喹唑啉类抑制剂的制备及其应用的制作方法
1.本发明属于药物合成领域,具体涉及一种新型kras
g12c
抑制剂及其制备方法与用途。
背景技术:
2.本发明通常涉及新的化合物及其制备方法以及作为kras
g12c
抑制剂(例如用于治疗癌症)的用途。
3.ras代表一组紧密相关的189个氨基酸(分子量21kda)的单体球状蛋白,其与质膜相关,并结合gdp或gtporas作为分子开关。当ras含有结合的gdp时,它处于静止或关闭状态,并且处于“非活动状态”。响应细胞暴露于某些促进生长刺激时,ras被诱导将其结合的gdp转换成gtp。与gtp结合后,ras被“打开”,并且能够与其它蛋白(其“下游目标”)相互作用和激活其它蛋白。ras蛋白本身具有非常低的内在能力,无法将gtp水解回gdp,从而使其自身处于关闭状态。关闭ras需要称为gtpase激活蛋白(gaps)的外在蛋白,该蛋白与ras相互作用并大大加快gtp向gdp的转化。ras中影响其与gap相互作用或将gtp转换回gdp的能力的任何突变都将导致蛋白质的活化时间延长,从而导致细胞信号延长,使其继续生长和分裂。因为这些信号导致细胞生长和分裂,所以过度活跃的ras信号可能最终导致癌症。
4.在结构上,ras蛋白包含一个g结构域,该结构域负责ras的酶促活性-鸟喋吟核背酸结合和水解(gtpase反应)。它还包含一个称为caax盒的c末端延伸,可进行翻译后修饰,并负责将蛋白质靶向膜。g结构域的大小约为21-25kda,它包含一个磷酸盐结合环(p-环)。p-环为核昔酸在蛋白质中结合的口袋,这是具有保守氨基酸残基((甘氨酸12、苏氨酸26和赖氨酸16))的结构域的刚性部分,对于核昔酸结合和水解至关重要。g域还包含所谓的switch i(残基30-40)和switch ii(残基60-76)区域,这两个区域都是蛋白质的动态部分,由于它们在能够在静止和负载状态间转换,其通常被称为“弹簧负载'机制。关键相互作用是苏氨酸35和甘氨酸60形成的氢键,具有gtp的y-磷酸酯,其分别使switch1和switch2区域保持其活性构象。gtp水解并释放出磷酸盐后,这两个松弛为非活性的gdp构象。
5.ras亚家族最著名的成员是hras,kras和nras,主要是因为它们与多种类型的癌症有关。ras的三个主要同工型(hras,nras或kras)基因中的任何一种突变都是人类肿瘤发生中最常见。发现人类肿瘤中约有30%携带ras基因突变o值得注意的是,kras突变在25-30%的肿瘤中检测到。相比之下,在nras和hras家族成员中发生的致癌突变率要低得多(分别为8%和3%)。在p环的残基g12和g13以及残基q61处发现了最常见的kras突变。g12c是kras基因的频繁突变(甘氨酸12突变为半胱氨酸)。已经在大约13%的癌症发生,大约43%的肺癌发生以及大约100%的myh相关性息肉病(家族性结肠癌综合征)中发现了这种突变。
6.作为前沿靶点,kras
g12c
突变蛋白受到了广泛关注。araxes(wellspring的子公司)分别在2013年和2016年开发了ars-853和arst620化合物。近年来,它还为kras
g12c
抑制剂申请了多项专利,例如w02016164675和w02016168540,mrs-853化合物显示出良好的细胞活力,但它们的药代动力学性能很差,这不适合用于评估动物模型在体内的药效学。ars-1620
对kras
g12c
具有高效率和选择性,可在体内实现快速,持续的靶标作用,从而诱导肿瘤消退。这项研究提供的体内证据表明ars-1620代表了新一代kras
g12c
特异性抑制剂,具有巨大的治疗潜力。wellspring宣布fda已批准ars-3248的ind应用。其它候选kras
g12c
抑制剂包括mirati公司的mrtx-849和boehringer ingelheim的bi-2852等。因此,尽管已在这个领域中取得进展,但在本领域中仍需要改进的治疗癌症的化合物和方法,例如通过抑制kras、hras或nras来治疗癌症。本发明满足此需要并提供其他相关优势。
