷却至室温,加入hcl水溶液(5%,10ml),搅拌0.5h后,添加二氯甲烷(5ml)。分离二氯甲烷相后,将水层用二氯甲烷萃取,并将合并的有机层用水洗涤直至中性,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=1∶1)柱层析得浅黄色油438.2mg,收率81%。1h nmr(400mhz,cdcl3)δ7.82(d,j=7.2hz,1h),7.77(s,1h),7.44-7.36(m,2h),6.51(s,1h),4.45-4.38(m,1h),4.17-4.08(m,2h),2.97-2.87(m,2h),2.57(s,3h).
13
c nmr(100mhz,cdcl3)δ198.2,159.8,137.6,136.7,133.9,129.3,128.8,127.4,69.4,53.6,41.1,26.8.mass spectrometry:hrms-esi(m/z):calcd for c
12h13
no3[m+h]
+
220.0974,found,220.0968.
[0119]
第二步,i-11的合成。将多聚甲醛(60mg,2.0mmol),盐酸二甲胺(163.1mg,2.0mmol)和i-11-b(328.6mg,1.5mmol)溶于乙醇(1ml),后加入35%的盐酸(5ul),混合物加热回流5h。反应结束后冷却至室温,用水稀释,再加入用饱和碳酸钾处理,并用乙酸乙酯萃取。有机相用无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
30∶1)柱层析得乳白色油174.0mg,收率42%。1h nmr(400mhz,cdcl3)δ7.83(d,j=7.6hz,1h),7.78(s,1h),7.43-7.34(m,2h),6.66(s,1h),4.02-4.37(m,1h),4.14-4.05(m,2h),3.12(t,j=7.2hz,2h),2.94-2.88(m,2h),2.70(t,j=7.2hz,2h),2.24(s,6h).
13
c nmr(100mhz,cdcl3)δ199.0,159.7,137.5,136.8,133.9,129.3,128.7,127.0,69.4,54.3,53.6,45.5,41.2,37.0.mass spectrometry:hrms-esi(m/z):calcd for c
15h21
n2o3[m+h]
+
277.1552,found,277.1546.
[0120]
实施例12:间位双取代化合物i-12的合成。
[0121]
第一步,中间体i-12-c的合成。在氩气氛下向100ml圆底烧瓶中加入2,4-二溴苯酚(i-12-a,1.26g,5.0mmol),4-氟苄脒盐酸盐(i-12-b,960.3mg,5.5mmol),氢氧化钾(841.7mg,15.0mmol)和碘化亚铜(95.2mg,0.5mmol),然后加入无水dmf(15ml),将混合物于120℃下反应16h。反应结束后,将反应混合物用水稀释,然后用乙酸乙酯萃取,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=100∶1)柱层析得白色固体1.13g,收率78%,
熔点129-131℃。1h nmr(400mhz,cdcl3)δ8.23(dd,j=8.0,5.6hz,2h),7.88(s,1h),7.49-7.41(m,2h),7.21(t,j=8.4hz,2h).
13
c nmr(100mhz,cdcl3)δ165.2(d,j=253.5hz),163.4,149.9,143.8,130.2(d,j=8.9hz),128.3,123.1,117.5,116.5(d,j=22.2hz),111.9.mass spectrometry:hrms-esi(m/z):calcd for c
13
h8brfno[m+h]
+
291.9973,found,291.9970.
[0122]
第二步,i-12的合成。化合物i-12-c(291.0mg,1.0mmol),4-二甲氨基哌啶(i-13-d,0.25ml,2.0mmol),pd2(dba)3(55.0mg,0.06mmol),rac-binap(112.1mg,0.18mmol),叔丁醇钾(480.5mg,5.0mmol)和甲苯(10ml)加入到100ml圆底烧瓶中,氩气置换气三次。加热至70℃反应24h后,将混合物冷却至室温并加入乙酸乙酯,过滤除去固体,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
20∶1)柱层析得淡黄色固体100.6mg,收率31%,熔点154-156℃。1h nmr(400mhz,cdcl3)δ8.20(dd,j=8.4,5.6hz,2h),7.33(d,j=8.8hz,1h),7.18(t,j=8.4hz,2h),6.94(s,1h),6.62(dd,j=8.8,1.6hz,1h),3.60(d,j=8.0hz,1h),3.29(d,j=7.6hz,1h),2.84(d,j=11.0hz,2h),2.31(s,3h),2.11(d,j=10.4hz,4h),1.51(dd,j=21.2,10.4hz,2h).
