一类高活性的过渡金属钴配合物、其制备方法及应用
1.本发明涉及聚烯烃催化剂技术领域,具体涉及一类制备高度线性聚乙烯蜡的高活性的过渡金属钴配合物、其制备方法及应用。
背景技术:
2.聚乙烯蜡是一种低分子量的聚乙烯合成蜡,一般指相对分子量小于10000的均聚聚乙烯,因其强度和韧性较差而不能作为单一材料进行加工,加之其与聚乙烯、聚氯乙烯、聚丙烯等树脂有良好的相容性,通常作为化工生产中的一种重要的加工助剂被广泛应用于pvc管材、薄膜、线缆和其他塑料橡胶产品中,在提高产品加工性能的同时改善成品外观。除此之外,聚乙烯蜡还具有熔点较高、硬度大、光泽度高、颜色雪白等特点。
3.目前国内生产聚乙烯蜡的主要方法是裂解法,即将高分子量的纯净聚乙烯或废旧聚乙烯塑料高温裂解为聚乙烯蜡,但是裂解过程难以控制,所得产物分子量分布较宽,多有黑点产生,质量难以控制,在低端应用中比较流行。为满足聚乙烯蜡的高端应用需求,设计和开发烯烃聚合催化剂,以乙烯为原料直接合成聚乙烯蜡是解决这一问题的关键。
4.可通过合成法制备聚乙烯蜡的催化剂主要有齐格勒-纳塔(z-n)催化剂、茂金属催化剂和后过渡金属催化剂。以镍、钯为代表的后过渡金属催化剂(如式1,a),通过调节催化剂结构和聚合条件,以精密调控所得聚合物分子量和微观结构;在此基础上,1998年,brookhart与gibbson分别报道了一类2,6-二烯胺基吡啶的铁、钴配合物(如式1,b),可以高活性的聚合乙烯,获得高度线性聚烯烃。自此以后,越来越多的研究集中于后过渡金属催化剂的制备和改性,如式1中的c、d类。
[0005][0006]
发明人课题组一直致力于烯烃聚合催化剂的设计开发和催化工艺的探究,围绕着后过渡金属类催化剂与n^n、n^n^n、n^o等配体做了大量工作。研究中发现,通过调控配体的立体和电子效应能够控制其乙烯催化的性能,在保证较高催化活性的同时进一步加强热稳
定性,获得了国际同行的关注与认可。在经典的吡啶二亚胺类n^n^n铁、钴配合物的基础上,通过使吡啶一侧或两侧成环来固定c=n双键,以提高催化体系的催化活性和热稳定性,以mao为助催化剂,最佳反应温度60℃下催化活性可维持在106g(pe)(mol co)-1
h-1
水平上(式1,c,appl.catal.part a gen 2012,447,67
–
73;式1,d,dalton trans.2019,48,8175
–
8185;式1,e,dalton trans.2019,48,2582
–
2591),得到相对分子量为1000-5000的聚乙烯蜡。这些研究结果为我们设计高活性、高热稳定性的催化剂起到了很好的借鉴作用,同时也为进一步的研究奠定了良好的基础。
[0007]
后过渡金属配合物作为新型烯烃聚合催化剂,依然存在相关基础研究的难点和推进工业化的制约因素,例如后过渡金属配合物的热稳定性能较差,使所得聚合物性质随温度变化而变化,对聚合过程温度的控制有较高的要求,因此,除了对制备条件和效率的改善,获得更高活性和高热稳定性的催化剂仍然是重点研究内容之一,亦是能否推进后过渡金属配合物烯烃聚合催化剂走向工业化的关键。
技术实现要素:
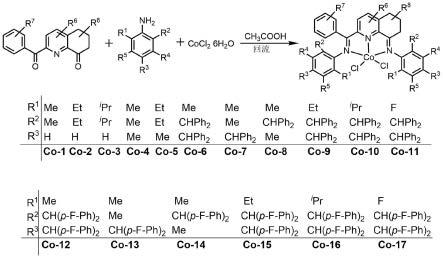
[0008]
为改善现有技术中存在的问题,本发明提供一种过渡金属钴配合物,所述过渡金属钴配合物具有如式(i)所示的结构:
[0009][0010]
式(i)中,
[0011]
r1、r2、r3均选自h、f、cl、br、i、无取代的c
1-6
烷基或c
1-6
烷氧基、被一个或多个ra取代的c
1-6
烷基或c
1-6
烷氧基、c
3-10
环烷基、c
3-10
环烷基氧基、芳基、芳基氧基或c
1-6
亚烷基芳基中至少一种,且每一个r1、r2、r3为相同或不同;
[0012]
r4、r5均选自h、f、cl、br、i,被一个或多个rb取代的下列基团:c
1-6
烷基、c
1-6
烷氧基、c
3-10
环烷基、c
3-10
环烷基氧基、芳基、芳基氧基或c
1-6
亚烷基芳基,且每一个r4、r5为相同或不同;
[0013]
r6、r7、r8均选自h、f、cl、br、i、无取代的c
1-6
烷基或c
1-6
烷氧基、被一个或多个rc取代的c
1-6
烷基或c
1-6
烷氧基、c
3-10
环烷基、c
3-10
环烷基氧基、芳基、芳基氧基或c
1-6
亚烷基芳基中至少一种,且每一个r6、r7、r8为相同或不同;
[0014]
x选自f、cl、br、i,且两个x为相同或不同;
[0015]
ra选自h、f、cl、br、i,无取代或任选被一个或多个rc取代的c
1-6
烷基、c
1-6
烷氧基、c
3-10
环烷基、c
3-10
环烷基氧基、芳基或芳基氧基,且每一个ra相同或不同;
[0016]
rb选自h、f、cl、br、i,无取代或任选被一个或多个rc取代的c
1-6
烷基、c
1-6
烷氧基、c3-10
环烷基、c
3-10
环烷基氧基、芳基或芳基氧基,且每一个rb相同或不同;
[0017]
