一种阿法替尼杂质的制备方法与流程
1.本发明属于化学制药领域,具体涉及一种阿法替尼杂质的制备方法。
背景技术:
2.马来酸阿法替尼(afatinib)是由德国勃林格殷格翰公司研制开发并生产的口服分子靶向药物,马来酸阿法替尼对表皮生长因子受体(egfr)和人表皮生长因子受体2(her2)具有强效、不可逆的双重抑制作用,适用于晚期非小细胞肺癌(nsclc)的一线治疗及her2阳性的晚期乳腺癌患者,也是治疗egfr-tkis耐药的二、三线靶点药物。
3.阿法替尼在生产和存储的过程中,会产生工艺杂质、降解杂质等,影响药物的质量,因此,研究阿法替尼的杂质,并开发简单高效的杂质定向合成路线,以便获得高纯度的杂质对照品,用以进行药物质量检测工作,确保药物品质,极其重要。阿法替尼的主要碱性降解杂质为式j和式11所示化合物,常用于阿法替尼药物产品的质量研究。
[0004][0005]
4-[(3-氯-4-氟苯基)氨基]-6-{[4-羟基-1-氧代-2-丁烯-1-基]氨基-7-((s)-四氢呋喃-3-基氧基)-喹唑啉即式j所示化合物,是马来酸阿法替尼的一种主要碱性降解杂质。目前虽然有相关文献报道该杂质的合成方法,但是经实验验证发现存在最终产物结构与真实情况不相符的问题。如中国专利文献cn106916147a公开了一种阿法替尼杂质标准品的制备方法,上述方法使用阿法替尼api在碱性条件下水解合成该杂质,但其合成方法经验证,最终产物其结构为1-(4-((3-氯-4-氟苯基)氨基)-7-(((s)-四氢呋喃-3-基)氧基)喹唑啉-6-基)-5-羟基吡咯烷-2-酮,即式11所示化合物,该专利文献中提供的核磁图谱在化学位移1.5-3之间存在6个亚甲基氢与式11结构化合物表征一致,而并非合成得到式j所示化合物(式j所示化合物在化学位移1.5-3之间仅存在2个亚甲基氢,而与羟基连接的亚甲基氢会在更低场的位置)。上述结论在期刊《a controlled,efficient and robust process for the synthesis of an epidermal growth factor receptor inhibitor:afatinib dimaleate》[chem rep,2019,1(1):3-12(doi:10.25082/cr.2019.01.001)]以及《马来酸阿法替尼原料药降解杂质的制备与结构推测》(化学世界,2019,60(3),177-181)中也得到了验证。
[0006]
本发明提供了杂质j的全新合成方法,且图谱表征与结构一致,对马来酸阿法替尼的质量控制和杂质研究具有重要意义。
技术实现要素:
[0007]
本发明提供一种式j所示的化合物的制备方法,包括以下步骤:
[0008]
1)化合物a或其盐在化合物a1作用下发生水解反应,得到化合物b;
[0009][0010]
2)化合物c与化合物c1反应得到化合物d;
[0011][0012]
3)所述化合物b与所述化合物d反应制备得到化合物e;
[0013][0014]
4)所述化合物e与化合物e1发生还原反应得到所述式j所示的化合物;
[0015][0016]
根据本发明的实施方案,步骤1)中所述化合物a1可以为酸,所述酸可以为无机酸,例如(浓)盐酸和/或(浓)硫酸,优选(浓)盐酸;
[0017]
根据本发明的实施方案,所述化合物a1的浓度可以为1-12mol/l,例如为6-12mol/l,示例性为2mol/l、3mol/l、4mol/l、5mol/l、6mol/l、7mol/l、8mol/l;
[0018]
根据本发明的实施方案,步骤1)中化合物a的盐可以为化合物a的马来酸盐(即马来酸阿法替尼);
[0019]