13
c nmr(100mhz,cdcl3)δ164.7(d,j=252.3hz),162.5,145.1,144.0,143.4,129.6(d,j=8.9hz),123.9(d,j=3.1hz),116.2(d,j=22.2hz),113.3,110.9,102.2,54.850.5,46.4,32.6.mass spectrometry:hrms-esi(m/z):calcd for c
19h21
fn3o[m+h]
+
326.1669,found,326.1663.
[0123]
实施例13:间位双取代化合物i-13的合成。
[0124]
第一步,中间体i-13-c的合成。将4-氟苯甲酰氯(i-13-a,1.30ml,11.0mmol)缓慢滴加入冰浴下的6-溴-2-氨基吡啶(i-13-b,1.73g,10.0mmol)和三乙胺(2.8ml,20mmol)的二氯甲烷(50ml)溶液中。反应升至室温搅拌1h。加水淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=10∶1)柱层析得白色固体2.51g,收率85%,熔点126-128℃。1h nmr(400mhz,cdcl3)δ8.60(s,1h),8.33(d,j=8.0hz,1h),7.94(t,j=8.0hz,2h),7.62(t,j=8.0hz,1h),7.26(d,j=8.0hz,1h),7.18(t,j=8.0hz,2h).
13
c nmr(100mhz,cdcl3)δ165.4(d,j=252.0hz),164.6,151.6,140.9,130.0(d,j=3.1hz),129.8(d,j=9.2hz),123.9,116.2(d,j=22.1hz),112.6.mass spectrometry:hrms-esi(m/z):calcd for c
12
h9brfn2o[m+h]
+
294.9882,found,294.9881.
[0125]
第二步,i-13的合成。化合物i-13-c(439.5mg,1.5mmol),4-二甲氨基哌啶(i-13-d,0.53ml,4.5mmol),pd2(dba)3(27.5mg,0.03mmol),rac-binap(56.1mg,0.09mmol),叔丁醇钾(404.0mg,3.6mmol)和甲苯(8ml)加入到100ml圆底烧瓶中,氩气置换气三次。加热至70℃反应24h后,将混合物冷却至室温并加入乙酸乙酯,过滤除去固体,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
30∶1)柱层析得淡黄色固体127.4mg,收率37%,熔点105-106℃。1h nmr(400mhz,cdcl3)δ8.23(s,1h),7.93(dd,j=8.4,5.4hz,2h),7.65(d,j=8.0hz,1h),7.56(t,j=8.0hz,1h),7.17(t,j=8.4hz,2h),6.45(d,j=8.4hz,1h),4.42(d,j=12.8hz,2h),3.00(t,j=10.2hz,1h),2.83(t,j=12.4hz,2h),2.61(s,6h),2.14(d,j=11.6hz,2h),1.72-1.60(m,2h).
13
c nmr(100mhz,cdcl3)δ164.5,157.5,149.7,140.3,130.9,129.6(d,j=9.0hz),116.0(d,j=21.8hz),103.3,103.1,100.0,63.5,44.3,40.2,26.3.mass spectrometry:hrms-esi(m/z):calcd for c
19h24
fn4o[m+h]
+
343.1934,found,343.1928.