rc选自h、f、cl、br、i、c
1-6
烷基、c
1-6
烷氧基、c
3-10
环烷基、c
3-10
环烷基氧基、芳基或芳基氧基,且每一个rc相同或不同。
[0018]
根据本发明的实施方案,式(i)中,每一个r1、r2、r3、r4、r5、r6、r7、r8相同或不同,各自独立地选自h、f、cl、br、i、c
1-3
烷基、或c
1-3
亚烷基芳基;
[0019]
每一个x相同或不同,各自独立地选自cl、br。
[0020]
作为实例,本发明所述式(i)所示的过渡金属钴配合物包括但不限于具有如下基团定义的配合物:
[0021]
配合物co-1:其中r1=me,r2=me,x选自cl,其他基团为h;
[0022]
配合物co-2:其中r1=et,r2=et,x选自cl,其他基团为h;
[0023]
配合物co-3:其中r1=ipr,r2=ipr,x选自cl,其他基团为h;
[0024]
配合物co-4:其中r1=me,r2=me,r3=me,x选自cl,其他基团为h;
[0025]
配合物co-5:其中r1=et,r2=et,r3=me,x选自cl,其他基团为h;
[0026]
配合物co-6:其中r1=me,r2=chph2,r3=chph2,x选自cl,其他基团为h;
[0027]
配合物co-7:其中r1=me,r2=me,r3=chph2,x选自cl,其他基团为h;
[0028]
配合物co-8:其中r1=me,r2=chph2,r3=me,x选自cl,其他基团为h;
[0029]
配合物co-9:其中r1=et,r2=chph2,r3=chph2,x选自cl,其他基团为h;
[0030]
配合物co-10:其中r1=ipr,r2=chph2,r3=chph2,x选自cl,其他基团为h;
[0031]
配合物co-11:其中r1=f,r2=chph2,r3=chph2,x选自cl,其他基团为h;
[0032]
配合物co-12:其中r1=me,r2=ch(p-f-ph)2,r3=ch(p-f-ph)2,x选自cl,其他基团为h;
[0033]
配合物co-13:其中r1=me,r2=me,r3=ch(p-f-ph)2,x选自cl,其他基团为h;
[0034]
配合物co-14:其中r1=me,r2=ch(p-f-ph)2,r3=me,x选自cl,其他基团为h;
[0035]
配合物co-15:其中r1=et,r2=ch(p-f-ph)2,r3=ch(p-f-ph)2,x选自cl,其他基团为h;
[0036]
配合物co-16:其中r1=ipr,r2=ch(p-f-ph)2,r3=ch(p-f-ph)2,x选自cl,其他基团为h;
[0037]
配合物co-17:其中r1=f,r2=ch(p-f-ph)2,r3=ch(p-f-ph)2,x选自cl,其他基团为h。
[0038]
本发明还提供上述式(i)所示的过渡金属钴配合物的制备方法,包括如下步骤:
[0039]
将如式(ⅱ)所示的化合物、如式(ⅲ)所示的苯胺类化合物和化合物cox2进行络合反应,得到所述式(i)所示的配合物;
[0040][0041]
其中,r1、r2、r3、r4、r5、r6、r7、r8、x具有上文所述定义。优选地,所述化合物cox2选自含钴的卤化物、或卤化物的水合物、溶剂合物,例如可以为cocl2或cocl2·
6h2o。
[0042]
优选地,所述反应优选在无氧条件下进行,例如在惰性气体如氮气的保护条件下进行。
[0043]
优选地,所述化合物cox2与所述式(ⅱ)所示的化合物的摩尔比可以为1:1~1.5,优选为1:1~1.3,进一步优选为1:1.1。
[0044]
优选地,所述化合物cox2与所述式(ⅲ)所示的化合物的摩尔比可以为1:2~3.2,优选为1:2~2.8,进一步优选为1:2.4。
[0045]
优选地,所述反应的温度选自100~120℃,优选为110~120℃;所述反应时间为4~9小时,优选5~7小时。
[0046]
优选地,所述反应可以在有机溶剂中进行,所述有机溶剂可以选自酸类溶剂。优选地,所述酸类溶剂选自冰醋酸。
[0047]
根据本发明的实施方案,所述方法还包括将所得式(i)所示的配合物进行纯化,所述纯化方法包括如下步骤:
[0048]
a)将所得式(i)所示的化合物用真空泵抽除溶剂,然后溶于有机溶剂(如无水乙醚)中,进行沉淀;
[0049]
b)经步骤a)沉淀后进行固液分离,对固相用无水乙醚洗涤并干燥。
[0050]
本发明还提供所述式(i)所示的过渡金属钴配合物的用途,其用于催化烯烃聚合反应,优选用于催化乙烯聚合反应。
[0051]
本发明还提供一种催化剂组合物,其特征在于,所述催化剂组合物包括主催化剂以及任选的助催化剂,其中,所述主催化剂选自式(i)所示的过渡金属钴配合物。
[0052]
根据本发明的实施方案,所述助催化剂可以选自铝氧烷、烷基铝和氯化烷基铝中的一种或多种。
[0053]
优选地,所述铝氧烷可以选自甲基铝氧烷(mao)或三异丁基铝改性的甲基铝氧烷(mmao)中的一种或两种。