7.简而言之,本发明提供了能够调节g12c突变kras、hras和/或nras蛋白的化合物,包括其立体异构体、药物可接受的盐、互变异构体和前药。在一些情况中,所述化合物充当能够与kras、hras或nras g12c突变蛋白的12位置处的半胱氨酸残基形成共价键的亲电子剂。还提供了使用此类化合物治疗诸如癌症的多种疾病或病况的方法。
技术实现要素:
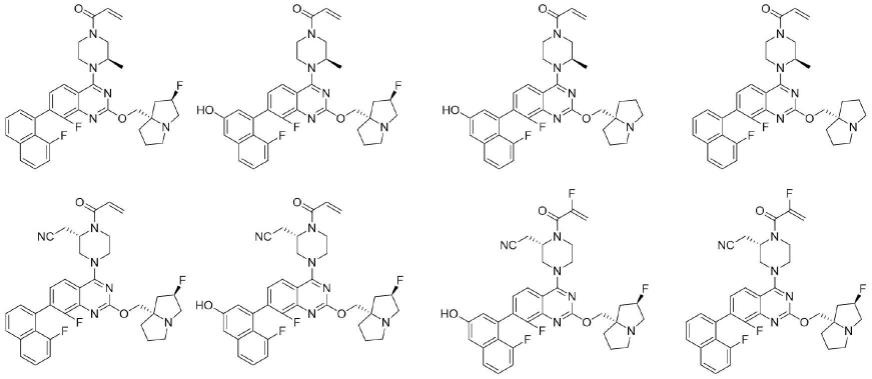
8.一种具有通式(i)所示的化合物、其立体异构体、可药用的盐、多晶型物或异构体,其中通式(i)所示的化合物结构如下:
[0009][0010]
其中,
[0011]
每个x1在每次出现时独立地选自n,cr5;
[0012]
每个r1和r4在每次出现时独立地选自氘、卤素、氧代、-c
1-6
烷基、-c
2-6
烯基、-c
2-6
炔基、-c
1-6
亚烷基-(卤素)
1-3
、c
1-6
杂烷基、-cn、-or6、-c
1-6
亚烷基-(or6)
1-3
、-o-c
1-6
亚烷基-(卤素)
1-3
、-sr6、-s-c
1-6
亚烷基-(卤素)
1-3
、-nr6r7、-c1-6亚烷基-nr6r7、-c(=o)r6、-c(=o)or6、-oc(=o)r6、-c(=o)nr6r7、-nr6c(=o)r7、-s(o)2nr6r7或-c
3-6
碳环基;每个r
12
独立地可选地被1、2、3、4、5或6个选自氘、卤素、-c
1-6
烷基、-c
1-6
烷氧基、氧代、-or6、-nr6r7、-cn、-c(=o)r6、-c(=o)or6、-oc(=o)r6、-c(=o)nr6r7、-nr6c(=o)r7或-s(o)2nr6r7的取代基取代或不取代;
[0013]
r2,r3和r5独立地选自h、d、氰基、卤素、c
1-6
烷基、cooh、nhcoh、conh2、oh或-nh2;
[0014]
u独立地选自-c
0-4
烷基-、-cr8r
9-、-c
1-2
烷基(r8)(oh)-、-c(o)-、-cr8r9o-、-ocr8r
9-、-scr8r
9-、-cr8r9s-、-nr
8-、-nr8c(o)-、-c(o)nr
8-、-nr8c(o)nr
9-、-cf
2-、-o-、-s-、-s(o)
m-、-nr8s(o)
m-、-s(o)mnr
8-;
[0015]
y不存在或选c
3-8
环烷基、3-8元杂环烷基、5-12元稠烷基、5-12元稠杂环基、5-12元螺环基、5-12元螺杂环基、芳香基或者杂芳香基,其中所述环烷基、杂环烷基、螺环基、稠环基、稠杂环基、螺杂环基、芳香基或者杂芳香基任选被一个或多个g1所取代;
[0016]
z独立地选自氰基、-nr
10
cn、
[0017]
键c为双键或者三键;
[0018]
当c为双键时,ra、rb和rc各自独立地选自h、氘、氰基,卤素、c
1-6
烷基、c
3-6
环烷基或3-6元杂环基。其中所述烷基,环烷基和杂环基任选被1个或多个g2所取代;
[0019]
ra和rb或rb和rc任选与它们连接的碳原子共同形成一任选含有杂原子的3-6元环;
[0020]
当键c为三键时,ra和rc不存在,rb独立选自h、氘、氰基,卤素、c
1-6
烷基、c
3-6
环烷基或3-6元杂环基被一个或多个g3所取代;
[0021]r10
独立地选自h、氘、c
1-6
烷基、c
3-6
环烷基或3-6元杂环基,其中所述烷基,环烷基和杂环基任选被1个或多个g4所取代;
[0022]
g1、g2、g3和g4各自独立选自氘、氰基,卤素、c
1-6
烷基、c
2-6
烯基、c
2-6
炔基、c
3-8
环烷基或3-8元杂环基、c
6-10
芳基、5-10元杂芳香基、-or
11
、-oc(o)nr
11r12
、-c(o)or
11
、-c(o)nr
11r12
、-c(o)r
11
、-nr
11r12
、-nr
11
c(o)r
12
、-nr