[0126]
实施例14:间位双取代化合物i-14的合成。
[0127]
第一步,中间体i-14-b的合成。将间苯二甲酸单甲酯(i-14-a,900.8mg,5.0mmol)和4-氟苯胺(i-9-a,0.48ml,5.0mmol)的dmf(9ml)混合物冷却至0℃,后加入hatu(1.90g,5.0mmol),diea(1.65ml,10mmol),在0℃下继续搅拌1h。减压下除去挥发物,混合物用乙酸乙酯稀释,并用饱和柠檬酸水溶液洗涤。分离有机层,依次用饱和碳酸钠水溶液、盐水洗涤,无水硫酸钠干燥。减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=10∶1)柱层析得白色粉末1.18g,收率87%。1h nmr(400mhz,cdcl3)δ8.46(t,j=1.2hz,1h),8.19(dt,j=7.6,1.2hz,1h),8.11(dt,j=7.6,1.2hz,1h),8.04(s,1h),7.65-7.59(m,2h),7.56(t,j=7.6hz,1h),7.09-7.03(m,2h),3.94(s,3h).
13
c nmr(100mhz,cdcl3)δ166.4,164.9,159.8(d,j=242.4hz),135.2,133.8,132.9,132.2,130.8,129.3,127.6,122.4(d,j=7.8hz),115.9(d,j=22.4hz),52.6.
[0128]
第二步,中间体i-14-c的合成。将化合物i-14-b(546.2mg,2.0mmol)和氢氧化钠(160mg,4.0mmol)溶解在水(3.0ml),四氢呋喃(3.0ml)和甲醇(3.0ml)混合溶液中室温搅拌1h。反应结束后减压下除去挥发物,将混合物用甲基叔丁基醚洗涤两次,然后用1m盐酸将水层调节至ph=4-5,析出大量白色固体,过滤得4-(2-(苯基磺酰基)乙基)苯甲酸(i-14-c)白色粉末447.1mg,产率86%。1h nmr(400mhz,dmso-d6)δ13.28(s,1h),10.51(s,1h),8.52(s,1h),8.19(d,j=7.6hz,1h),8.14(d,j=7.6hz,1h),7.84-7.77(m,2h),7.67(t,j=7.6hz,1h),7.2-7.17(m,2h).
13
c nmr(100mhz,dmso-d6)δ166.9,164.7,158.4(d,j=240.5hz),135.4(d,j=2.1hz),135.2,132.3,132.0,131.1,128.9,128.5,122.4(d,j=7.9hz),115.2(d,j=22.1hz).
[0129]
第三步,i-14的合成。将i-14-c(259.1mg,1.0mmol)和1-甲基哌嗪(i-2-b,0.12ml,1.1mmol)的dmf(6ml)溶液冷却至0℃,加入edci盐酸盐(287.6mg,1.5mmol),hobt(202.7mg,1.5mmol),diea(0.50ml,3.0mmol)。混合物在0℃下反应2h。减压除去挥发物,用二氯甲烷萃取,有机相用饱和碳酸钠水溶液洗涤,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
30∶1)柱层析得淡黄色固体294.6mg,收率86%,熔点129-131℃。1h nmr(400mhz,cdcl3)δ9.31(s,1h),7.82(dt,j=7.2,1.6hz,1h),7.77(s,1h),7.73-7.67(m,2h),7.39-7.30(m,2h),7.06-6.99(m,2h),3.78(s,2h),3.37(s,2h),2.45(s,2h),2.31(s,5h).
13
c nmr(100mhz,cdcl3)δ169.7,165.3,159.4(d,j=243.6hz),135.4(d,j=19.7hz),134.5(d,j=2.5hz),129.7,129.0,128.9,125.6,122.3(d,j=7.7hz),115.6(d,j=22.4hz),55.2,54.7,47.8,46.0,42.3.mass spectrometry:hrms-esi(m/z):calcd for c
19h21
fn3o2[m+h]
+
342.1618,found,342.1613.