[0054]
优选地,当所述催化剂组合物还包括上述助催化剂时,所述助催化剂中的金属al与式(i)所示的配合物的中心金属co的摩尔比为(500~4000):1,优选摩尔比为(1000~3500):1,例如可以为1000:1、1500:1、1750:1、2000:1、2250:1、2500:1、2750:1、3250:1。
[0055]
优选地,所述助催化剂为甲基铝氧烷(mao)时,甲基铝氧烷(mao)中的金属al与式(i)所示的配合物的中心金属co的摩尔比为(1500~3500):1,更优选摩尔比为2000:1。
[0056]
优选地,所述助催化剂为三异丁基铝改性的甲基铝氧烷(mmao)时,三异丁基铝改
性的甲基铝氧烷(mmao)中的金属al与式(i)所示的配合物的中心金属co的摩尔比为(1500~3000):1,更优选摩尔比为2250:1。
[0057]
本发明还提供一种聚乙烯的制备方法,包括:在上述催化剂组合物的作用下,使乙烯进行聚合反应。
[0058]
优选地,所述聚合反应的温度为30~90℃,例如可以是30℃、40℃、50℃、60℃、70℃、80℃、90℃;所述聚合反应的时间5~120min,例如可以是5min、10min、15min、30min、45min、60min;所述聚合反应的压力为0.3~20atm,例如可以是1atm、5atm或10atm。
[0059]
优选地,所述聚合反应的溶剂可以选自甲苯、邻二甲苯、二氯甲烷、乙醇、四氢呋喃、己烷或环己烷中的一种或几种。
[0060]
优选地,所述聚合反应优选在乙烯气氛下进行。
[0061]
本发明还提供上述催化剂组合物在催化烯烃聚合反应,特别是乙烯聚合反应中的用途。
[0062]
本发明具有以下优点:
[0063]
1.本发明提供了制备聚乙烯蜡的高活性、高热稳定的过渡金属钴配合物的制备方法。该类化合物的制备过程均具有反应条件温和、周期短、操作条件简单等优点。
[0064]
2.本发明提供了一类制备聚乙烯蜡的高活性的过渡金属钴配合物。该类配合物含有f、甲基、乙基、异丙基,及大体积取代基二苯甲基和二(4-氟苯基)甲基,表现出单一催化活性中心,可以通过改变催化剂结构和聚合反应条件实现对聚合物分子量的精密调控,具有突出的催化活性高、热稳定性、成本低等优点。甲基铝氧烷(mao)或者三异丁基铝改性的甲基铝氧烷(mmao)为助催化剂时,50℃条件下,其最佳催化活性分别为27.06
×
106g(pe)
·
(mol co)-1
·
h-1
、20.10
×
106g(pe)
·
(mol co)-1
·
h-1
,催化活性高,即使在70℃高温时,催化活性仍然能保持在2.61
×
106g(pe)
·
(mol co)-1
·
h-1
,热稳定性高,符合工业生产的操作温度,具有进一步工业化的应用前景。
[0065]
3.本发明提供了高热稳定的过渡金属钴配合物的用途。作为新型烯烃催化剂应用于乙烯聚合反应。此类配合物具有以下结构特点:在吡啶环的一侧引入亚苄基,另一侧引入六元环,同时固定两个c=n键来固化骨架以提高其热稳定性;苯胺上取代基团在吸供电子能力、空间位阻大小方面进行系统变化,2位取代基在甲基、乙基、异丙基、大体积二苯甲基以及二(4-氟苯基)甲基间变化,4位取代基在甲基、大体积二苯甲基以及二(4-氟苯基)甲基间变化,6位取代基在氟、甲基、乙基、异丙基间变化。在这些结构的协同作用下,特别是强吸电子氟取代基的存在,有利地稳定了中心金属更强的正电特点,更强的路易斯酸性,提高了乙烯插入的几率,保证了该体系的高催化活性及稳定性。例如在50℃条件下,co-13催化乙烯聚合的活性可高达27.06
×
106g(pe)
·
(mol co)-1
·
h-1
,且所得聚乙烯重均分子量mw小,多在0.7-1.7kg
·
mol-1
之间波动,属于典型的聚乙烯蜡。
[0066]
4.本发明提供的制备聚乙烯的方法操作简单且反应条件温和,可用于制备高度线性聚乙烯。所得聚合物分子量在0.5-318.7kg
·
mol-1
之间波动,其中通过调控苯胺上的取代基可以获得不同相对分子量分布的端基为双键的聚乙烯蜡,可以用作特种高端聚乙烯蜡,如食品级聚乙烯蜡,也可用作共聚单体,在生产长链共聚物、功能性聚合物和涂层材料方面显示出潜在应用。
[0067]
5.在本发明设计并合成的含大体积二苯甲基以及二(4-氟苯基)甲基的过渡金属
钴配合物中,由于邻位的二苯甲基以及二(4-氟苯基)甲基的空间位阻作用,使得芳基亚胺平面与配位平面基本处于垂直位置,可以对金属活性中心形成有效保护。因此,本发明中所述配合物活性高,性质稳定。
附图说明
[0068]
图1为本发明实施例1-17制备过渡金属钴配合物的反应流程图。
[0069]
图2为实施例13制备的过渡金属钴配合物co-13晶体结构示意图。
[0070]
图3为实施例31中所得聚合物升温核磁氢谱、碳谱图。
[0071]
图4为实施例48中所得聚合物升温核磁氢谱、碳谱图。
具体实施方式
[0072]