11
c(o)nr
12r13
、-s(o)
mr11
或-nr
11
s(o)
mr12
,其中所述烷基、烯基、炔基、环烷基、杂环烷基、芳香基、杂芳香基任选被1个或多个氘、氰基,卤素、c
1-6
烷基、c
2-6
烯基、c
2-6
炔基、c
3-8
环烷基或3-8元杂环基、c
6-10
芳基、5-10元杂芳香基、-or
14
、-oc(o)nr
14r15
、-c(o)or
14
、-c(o)nr
14r15
、-c(o)r
14
、-nr
14r15
、-nr
14
c(o)r
15
、-nr
14
c(o)nr
15r16
、-s(o)
mr14
或-nr
14
s(o)
nr15
的取代基所取代;r8、r9、r
11
、r
12
、r
13
、r
14
和r
15
各自独立选自氢、氘、氰基、卤素、c
1-6
烷基、c
3-8
环烷基或3-8元单环杂环基、单环杂芳香基或者苯基;
[0023]
且m为1或2。
[0024]
在一些实施方式中,式(i)所述的化合物或者其异构体、溶剂合物或其前体,或它们的药学上可接受的盐选自以下化合物、其异构体、溶剂合物或其前体,或它们的药学上可接受的盐:
[0025][0026]
另一方面,本发明还提供药物组合物,其包含式(i)所示化合物或其药学可接受的盐和药学上可接受的辅料。
[0027]
另一方面,本发明涉及治疗哺乳动物中与kras g12c查关疾病的方法,包括对需要
该治疗的哺乳动物,优选人类,给予治疗有效量的式(i)所示化合物或其药学可接受的盐、或其药物组合物。
[0028]
另一方面,本发明涉及式(i)所示化合物或其药学可接受的盐预防或治疗kras g12c相关疾病的药物中的用途。
[0029]
另一方面,本发明涉及预防或治疗kras g12c相关疾病的式(i)所示化合物或其药学可接受的盐、或其药物组合物。
[0030]
具体实施方法
[0031]
本发明还提供制备所述化合物的方法。本发明通式(i)所述化合物的制备,可通过以下示例性方法和实施例完成,但这些方法和实施例不应以任何方式被认为是对本发明范围的限制。也可地本领域技术人员所知的合成技术合成本发明所述的化合物,或者综合使用本领域已知方法和本发明所述的方法。每步应所得的产物用本领域已知的分离技术得到,包括但不限于萃取、过滤、蒸馏、结晶、色谱分离等。合成所需要的起始原料和化学试剂可以根据文献(reaxys)常规合成或购买。
[0032]
除非另有说明,温度是摄氏温度。试剂购自chemblocks inc、astatech inc或麦克林等商业供应商,并且这些试剂可直接使用无需进一步纯化,除非另有说明。
[0033]
除非另有说明,下列反应在室温、无水溶剂中、氮气或气的正压下或使用干燥管进行;玻璃器皿烘干和/或加热干燥。
[0034]
除非另有说明,柱色谱纯化使用青岛海洋化工厂的200-300目硅胶;制备薄层色谱使用烟台市化学工业研究所生产的薄层色谱硅胶预制板(hsgf254);ms的测定用therno lcd fleet型(esi)液相色谱-质谱联用仪。
[0035]
核磁数据(1h nmr)使用brukeravance-400mhz或varian oxford-400hz核磁仪,核磁数据使用的溶剂有cdcl3、cd3od、d2o、dms-d6等,以四甲基硅烷(0.000ppm)为基准或以残留溶剂为基准(cdcl3:7.26ppm;cd3od:3.31ppm;d2o:4.79ppm;d6-dmso:2.50ppm)当标明峰形多样性时,以下简写表示不同峰形:s(单峰)、d(双重峰)、t(三重峰)、q(四重峰)、m(多重峰)、br(宽峰)、dd(双双重峰)、dt(双三重峰)。如果给出了耦合常数,则以hertz(hz)为单位。
[0036]
实施例1
[0037]
7-(8-氟萘基)-8-氟-4-(((r)-4-丙烯酰基-2-甲基哌嗪)-1-基)-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉
[0038]
[0039][0040]
第一步:7-溴-8-氟-2,4-喹唑啉二酮的制备
[0041][0042]
将3-氟-4-溴-2-氨基苯甲酸(11.7g,0.05mol)和尿素(45g,0.75mol)加热到150℃,搅拌反应12小时,然后降温至95℃,然后加入200ml水,搅拌半小时过滤,用乙酸打浆,然后干燥得到黄色固体7-溴-8-氟-2,4-喹唑啉二酮(11.88g,87%)。
[0043]
lc/ms(esi):m/z=274[m+h]
+
.
[0044]
第二步:7-溴-8-氟-2,4-二氯喹唑啉的制备
[0045][0046]
将7-溴-8-氟-2,4-喹唑啉二酮(10.92g 40mmol)溶于pocl3(100ml)中,加入少量n,n-二甲苯胺,加热回流搅拌反应10h。然后倒入冰水中淬灭,过滤得到固体产品,水洗,干燥得到粗品黄色固体7-溴-8-氟-2,4-二氯喹唑啉1f(9.94g,84%),无需再纯化进行下一反应。
[0047]
lc/ms(esi):m/z=297[m+h]
+
.
[0048]
第三步:2-氯-7-溴-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))喹唑啉的制备
[0049][0050]
将7-溴-8-氟-2,4-二氯喹唑啉(1.18g,4mmol)、(r)-4-boc-2-甲基哌嗪(0.88g,
4.4mmol)、碳酸钾(0.88g,6.4mmol)催化量碘化钾和dmf(80ml)混合,加热到120℃,搅拌反应4小时。冷却至室温,减压蒸溶,得到黄色固体2-氯-7-溴-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))喹唑啉1g(1.51g,82%),
[0051]
lc/ms(esi):m/z=460[m+h]
+
。
[0052]
第四步:6-氯-7-溴-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉的制备
[0053][0054]
将2-氯-7-溴-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))喹唑啉(275mg,0.6mmol)、(2r,8s)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲醇(106mg,0.66mmol)、碳酸钾(124mg,0.90mmol)催化量碘化钾和dmf(20ml)混合,加热到120℃,搅拌反应4小时。冷却至室温,减压蒸溶,柱层析得到黄色固体7-溴-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉1h(310mg,89%)。
[0055]
lc/ms(esi):m/z=583.2[m+h]
+
。
[0056]
第五步:7-(8-氟萘基)-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉的制备
[0057][0058]
将7-溴-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉1e(175mg,0.3mmol)、8-氟萘-1-硼酸(57mg,0.3mmol)、三(二亚苄基丙酮)二钯(0.026g,0.027mmol)、碳酸铯、1,4-二氧六环(6ml)和水(1.5ml)混合后,然后回流加热到120℃,搅拌反应16小时。将反应物冷却至室温并搅拌过夜,得到淡黄色沉淀物。用水(2ml)稀释反应混合物,并通过过滤收集固体。干燥得到黄色固体7-(8-氟萘基)-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉1f(145mg,75%),无需再纯化进行下一反应。
[0059]
lc/ms(esi):m/z=648.3[m+h]
+
.