[0130]
实施例15:间位双取代化合物i-15的合成。
[0131]
第一步,中间体i-15-b的合成。将邻苯二甲酸酐(i-15-a,1.48g,10.0mmol)和2-氨基-6-溴吡啶(i-9-b,1.73g,10.0mmol)在乙酸(60ml)中回流4h。冷却至室温,加入水析出大量白色固体,过滤得到白色固体2.47g,收率82%,熔点193-194℃。1h nmr(400mhz,cdcl3)δ7.97(dt,j=7.2,3.6hz,2h),7.84-7.80(m,2h),7.75(t,j=7.8hz,1h),7.57(dd,j=7.8,0.8hz,1h),7.42(dd,j=7.8,0.8hz,1h).
13
c nmr(100mhz,cdcl3)δ166.2,146.0,141.1,140.3,134.9,131.7,128.2,124.2,121.1.mass spectrometry:hrms-esi(m/z):calcd for c
13
h8brn2o2[m+h]
+
302.9769,found,302.9764.
[0132]
第二步,中间体i-15-c的合成。在干燥的100ml圆底烧瓶中加入30ml 1,4-二氧六
环,后加入化合物i-15-b(1.21g,4.0mmol),i-1-c(672.9mg,4.0mmol),三乙胺(1.1ml,8.0mmol)和pd(pt-bu3)2(204.4mg,0.4mmol,10mol%),氩气置换气三次。混合物于70℃加热反应24h。冷却至室温,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=2∶1)柱层析得白色固体975.2mg,收率63%,熔点220-221℃。1h nmr(400mhz,cdcl3)δ8.01-7.89(m,5h),7.81(dd,j=5.2,3.2hz,2h),7.68(d,j=14.8hz,1h),7.61(t,j=7.6hz,1h),7.53(t,j=7.6hz,2h),7.49-7.45(m,2h),7.43(s,1h).
13
c nmr(100mhz,cdcl3)δ166.5,151.3,146.7,140.1,139.6,139.5,134.9,133.8,133.1,131.7,129.5,128.1,125.0,124.2,123.4.mass spectrometry:hrms-esi(m/z):calcd for c
21h15
n2o4s[m+h]
+
391.0753,found,391.0744.
[0133]
第三步,中间体i-15-d的合成。向100ml圆底烧瓶中加入化合物i-15-c(780.1mg,2.0mmol),10%钯碳(425.7mg,0.4mmol),后加入25ml甲醇。用氢气置换气5次,室温下反应24h
[181]
。然后将混合物经硅藻土垫过滤并旋蒸溶剂,得到白色固体776.3mg,当量收率。直接进行下一步操作。
[0134]
将上一步产物(392.1mg,1.0mmol)和水合肼(0.39ml)的乙醇(5ml)溶液加热回流2h。将反应混合物冷却至室温,经硅藻土垫过滤。减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=1∶1)柱层析得白色固体246.4mg,产率94%,熔点94-96℃。1h nmr(400mhz,cdcl3)δ7.91(d,j=7.6hz,2h),7.63(t,j=7.2hz,1h),7.54(t,j=7.2hz,2h),7.28(t,j=7.6hz,1h),6.43(d,j=7.2hz,1h),6.30(d,j=8.0hz,1h),4.67(s,2h),3.58-3.49(m,2h),3.04-2.95(m,2h).
13
c nmr(100mhz,cdcl3)δ158.4,155.3139.1,138.4,133.7,129.3,128.1,112.7,106.9,55.4,30.6.mass spectrometry:hrms-esi(m/z):calcd for c
13h15
n2o2s[m+h]
+
263.0854,found,263.0850.