下面结合具体实施例对本发明作进一步阐述,但本发明并不限于以下实施例。所述方法如无特别说明均为常规方法。所述原材料如无特别说明均能从公开商业途径而得。
[0073]
所用甲基铝氧烷(简称mao)和改性甲基铝氧烷(简称mmao)均购自美国akzonobel公司。下述实施例18~51中,al/co的定义为助催化剂mao或mmao中的金属al与配合物中co的摩尔比。
[0074]
实施例1.制备2-(1-(2,6-二甲基苯胺基)亚苄基)-8-(2,6-二甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-1
[0075]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、81mg(0.66mmol)2,6-二甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到97mg黄绿色粉末,即为co-1,产率:61%。
[0076]
结构确证数据如下:
[0077]
ft-ir(cm-1
):2954(w),2917(m),2869(w),1622(s),1566(s),1469(s),1446(s),1381(m),1322(m),1267(vs),1201(m),1144(w),1106(w),981(m),870(m),779(s),728(m),700(vs).
[0078]
元素分析:c
32h31
cl2con3(587.45)理论值:c,65.43;h,5.32;n,7.15%.实验值:found:c,64.81;h,4.90;n,6.99%.
[0079]
实施例2.制备2-(1-(2,6-二乙基苯胺基)亚苄基)-8-(2,6-二乙基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-2
[0080]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、99mg(0.66mmol)2,6-二乙基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到84mg黄绿色粉末,即为co-2,产率:46%。
[0081]
结构确证数据如下:
[0082]
ft-ir(cm-1
):2966(m),1622(s),1570(s),1447(s),1322(m),1266(vs),1187(m),1109(m),1062(m),981(m),780(s),730(s),699(vs).
[0083]
元素分析:c
36h39
cl2con3(673.63)理论值:c,67.75;h,6.73;n,6.24%.实验值:c,67.45;h,6.23;n,6.33%.
[0084]
实施例3.制备2-(1-(2,6-二异丙基苯胺基)亚苄基)-8-(2,6-二异丙基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-3
[0085]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、117mg(0.66mmol)2,6-二异丙基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到103mg黄绿色粉末,即为co-3,产率:55%。
[0086]
结构确证数据如下:
[0087]
ft-ir(cm-1
):3067(w),2965(m),2920(w),2873(m),1619(m),1563(s),1449(s),1324(s),1267(s),1185(s),1055(s),982(m),802(s),778(m),730(m),699(m).
[0088]
元素分析:c
40h47
cl2con3(699.67)理论值:c,68.67;h,6.77;n,6.01%.实验值:c,68.17;h,6.97;n,5.88%.
[0089]
实施例4.制备2-(1-(2,4,6-三甲基苯胺基)亚苄基)-8-(2,4,6-三甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-4
[0090]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、90mg(0.66mmol)2,4,6-三甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到87mg黄绿色粉末,即为co-4,产率:52%。
[0091]
结构确证数据如下:
[0092]
ft-ir(cm-1
):2972(w),2908(m),1626(s),1592(s),1477(s),1447(s),1382(m),1321(m),1267(vs),1140(w),1066(w),982(m),854(m),775(s),733(m),699(vs).