根据本发明的实施方案,步骤1)中所述化合物a或其盐与化合物a1的质量体积比(g:ml)可以为1:(0.5-3),例如为1:(0.8-2),示例性为1:2;
[0020]
根据本发明的实施方案,步骤1)所述水解反应的溶剂选自四氢呋喃、2-甲基四氢
呋喃、甲苯、二氯甲烷、氯仿、二氧六环、丙酮、甲醇、乙醇和异丙醇中的至少一种;
[0021]
根据本发明的实施方案,步骤1)中所述化合物a或其盐与步骤1)所述水解反应的溶剂的质量体积比(g:ml)可以为1:(1-10),例如为1:(2-5),示例性为1:2;
[0022]
根据本发明的实施方案,步骤1)中所述水解反应的温度为60-80℃,例如65-75℃,示例性为70℃;
[0023]
根据本发明的实施方案,步骤1)中所述水解反应的时间为3-8小时,例如4-7小时,示例性为5小时。
[0024]
根据本发明的实施方案,步骤1)还包括水解反应完成后,对反应液进行后处理,得到所述化合物b。例如,所述后处理包括:将所述反应液降温(优选降至室温),倒入冰水中,将混合液调节至碱性(优选ph=8-9),搅拌后加入二氯甲烷萃取,合并萃取得到的有机相,所述有机相经洗涤、干燥、纯化后,得到所述化合物b。优选地,所述将混合液调节至碱性可以向混合液中加入氢氧化钠水溶液。
[0025]
根据本发明的实施方案,步骤2)中所述化合物c1可以为草酰氯、氯化亚砜、三氯氧磷和三氯化磷中的至少一种,优选氯化亚砜;
[0026]
根据本发明的实施方案,步骤2)中所述化合物c与化合物c1的摩尔比可以为1:(1-5),例如为1:(2-3),示例性为1:3;
[0027]
根据本发明的实施方案,步骤2)所述反应的溶剂选自四氢呋喃、甲苯、二氯甲烷、氯仿和二氯乙烷中的至少一种;
[0028]
根据本发明的实施方案,步骤2)中所述化合物c与步骤2)所述反应的溶剂的质量体积比(g:ml)可以为1:(5-20),例如为1:(8-12),示例性为1:10。
[0029]
根据本发明的实施方案,步骤2)所述反应还在催化剂存在下进行。例如,所述催化剂选自dmf;
[0030]
根据本发明的实施方案,步骤2)中所述反应的温度为60-80℃,例如65-75℃,示例性为70℃;
[0031]
根据本发明的实施方案,步骤2)中所述反应的时间为3-8小时,例如4-7小时,示例性为5小时;
[0032]
根据本发明的实施方案,步骤2)包括:将所述化合物c加入到溶剂中,在惰性气氛(优选氮气气氛)中将体系降至-5~2℃(优选0℃),向体系中加入(优选为滴加)所述化合物c1,所述化合物c1加入完成后,再向体系中加入(优选为滴加)催化剂,升温搅拌反应。
[0033]
根据本发明的实施方案,步骤2)还包括对反应完成后的反应液进行后处理。例如,所述后处理包括将所述反应液降温(优选降至室温),浓缩至干,得到所述化合物d。
[0034]
根据本发明的实施方案,步骤3)中所述化合物b与化合物d的摩尔比可以为1:(1-5),例如为1:(2-3),示例性为1:3;
[0035]
根据本发明的实施方案,步骤3)所述反应的溶剂选自四氢呋喃、2-甲基四氢呋喃、甲苯、二氯甲烷和氯仿的至少一种;
[0036]
优选地,步骤3)包括:将所述化合物b与步骤3)所述反应的溶剂混合,得到所述化合物b的溶液;在将化合物d与步骤3)所述反应的溶剂混合,得到所述化合物d的溶液;而后将所述化合物d的溶液滴加入所述化合物b的溶液中,搅拌反应;
[0037]
优选地,所述化合物b的溶液与所述化合物d的溶液的体积比为1:(0.8-1.5),优选
为1:1;
[0038]
优选地,将所述化合物b的溶液降温至-30℃~-10℃(优选降温至-20℃),向其中滴加所述化合物d的溶液;
[0039]
优选地,待所述化合物d的溶液滴加完成后,搅拌升至室温,反应1-4小时;
[0040]