[0135]
第四步,i-15的合成。在氩气氛下向圆底烧瓶中加入1-甲基吡咯烷-3-甲酸(i-15-e,129.2mg,1.0mmol)和i-15-d(193.4mg,0.7mmol),然后加入二氯甲烷(4ml)和吡啶(0.28ml,3.5mmol),之后缓慢加入三氯氧磷(0.15ml,1.58mmol)以避免产生过多的热量。将混合物室温搅拌30min,然后用水淬灭。分离各层,水溶液用二氯甲烷萃取,合并有机相用饱和碳酸氢钠及饱和氯化铵洗涤,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
30∶1)柱层析得淡黄色油109.7mg,收率42%。1h nmr(400mhz,cdcl3)δ9.24(s,1h),7.96(d,j=8.0hz,1h),7.92-7.87(m,2h),7.62(t,j=7.6hz,1h),7.53(td,j=8.0,2.8hz,4h),6.79(d,j=7.6hz,1h),3.60-3.54(m,2h),3.11-3.05(m,2h),3.00-2.93(m,2h),2.49(dd,j=9.6,6.8hz,1h),2.45(s,3h),2.37-2.31(m,1h),2.30-2.19(m,2h),2.11-2.01(m,1h).
13
c nmr(101mhz,cdcl3)δ174.9,155.3,151.5,139.2,138.9,133.8,129.3,128.17,118.6,112.1,59.5,55.1,45.9,41.5,30.6,29.8,29.3.mass spectrometry:hrms-esi(m/z):calcd for c
19h24
n3o3s[m+h]
+
374.1538,found,374.1533.
[0136]
实施例16:间位双取代化合物i-16的合成。
[0137]
第一步,中间体i-16-b的合成。将对氟苯硼酸(i-16-a,699.6mg,5.0mmol)加入到6-溴吡啶-2-胺(i-13-b,865.1mg,5.0mmol)的甲醇(2.5ml)和甲苯(25ml)的混合溶液中,然后加入碳酸钠水溶液(1059.9mg,10.0mmol,5ml),及四(三苯基膦)钯(0)(173.4mg,0.15mmol),氩气置换气三次,在110℃下加热过夜。将混合物冷却至室温并用水稀释,用乙酸乙酯萃取,将合并的有机层用水洗涤并,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=6∶1)柱层析得白色固体573.9mg,收率61%。1h nmr(400mhz,cdcl3)δ7.98-7.86
(m,2h),7.45(t,j=8.0hz,1h),7.12(t,j=8.8hz,2h),7.01(d,j=7.4hz,1h),6.40(d,j=8.0hz,1h),4.67(s,2h).
13
c nmr(100mhz,cdcl3)δ163.2(d,j=247.5hz),158.4,155.0,138.5,135.9(d,j=3.0hz),128.6(d,j=8.2hz),115.4(d,j=21.4hz),110.5,107.1.mass spectrometry:hrms-esi(m/z):calcd for c
11h10
fn2[m+h]
+
189.0828,found,189.0823.
[0138]
第二步,中间体i-16-d的合成。在氩气氛下向圆底烧瓶中加入4-环己酮羧酸(i-16-c,106.6mg,0.75mmol)和i-16-b(94.0mg,0.5mmol),然后加入二氯甲烷(4ml)和吡啶(0.20ml,2.5mmol),之后缓慢加入三氯氧磷(70ul,0.75mmol)以避免产生过多的热量。将混合物室温搅拌30min,然后用水淬灭。分离各层,水溶液用二氯甲烷萃取,合并有机相用饱和碳酸氢钠及饱和氯化铵洗涤,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=5∶1)柱层析得淡黄色油77.5mg,收率50%。1h nmr(400mhz,cdcl3)δ8.72(s,1h,nh),8.17(d,j=8.0hz,1h),7.94-7.87(m,2h),7.79(t,j=8.0hz,1h),7.44(d,j=7.6hz,1h),7.14(t,j=8.4hz,2h),2.62(t,j=10.2hz,1h),2.54(t,j=3.6hz,1h),2.50(t,j=3.4hz,1h),2.35-2.24(m,2h),2.20-2.12(m,2h),2.10-1.99(m,2h).
13
c nmr(100mhz,cdcl3)δ209.8,173.0,163.7(d,j=249.0hz),155.0,151.3,139.7,134.7(d,j=2.9hz),128.8(d,j=8.3hz),116.6,115.9(d,j=21.7hz),112.5,43.7,39.9,29.1.mass spectrometry:hrms-esi(m/z):calcd for c
18h18
fn2o2[m+h]
+
313.1352found,313.1347.