[0093]
元素分析:c
34h35
cl2con3(615.51)理论值:c,66.35;h,5.73;n,6.83%.实验值:c,61.13;h,5.35;n,6.30%.
[0094]
实施例5.制备2-(1-(2,6-二乙基-4-甲基苯胺基)亚苄基)-8-(2,6-二乙基-4-甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-5
[0095]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、108mg(0.66mmol)2,6-二乙基-4-甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到75mg黄绿色粉末,即为co-5,产率:41%。
[0096]
结构确证数据如下:
[0097]
ft-ir(cm-1
):2967(m),2928(m),1622(s),1576(s),1451(s),1321(m),1266(vs),1183(m),1103(m),1062(m),981(m),780(s),698(vs).
[0098]
元素分析:c
38h43
cl2con3(671.62)理论值:c,67.96;h,6.45;n,6.26%.实验值:c,67.26;h,5.95;n,6.00%.
[0099]
实施例6.制备2-(1-(2,4-二二苯甲基-6-甲基苯胺基)亚苄基)-8-(2,4-二二苯甲
基-6-甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-6
[0100]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、291mg(0.66mmol)2,4-二二苯甲基-6-甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到165mg黄绿色粉末,即为co-6,产率:50%。
[0101]
结构确证数据如下:
[0102]
ft-ir(cm-1
):3665(w),2970(m),1598(m),1559(s),1531(s),1475(m),1449(s),1266(m),1075(m),1033(m),744(m),700(s).
[0103]
元素分析:c
82h67
cl2con3(1224.29)理论值:c,80.45;h,5.52;n,3.43%.实验值:c,73.52;h,5.20;n,3.25%.
[0104]
实施例7.制备2-(1-(4-二苯甲基-2,6-二甲基苯胺基)亚苄基)-8-(4-二苯甲基-2,6-二甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-7
[0105]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、190mg(0.66mmol)4-二苯甲基-2,6-二甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到120mg黄绿色粉末,即为co-7,产率:48%。
[0106]
结构确证数据如下:
[0107]
ft-ir(cm-1
):3669(w),3054(w),2967(m),1604(s),1563(s),1500(s),1447(m),1268(w),1225(s),1158(m),1034(m),833(m),699(s).
[0108]
元素分析:c
58h51
cl2con3(919.90)理论值:c,75.73;h,5.59;n,4.57%.实验值:c,70.18;h,5.29;n,4.25%.
[0109]
实施例8.制备2-(1-(2-二苯甲基-4,6-二甲基苯胺基)亚苄基)-8-(2-二苯甲基-4,6-二甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-8
[0110]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、190mg(0.66mmol)2-二苯甲基-4,6-二甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到132mg黄绿色粉末,即为co-8,产率:53%。
[0111]
结构确证数据如下:
[0112]
ft-ir(cm-1
):3664(w),2969(m),2157(w),2025(w),1972(w),1599(m),1505(s),1447(m),1266(m),1225(s),1159(m),1075(m),1036(m),840(s),699(s).
[0113]
元素分析:c
58h51
cl2con3(919.90)理论值:c,75.73;h,5.59;n,4.57%.实验值:c,71.75;h,5.29;n,4.27%.
[0114]
实施例9.制备2-(1-(2,4-二二苯甲基-6-乙基苯胺基)亚苄基)-8-(2,4-二二苯甲基-6-乙基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-9
[0115]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、300mg(0.66mmol)2,4-二二苯甲基-6-乙基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml
冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到196mg黄绿色粉末,即为co-9,产率:58%。
[0116]
结构确证数据如下:
[0117]
ft-ir(cm-1
):3057(w),3025(w),2361(m),1596(m),1494(s),1447(s),1266(m),1031(m),744(s),699(s).
[0118]
元素分析:c
84h71
cl2con3(1252.35)理论值:c,80.56;h,5.71;n,3.36%.实验值:c,77.71;h,5.52;n,3.25%.
[0119]
实施例10.制备2-(1-(2,4-二二苯甲基-6-异丙基苯胺基)亚苄基)-8-(2,4-二二苯甲基-6-异丙基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-10
[0120]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、309mg(0.66mmol)2,4-二二苯甲基-6-异丙基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到142mg黄绿色粉末,即为co-10,产率:41%。
[0121]
结构确证数据如下:
[0122]
ft-ir(cm-1
):3669(w),2967(m),1599(m),1499(s),1447(s),1266(m),1224(m),1077(m),1037(m),829(m),742(m),699(s).
[0123]
元素分析:c
86h75
cl2con3(1280.40)理论值:c,80.67;h,5.90;n,3.28%.实验值:c,77.97;h,5.89;n,3.29%.