根据本发明的实施方案,步骤3)中所述化合物b与步骤3)反应体系中总溶剂的质量体积比(g:ml)可以为1:(5-20),例如为1:(8-12),示例性为1:10;
[0041]
根据本发明的实施方案,步骤3)所述反应在无氧氛围中进行,例如氮气氛围中进行;
[0042]
根据本发明的实施方案,步骤3)还包括反应完成后,经后处理得到所述化合物e;例如,所述后处理包括向反应液中加水淬灭,搅拌、过滤、纯化,得到所述化合物e。
[0043]
根据本发明的实施方案,步骤4)中所述化合物e1可以选自硼氢化钠、醋酸硼氢化钠、氰基硼氢化钠、硼氢化钾和四氢铝锂中的至少一种,优选硼氢化钠;
[0044]
根据本发明的实施方案,步骤4)中所述化合物e和化合物e1的摩尔比可以为1:(0.5-5),例如为1:(0.8-3),示例性为1:1.8;
[0045]
根据本发明的实施方案,步骤4)所述反应的溶剂为二氯甲烷、三氯甲烷、四氢呋喃、2-甲基四氢呋喃、二氧六环和乙腈中的至少一种;
[0046]
根据本发明的实施方案,步骤4)所述化合物e与步骤4)所述反应的总溶剂的质量体积比(g:ml)可以为1:(2-15),例如为1:(3-10),示例性为1:10、1:6;
[0047]
根据本发明的实施方案,步骤4)所述反应在酯化试剂存在下进行,所述酯化试剂与羧酸反应得到活化酯,所述酯化试剂可以选自氯甲酸甲酯、氯甲酸乙酯、氯甲酸异丙酯、氯甲酸苄酯、氯甲酸苯酯、溴甲酸甲酯、溴甲酸乙酯、溴甲酸异丙酯、溴甲酸苄酯、溴甲酸苯酯中的至少一种;优选地,所述化合物e与所述酯化试剂的摩尔比可以为1:(0.5-5),例如1:(0.8-3),示例性为1:1.2;
[0048]
根据本发明的实施方案,步骤4)所述反应的过程包括:将所述化合物e加入到步骤4)所述反应的溶剂中,降温至-5~2℃(优选0℃),向体系中加入氯甲酸乙酯,而后向体系中加入(优选为滴加)所述化合物e1的溶液,待所述化合物e1的溶液加完后,搅拌条件下于室温反应。优选地,所述化合物e1的溶液为所述化合物e1的thf和水的混合溶液。
[0049]
根据本发明的实施方案,步骤4)还包括反应完成后,对反应液进行后处理,得到所述化合物j。例如,所述后处理包括将反应液加入到饱和氯化铵溶液中,搅拌后萃取(优选加入ea萃取),收集有机相,所述有机相经洗涤、干燥、纯化,得到所述化合物j。
[0050]
作为一个实例,采用如下方法制备所述式j所示的化合物,该制备方法包括如下步骤:
[0051][0052]
1)化合物a或其盐与浓盐酸作用下发生酰胺水解反应得到化合物b;
[0053]
2)化合物c在氯化亚砜和催化剂dmf作用下反应得到化合物d;
[0054]
3)所述化合物b与化合物d作用得到化合物e;
[0055]
4)所述化合物e通过硼氢化钠还原得到所述式j所示的化合物。
[0056]
本发明还提供所述制备方法在药物工艺研究中的应用,其可用于阿法替尼杂质研究。
[0057]
本发明中所述的“室温”指温度为15-36℃,例如为25℃。
[0058]
有益效果
[0059]
本发明为阿法替尼或其盐的碱降解杂质j的合成提供了新思路,本发明提供的合成方法反应步数少、工艺简单、起始物料易得、目标产物收率高。本发明操作真实、数据可靠,真正能够制备得到杂质j,且步骤更加简短,所需物料易于保存,无危害,产品易纯化,且收率和纯度较高能够满足对该杂质的制备需求,对阿法替尼或其盐杂质的研究具有重要意义。
附图说明
[0060]
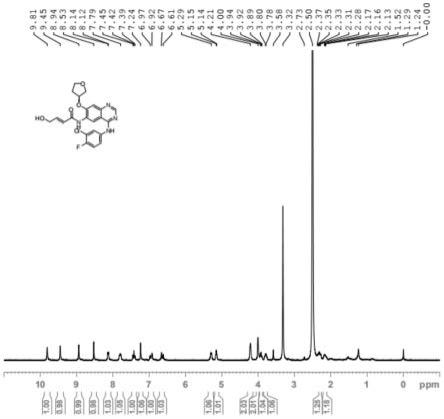
图1为实施例4中杂质j纯品的1h nmr谱图,溶剂为氘代dmso;
[0061]
图2为实施例4中杂质j纯品的hplc液相谱图;
[0062]