[0139]
第三步,i-16的合成。氩气保护下,将中间体i-16-d(200.0mg,0.64mmol)和二甲胺(0.97ml,1.93mmol,2m/thf)加入到二氯甲烷中,反应10min后,加入三乙酰氧基硼氢化钠(542.6mg,2.56mmol)反应4h。用饱和碳酸氢钠淬灭反应,分离有机层,水层用二氯甲烷萃取3次合并有机相,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
30∶1)柱层析得白色固体105.3mg,收率48%,熔点83-85℃。1h nmr(400mhz,cdcl3)δ8.37(d,j=7.6hz,1h),8.16(d,j=8.0hz,1h),7.94-7.87(m,2h),7.72(t,j=8.0hz,1h),7.37(d,j=7.6hz,1h),7.10(t,j=8.8hz,2h),2.47-2.37(m,1h),2.22(s,6h),2.16-2.10(m,1h),2.09-1.99(m,2h),1.86-1.76(m,2h),1.64-1.47(m,4h).
13
c nmr(100mhz,cdcl3)δ174.2,163.5(d,j=248.6hz),154.7,151.5,139.3,134.95(d,j=3.1hz),128.65(d,j=8.4hz),116.0,115.68(d,j=21.6hz),112.3,61.34,43.6,42.5,26.7,25.3.mass spectrometry:hrms-esi(m/z):calcd for c
20h25
fn3o[m+h]
+
342.1982found,342.1976.
[0140]
实施例17:间位双取代化合物i-17的合成。
[0141]
第一步,中间体i-17-b的合成。将i-13-c(588.0mg,2.0mmol),1,4-二氧杂-螺[4,5]癸-7-烯-8-硼酸频哪醇酯(i-17-a,745.2mg,2.8mmol)和四(三苯基膦基)钯(0)(115.6mg,0.1mmol)加入1,4-二氧六环(20ml)中,后加入碳酸钠的水溶液(1.06g,5.0mmol,2.5ml),然后氩气置换气三次,加热至100℃反应24h。反应结束冷却至室温并用水稀释,用二氯甲烷萃取三次,将水层酸化至约ph=7,用二氯甲烷再萃取两次,合并的有机层用盐水洗涤,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=7∶1)柱层析得白色固体658.3mg,收率93%,熔点186-188℃。1h nmr(400mhz,cdcl3)δ8.47(s,1h),8.18(d,j=8.0hz,1h),7.97-7.91(m,2h),7.70(t,j=8.0hz,1h),7.18(td,j=11.6,5.2hz,3h),6.59-6.55(m,1h),4.03(s,4h),2.74(td,j=6.4,2.0hz,2h),2.52(s,2h),1.93(t,j=6.4hz,2h).
13
c nmr(100mhz,cdcl3)δ165.2(d,j=252.0hz),164.7,156.6,150.4,138.9,135.7,130.7(d,j=3.1hz),129.8(d,j=9.2hz),125.7,116.1(d,j=22.0hz),115.7,112.1,
nmr(400mhz,dmso-d6)δ13.10(s,1h),8.28-8.13(m,2h),7.78(s,1h),7.54(d,j=7.2hz,1h),7.39(t,j=8.8hz,2h),7.32(dd,j=8.4,1.6hz,1h).
13
c nmr(100mhz,dmso-d6)δ163.3(d,j=248.0hz),151.6,129.0(d,j=8.7hz),126.3(d,j=2.9hz),125.0,116.1(d,j=22.0hz),114.3.mass spectrometry:hrms-esi(m/z):calcd for c
13
h9brfn2[m+h]
+
290.9933found,290.9929.