[0124]
实施例11.制备2-(1-(2,4-二二苯甲基-6-氟苯胺基)亚苄基)-8-(2,4-二二苯甲基-6-氟苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-11
[0125]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、293mg(0.66mmol)2,4-二二苯甲基-6-氟苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到163mg黄绿色粉末,即为co-11,产率:49%。
[0126]
结构确证数据如下:
[0127]
ft-ir(cm-1
):3667(w),2969(m),1600(m),1557(s),1527(m),1472(s),1450(s),1425(m),1268(m),1031(m),745(m),700(s).
[0128]
元素分析:c
80h61
cl2con3(1232.22)理论值:c,77.98;h,4.99;n,3.41%.实验值:c,76.29;h,4.89;n,3.12%.
[0129]
实施例12.制备2-(1-(2,4-二(二(4-氟苯基)甲基)-6-甲基苯胺基)亚苄基)-8-(2,4-二(二(4-氟苯基)甲基)-6-甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-12
[0130]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、338mg(0.66mmol)2,4-二(二(4-氟苯基)甲基)-6-甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到222mg黄棕色粉末,即为co-12,产率:60%。
[0131]
结构确证数据如下:
[0132]
ft-ir(cm-1
):3055(w),2966(w),1597(m),1494(s),1447(s),1266(m),1246(m),1032(w),744(m),698(s).
[0133]
元素分析:c
82h59
cl2cof8n3(1368.22)理论值:c,71.98;h,4.35;n,3.07%.实验值:c,70.62;h,4.38;n,2.95%.
[0134]
实施例13.制备2-(1-(4-二(4-氟苯基)甲基-2,6-二甲基苯胺基)亚苄基)-8-(4-二(4-氟苯基)甲基-2,6-二甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-13
[0135]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、214mg(0.66mmol)4-二(4-氟苯基)甲基-2,6-二甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到150mg黄棕色粉末,即为co-13,产率:56%。
[0136]
co-13晶体结构示意图如图2所示。
[0137]
由图可知,配合物co-13的中心金属co采用五配位方式,分别与三个氮原子n1,n2,n3和两个氯原子cl1,cl2相连,呈扭曲的四方锥结构。其中三个氮原子与cl1原子形成四方锥底,cl2占据四方锥顶。由于空间效应,co原子与锥顶cl2原子间距约基底各原子与co原子的间距n(1)
–
co(1)、n(3)
–
co(1)、n(2)
–
co(1)以及cl(1)
–
co(1)依次为和
[0138]
结构确证数据如下:
[0139]
ft-ir(cm-1
):3669(w),3055(w),2967(m),1603(s),1562(s),1500(s),1446(m),1267(w),1225(s),1158(m),1033(m),833(m),699(s).
[0140]
元素分析:c
58h47
cl2cof8n3(991.86)理论值:c,70.24;h,4.78;n,4.24%.实验值:c,70.35;h,4.81;n,4.12%.
[0141]
实施例14.制备2-(1-(2-二(4-氟苯基)甲基-4,6-二甲基苯胺基)亚苄基)-8-(2-二(4-氟苯基)甲基-4,6-二甲基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-14
[0142]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、214mg(0.66mmol)2-二(4-氟苯基)甲基-4,6-二甲基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到129mg黄棕色粉末,即为co-14,产率:48%。
[0143]
结构确证数据如下:
[0144]
ft-ir(cm-1
):3664(w),2968(m),2157(w),2024(w),1972(w),1599(m),1504(s),1447(m),1266(m),1225(s),1158(m),1075(m),1036(m),840(s),700(s).
[0145]
元素分析:c
58h47
cl2cof8n3(991.86)理论值:c,70.24;h,4.78;n,4.24%.实验值:c,66.89;h,4.65;n,3.91%.
[0146]
实施例15.制备2-(1-(2,4-二(二(4-氟苯基)甲基)-6-乙基苯胺基)亚苄基)-8-(2,4-二(二(4-氟苯基)甲基)-6-乙基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-15
[0147]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、347mg(0.66mmol)2,4-二(二
(4-氟苯基)甲基)-6-乙基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到186mg黄棕色粉末,即为co-15,产率:53%。
[0148]
结构确证数据如下:
[0149]
ft-ir(cm-1
):3667(w),2969(m),1601(m),1505(s),1435(m),1224(s),1157(w),1094(m),1048(m),829(s),699(m).
[0150]
元素分析:c
84h63
cl2cof8n3(1296.27)理论值:c,72.26;h,4.55;n,3.01%.实验值:c,71.82;h,4.71;n,2.71%.
[0151]
实施例16.制备2-(1-(2,4-二(二(4-氟苯基)甲基)-6-异丙基苯胺基)亚苄基)-8-(2,4-二(二(4-氟苯基)甲基)-6-异丙基苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-16
[0152]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、356mg(0.66mmol)2,4-二(二(4-氟苯基)甲基)-6-异丙基苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到212mg黄绿色粉末,即为co-16,产率:55%。
[0153]
结构确证数据如下:
[0154]
ft-ir(cm-1
):3662(w),2967(m),1600(m),1506(s),1447(m),1265(s),1227(s),1158(m),833(s),700(m).