[0060]
第六步:7-(8-氟萘基)-8-氟-4-(((r)-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉的制备
[0061][0062]
将7-(8-氟萘基)-8-氟-4-(((r)-4-boc-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉(129mg,0.2mmol)溶于1ml乙酸乙酯和1n hcl的1,4-二氧六环溶液2ml。室温下搅拌2小时,反应液用1n氢氧化钠溶液中和,乙酸乙酯萃取。所得有机相再用饱和碳酸氢钠和饱和食盐水洗涤,无水硫酸钠干燥,有机相减压蒸干。得到化合物7-(8-氟萘基)-8-氟-4-(((r)-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉(94mg,产率86%),直接用于下一步。
[0063]
lc/ms(esi):m/z=548.3[m+h]
+
。
[0064]
第七步:7-(8-氟萘基)-4-(((r)-4-丙烯酰基-2-甲基哌嗪)-1-基))-8-氟-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉的制备
[0065][0066]
于反应瓶中加入7-(8-氟萘基)-8-氟-4-(((r)-2-甲基哌嗪)-1-基))-2-(((2r,7as)-2-氟四氢-1h-吡呤环-7a(5h)-基)甲氧基)喹唑啉1g(82mg,0.15mmol),三乙胺(20.4mg,0.2mmol),4ml四氢呋喃,冰水浴冷却后缓慢滴加2-丙烯酰氯(18mg,0.2mmol)的0.5ml四氢呋喃溶液。加完后继续搅拌4小时。反应液用甲醇淬灭反应并减压蒸干。残余物通过柱层析纯化,得到化合物1(43mg,产率48%)为黄色固体。
[0067]
lc/ms(esi):m/z=602.3[m+h]
+
.
[0068]
实施例2-12参照化合物1的制备方法与相应的中间体制备
[0069]
[0070]
[0071][0072]
实施例13生物活性测试
[0073]
以下结合测试试例进一步描述解释本发明,但这些实施并非意味着限制本发明的范围。
[0074]
一、肿瘤细胞增殖抑制实验
[0075]
1、实验方法
[0076]
将h358(kras g12c突变)细胞消化离心重悬后用scepter自动细胞计数仪测定细胞密度,将细胞稀释成每毫升含44,000个细胞的溶液,调整密度后的细胞溶液以每孔90微升加入96孔培养板中。将96孔板置于37℃、5%co2培养箱中,待细胞培养24小时后,加入不同浓度的待测化合物细胞在10%胎牛血清存在下与化合物一起培养72小时,使用cell titer-glo发光细胞活力检测试剂盒详见厂家说明书)测定atp的含量来评估细胞生长抑制,简要来讲,每个孔中加入30微升cell titer-glo试剂,摇板10分钟,诱导细胞裂解,用
fluoroskanascentfl(thermo)检测记录萤光信号,从二甲基亚砜处理72小时的细胞得到最大的信号值。从单独的培养基(细胞数为零)得到最小信号值,抑制率%=(最大信号值化合物信号值)/(最大信号值一最小信号值
×
100%,使用graphpadprism5软件处理数据。通过s形剂量反应曲线拟合计算ic
50
值。其中“a”表示ic
50
≤10nm;“b”表示10《ic
50
≤100nm;“c”表示100《ic
50
≤1000nm;“d”表示1000nm《ic
50
[0077]
2、实验结果
[0078]
计算出上述实验中各化合物的1c
50
,结果如下表1所示
[0079]
表1、化合物对肿瘤细胞增殖的抑制活性ic
50
(nm)。
[0080]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1