[0147]
第二步,中间体i-18-d的合成。将i-18-c(580.0mg,2.0mmol),1,4-二氧杂-螺[4,5]癸-7-烯-8-硼酸频哪醇酯(i-17-a,745.2mg,2.8mmol)和四(三苯基膦基)钯(0)(115.6mg,0.1mmol)加入1,4-二氧六环(20ml)中,后加入碳酸钠的水溶液(1.06g,5.0mmol,2.5ml),然后氩气置换气三次,加热至100℃反应24h。反应结束冷却至室温并用水稀释,用二氯甲烷萃取三次,将水层酸化至约ph=7,用二氯甲烷再萃取两次,合并的有机层用盐水洗涤,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=5∶1)柱层析得白色固体609.6mg,收率87%。熔点208-210℃。1h nmr(400mhz,dmso-d6)δ12.89(s,1h),8.21(dd,j=8.8,5.6hz,2h),7.68-7.52(m,2h),7.40(t,j=8.8hz,2h),7.31(d,j=7.2hz,1h),6.01(s,1h),3.93(s,4h),2.63(s,2h),2.39(s,2h),1.84(t,j=6.4hz,2h).
13
c nmr(100mhz,dmso-d6)δ163.0(d,j=246.0hz),150.7,135.7,132.2,132.0,132.0(d,j=2.4hz),131.5(d,j=9.6hz),128.8,128.7,128.6,126.8(d,j=2.8hz),116.0(d,j=21.9hz),107.0,63.6,35.8,31.0,26.7,25.0.mass spectrometry:hrms-esi(m/z):calcd for c
21h20
fn2o2[m+h]
+
351.1509found,351.1504.
[0148]
第三步,中间体i-18-e的合成。向100ml圆底烧瓶中加入化合物i-18-d(560.2g,1.6mmol),10%钯碳(255.4mg,0.24mmol),后加入30ml 1,4-二氧六环。氢气置换气5次,室温搅拌72h。经硅藻土垫过滤,二氯甲烷洗涤,减压浓缩,得到白色固体485.1mg,收率86%。熔点97-99℃。1h nmr(400mhz,cdcl3)δ8.10(dd,j=8.8,5.2hz,2h),7.51(d,j=8.4hz,1h),7.41(s,1h),7.14(dd,j=8.4,1.2hz,1h),6.98(t,j=8.8hz,2h),3.98(s,4h),2.62(dd,j=15.2,7.2hz,1h),1.91-1.82(m,4h),1.82-1.65(m,4h).
13
c nmr(100mhz,cdcl3)δ163.8(d,j=250.7hz),151.7,142.0,129.0(d,j=8.6hz),126.4(d,j=2.9hz),122.6,116.3,116.2(d,j=22.0hz),108.7,64.38,64.35,43.6,35.3,32.1.mass spectrometry:hrms-esi(m/z):calcd for c
21h22
fn2o2[m+h]
+
353.1655found,353.1661.
[0149]
第四步,中间体i-18-f的合成。将i-18-e(528.2mg,1.5mmol)加入1n盐酸(9ml)及丙酮(17ml)的溶液中,室温,反应4h。用饱和碳酸氢钠水溶液中和,减压除去溶剂,残余物用乙酸乙酯和水萃取。将有机层用盐水洗涤,无水硫酸钠干燥,减压浓缩,经(v(石油醚)∶v(乙酸乙酯)=4∶1)柱层析得白色固体444.7mg,收率96%,熔点104-106℃。1h nmr(400mhz,cdcl3)δ8.12(dd,j=8.8,5.2hz,2h),7.53(d,j=8.4hz,1h),7.39(s,1h),7.13(dd,j=8.4,1.2hz,1h),7.04(t,j=8.8hz,2h),3.10(tt,j=12.0,3.2hz,1h),2.61-2.44(m,4h),2.28-2.17(m,2h),1.89(qd,j=12.8,4.8hz,2h).
13
c nmr(101mhz,cdcl3)δ212.1,163.9(d,j=251.1hz),151.8,140.0,129.0(d,j=8.3hz),126.3(d,j=3.1hz),122.5,116.3(d,j=21.9hz),42.9,41.6,34.6.mass spectrometry:hrms-esi(m/z):calcd for c
19h17
fn2o[m+h]
+
309.1403found,309.1397.