[0155]
元素分析:c
86h67
cl2cof8n3(1424.32)理论值:c,72.52;h,4.74;n,2.95%.实验值:c,69.56;h,4.77;n,2.82%.
[0156]
实施例17.制备2-(1-(2,4-二(二(4-氟苯基)甲基)-6-氟苯胺基)亚苄基)-8-(2,4-二(二(4-氟苯基)甲基)-6-氟苯胺基)-5,6,7-三氢喹啉合氯化钴配合物co-17
[0157]
将76mg(0.30mmol)2-苯甲酰基-6,7-二氢喹啉-8-酮、341mg(0.66mmol)2,4-二(二(4-氟苯基)甲基)-6-氟苯胺和64mg(0.27mmol)cocl2·
6h2o在氮气氛围下放入反应瓶中,加入10ml冰醋酸中。在120℃下,迅速溶解且溶液颜色转变成棕色,搅拌反应6h,以确保反应充分。使用冷阱将溶剂抽走,然后加入1ml二氯甲烷和10ml无水乙醚重结晶,有固体析出。通过过滤收集,并用大量乙醚(3
×
5ml)洗涤。得到212mg黄绿色粉末,即为co-17,产率:57%。
[0158]
结构确证数据如下:
[0159]
ft-ir(cm-1
):3667(w),2969(m),1600(m),1557(s),1527(m),1472(s),1450(s),1425(m),1268(m),1031(m),745(m),700(s).
[0160]
元素分析:c
80h53
cl2cof
10
n3(1376.14)理论值:c,69.82;h,3.88;n,3.05%.实验值:c,69.68;h,3.90;n,3.18%.
[0161]
实施例18.利用配合物co-17及助催化剂mao高压下联合催化乙烯聚合:
[0162]
a)在乙烯氛围下,将25ml的催化剂co-17(2μmol)的甲苯溶液注射到250ml装有机械搅拌的不锈钢高压釜中,接着加入25ml甲苯,加入所需量的2.1ml的助催化剂mao(1.46mol/l在甲苯中),继续加入甲苯使溶剂总体积为100ml。此时al/co=1500:1。机械搅拌开始,保持400转/分,当聚合温度达到50℃时,往反应釜中充入乙烯,聚合反应开始。在50
℃下保持10atm的乙烯压强,搅拌进行聚合反应30min。用10%盐酸酸化的乙醇溶液中和反应液,得到聚合物沉淀,用乙醇洗数次,真空50℃烘干至恒重,称重得9.49g聚合物,聚合活性:9.49
×
106g(pe)(mol co)-1
h-1
,所得聚合物分子量mw=2.0kg
·
mol-1
、分子量分布mw/mn=2.7(mw为聚合物的重均分子量,mn为聚合物的数均分子量,通过gpc测试所得),聚合物tm=119.7℃(tm为聚合物的熔融温度,通过dsc测试所得)。
[0163]
b)基本同本实施例中方法a),区别在于:2.4ml的助催化剂mao(1.46mol/l在甲苯中)使al/co=1750:1。聚合活性:10.05
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.8kg
·
mol-1
、分子量分布mw/mn=2.0,聚合物tm=120.5℃。
[0164]
c)基本同本实施例中方法a),区别在于:2.8ml的助催化剂mao(1.46mol/l在甲苯中)使al/co=2000:1。聚合活性:10.58
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=1.8kg
·
mol-1
、分子量分布mw/mn=2.0,聚合物tm=119.4℃。
[0165]
d)基本同本实施例中方法a),区别在于:3.1ml的助催化剂mao(1.46mol/l在甲苯中)使al/co=2250:1。聚合活性:10.56
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.7kg
·
mol-1
、分子量分布mw/mn=1.7,聚合物tm=119.4℃。
[0166]
e)基本同本实施例中方法a),区别在于:3.4ml的助催化剂mao(1.46mol/l在甲苯中)使al/co=2500:1。聚合活性:8.10
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=1.0kg
·
mol-1
、分子量分布mw/mn=2.2,聚合物tm=119.9℃。
[0167]
f)基本同本实施例中方法c),区别在于:聚合温度为40℃。聚合活性:6.52
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.9kg
·
mol-1
、分子量分布mw/mn=1.9,聚合物tm=120.4℃。
[0168]
g)基本同本实施例中方法c),区别在于:聚合温度为60℃。聚合活性:5.08
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=3.1kg
·
mol-1
、分子量分布mw/mn=2.2,聚合物tm=120.1℃。
[0169]
h)基本同本实施例中方法c),区别在于:聚合温度为70℃。聚合活性:2.61
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=1.5kg
·
mol-1
、分子量分布mw/mn=1.9,聚合物tm=123.6℃。
[0170]
实施例19.利用配合物co-1及mao联合催化加压下的乙烯聚合:
[0171]
基本同实施例18c),区别在于:主催化剂为co-1。聚合活性:16.18
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=2.7kg
·
mol-1
、分子量分布mw/mn=2.4,聚合物tm=123.3℃。
[0172]
实施例20.利用配合物co-2及mao联合催化加压下的乙烯聚合:
[0173]