图3为实施例4中杂质j纯品的高分辨质谱谱图。
具体实施方式
[0063]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0064]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0065]
实施例1化合物b的合成
[0066][0067]
具体步骤如下:首先,将10g(0.014mol)马来酸阿法替尼即化合物a的马来酸盐分批加入于20ml乙醇和20ml的浓盐酸的混合溶剂中,升温至70℃,搅拌5小时,tlc薄层色谱板监测(加氨水调至碱性,加甲醇稀释,展开剂:dcm:meoh=10:1),待原料基本反应完毕,将反应液降至室温,倒入冰水中,加入2n氢氧化钠水溶液调至ph=8-9,搅拌10min,加入二氯甲烷萃取三次,合并有机相,盐水洗涤,干燥旋干,干法硅胶柱层析纯化后得到化合物b,黄色固体4.6g,收率87.8%。
[0068]
实施例2化合物d的合成
[0069][0070]
具体步骤如下:首先,将4.27g(0.037mol)化合物c加入到40ml甲苯中,氮气置换,降温至0℃,滴加13.14g(0.11mol)氯化亚砜,滴毕,加入两滴dmf,升温至70℃,搅拌5小时,tlc薄层色谱板监测(加甲醇稀释,展开剂:pe:ea=1:1)反应,当化合物c原料完全消失后将反应液降至室温,直接浓缩至干,得到化合物d,黄色油状物5.43g,收率96%。
[0071]
实施例3化合物e的合成
[0072][0073]
具体步骤如下:将5.43g(0.036mol)化合物d加入到23ml thf中,氮气置换降温至-20℃,向反应体系中缓慢滴入4.5g(0.012mol)化合物b的23ml thf溶液,滴毕后缓慢升至室温下搅拌2小时,tlc薄层色谱板监测(加水和ea萃取,取ea相检测,展开剂:dcm:meoh=10:1),化合物b反应完毕。然后向反应液中加入5ml水淬灭,搅拌10min,过滤得到黄色固体,干法硅胶柱层析纯化后得到化合物e,黄色固体5.02g,收率73%。
[0074]
实施例4化合物j的合成
[0075][0076]
具体步骤如下,将5g化合物e加入到30ml thf中降温至0℃,然后滴加氯甲酸乙酯(1.3g),10分钟后将670mg硼氢化钠溶于15ml thf和15ml水的混合溶剂中并缓慢滴入上述溶液中,滴毕后缓慢升至室温下搅拌2小时,tlc薄层色谱板监测(加水和ea萃取,取ea相检测,展开剂:dcm:meoh=10:1),显示原料反应完毕。将反应液加入饱和氯化铵溶液中,搅拌30min,加入ea萃取,有机相盐水洗涤,干燥旋干,干法硅胶柱层析,ea:meoh=30:1冲出产品,富集浓缩得化合物j,黄色固体2.3g,hplc:99.09%,收率47%。
[0077]
如图1所示的1h nmr(500mhz,dmso-d6)δ9.81(s,1h),9.45(s,1h),8.94(s,1h),8.53(s,1h),8.12(dd,j=6.8,2.6hz,1h),7.79(m,1h),7.42(t,j=9.1hz,1h),7.24(s,1h),6.95(dt,j=15.3,3.7hz,1h),6.64(d,j=15.3hz,1h),5.29(m,1h),5.16(m,1h),4.28
–
4.15(m,2h),4.01(d,j=3.3hz,2h),3.93(q,j=7.7hz,1h),3.79(m,1h),2.36
–
2.28(m,1h),2.15(m,1h)。
[0078]
由图1、图2和图3的表征,可以证明化合物j合成成功。
[0079]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1