[0150]
第五步,i-18的合成。氩气保护下,将中间体i-18-f(215.7mg,0.70mmol)和二甲胺(1.05ml,2.1mmol,2m/thf)加入到二氯甲烷中,反应10min后,加入三乙酰氧基硼氢化钠
(593.4mg,2.8mmol)反应4h。用饱和碳酸氢钠淬灭反应,分离有机层,水层用二氯甲烷萃取3次合并有机相,无水硫酸钠干燥,减压浓缩,经(v(二氯甲烷)∶v(甲醇)=60/1
→
30∶1)柱层析得白色固体108.7mg,收率46%,熔点99-101℃,hplc纯度99.6%(甲醇∶水(含1%三乙胺)(80∶20))。1h nmr(400mhz,cdcl3)δ8.05(dd,j=8.4,5.2hz,2h),7.49(m,2h),7.19-7.14(m,1h),7.00(t,j=8.4hz,2h),2.73(t,j=9.8hz,1h),2.23(s,6h),2.13(s,1h),1.99-1.87(m,4h),1.68-1.58(m,2h),1.58-1.47(m,2h).
13
c nmr(100mhz,cdcl3)δ163.8(d,j=250.5hz),151.4,142.5,128.9(d,j=8.4hz),126.6(d,j=3.0hz),122.8,116.2(d,j=21.9hz),61.2,43.4,43.3,29.0,28.7.mass spectrometry:hrms-esi(m/z):calcd for c
21h25
fn3[m+h]
+
338.2033found,338.2027.
[0151]
实施例19:杀蚊幼虫活性的测定(culex pipiens pallens),测定程序如下:
[0152]
尖音库蚊淡色亚种(culex pipiens pallens),室内饲养的正常群体。选取10头3龄库蚊幼虫,置于配制好的所需浓度100ml烧杯中。将处理放入标准处理室内,72h后检查死亡率。以含有1ml试验溶剂的水溶液为空白对照。每个化合物重复3次。
[0153]
死亡率(%)=(施药死亡虫数/施药总虫数)
×
100
[0154]
校正死亡率(%)=[(施药死亡率-空白死亡率)/(1-空白死亡率)]
×
100
[0155]
表1 间位双取代化合物i-1~i-18的杀蚊幼虫活性测试结果:
[0156][0157]
从表中数据得出,间位双取代化合物i-1~i-18在处理剂量为10mg/l的浓度下都表现出不错的抗tmv活性,其中衍生物i-1、i-5、i-6、i-8、i-11~i-14、i-6、i-17在10mg/l的浓度下对蚊幼虫具有100%的抑制率。
[0158]
实施例12:抗菌活性测试,测定程序如下:
[0159]
离体杀菌测试,菌体生长速率测定法(平皿法):
[0160]
将一定量药剂溶解在适量丙酮内,然后用含有200mg/l乳化剂水溶液稀释至所需浓度,然后各吸取1ml药液注入培养皿内,再分别加入9ml培养基,摇匀后制成50μg/ml的含
药平板,以添加1ml灭菌水的平板做空白对照。用直径4mm的打孔器沿菌丝外缘切取菌盘,移至含药平板上。每处理重复三次。将培养皿放在24
±
1℃恒温培养箱内培养。48小时后调查各处理菌盘扩展直径,求平均值,与空白对照比较计算相对抑菌率。
[0161][0162]
表2 间位双取代化合物i-1~i-18的抗植物病菌活性测试结果:
[0163][0164]
间位双取代化合物i-1~i-18在测试浓度为50mg/l的条件下对14种被测试菌都表现出广谱的抑制活性,对苹果轮纹病原菌抑制活性突出,大部分化合物在50mg/l浓度下抑制率大于60%。化合物i-2、i-12、i-16对油菜菌核病菌有突出活性,50mg/l浓度下抑制率分别为83%、74%、85%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1