基本同实施例18c),区别在于:主催化剂为co-2。聚合活性:2.42
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=9.9kg
·
mol-1
、分子量分布mw/mn=2.3,聚合物tm=129.6℃。
[0174]
实施例21.利用配合物co-3及mao联合催化加压下的乙烯聚合:
[0175]
基本同实施例18c),区别在于:主催化剂为co-3。聚合活性:0.16
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=146.4kg
·
mol-1
、分子量分布mw/mn=1.7,聚合物tm=131.2℃。
[0176]
实施例22.利用配合物co-4及mao联合催化加压下的乙烯聚合:
[0177]
基本同实施例18c),区别在于:主催化剂为co-4。聚合活性:8.65
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=3.2kg
·
mol-1
、分子量分布mw/mn=2.1,聚合物tm=124.2℃。
[0178]
实施例23.利用配合物co-5及mao联合催化加压下的乙烯聚合:
co)-1
h-1
,聚合物分子量mw=193.7kg
·
mol-1
、分子量分布mw/mn=4.2,聚合物tm=134.4℃。
[0204]
实施例35.利用配合物co-17及助催化剂mmao高压下联合催化乙烯聚合:
[0205]
a)在乙烯氛围下,将25ml的催化剂co-17(2μmol)的甲苯溶液注射到250ml装有机械搅拌的不锈钢高压釜中,接着加入25ml甲苯,加入所需量的2.1ml的助催化剂mmao(1.93mol/l在正己烷中),继续加入甲苯使溶剂总体积为100ml。此时al/co=2000:1。机械搅拌开始,保持400转/分,当聚合温度达到50℃时,往反应釜中充入乙烯,聚合反应开始。在50℃下保持10atm的乙烯压强,搅拌进行聚合反应30min。用10%盐酸酸化的乙醇溶液中和反应液,得到聚合物沉淀,用乙醇洗数次,真空50℃烘干至恒重,称重得3.48g聚合物,聚合活性:3.48
×
106g(pe)(mol co)-1
h-1
,所得聚合物分子量mw=0.8kg
·
mol-1
、分子量分布mw/mn=2.1(mw为聚合物的重均分子量,mn为聚合物的数均分子量,通过gpc测试所得),聚合物tm=119.9℃(tm为聚合物的熔融温度,通过dsc测试所得)。
[0206]
b)基本同本实施例中方法a),区别在于:2.4ml的助催化剂mmao(1.93mol/l在正己烷中)使al/co=2250:1。聚合活性:6.64
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.7kg
·
mol-1
、分子量分布mw/mn=1.9,聚合物tm=117.8℃。
[0207]
c)基本同本实施例中方法a),区别在于:2.6ml的助催化剂mmao(1.93mol/l在正己烷中)使al/co=2500:1。聚合活性:4.00
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.8kg
·
mol-1
、分子量分布mw/mn=1.8,聚合物tm=119.2℃。
[0208]
d)基本同本实施例中方法a),区别在于:2.9ml的助催化剂mmao(1.93mol/l在正己烷中)使al/co=2750:1。聚合活性:4.77
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.9kg
·
mol-1
、分子量分布mw/mn=2.6,聚合物tm=119.6℃。
[0209]
e)基本同本实施例中方法a),区别在于:3.1ml的助催化剂mmao(1.93mol/l在正己烷中)使al/co=3000:1。聚合活性:3.51
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.8kg
·
mol-1
、分子量分布mw/mn=2.9,聚合物tm=119.7℃。
[0210]
f)基本同本实施例中方法b),区别在于:聚合温度为30℃。聚合活性:7.11
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=1.0kg
·
mol-1
、分子量分布mw/mn=2.1,聚合物tm=119.0℃。
[0211]
g)基本同本实施例中方法b),区别在于:聚合温度为40℃。聚合活性:8.38
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.8kg
·
mol-1
、分子量分布mw/mn=2.2,聚合物tm=117.9℃。
[0212]
h)基本同本实施例中方法b),区别在于:聚合温度为60℃。聚合活性:3.88
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=0.8kg
·
mol-1
、分子量分布mw/mn=1.8,聚合物tm=118.8℃。
[0213]
i)基本同本实施例中方法b),区别在于:聚合温度为70℃。聚合活性:2.07
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=1.1kg
·
mol-1
、分子量分布mw/mn=2.1,聚合物tm=119.8℃。
[0214]
实施例36.利用配合物co-1及mmao联合催化加压下的乙烯聚合:
[0215]
基本同实施例35g),区别在于:主催化剂为co-1。聚合活性:7.14
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=2.4kg
·
mol-1
、分子量分布mw/mn=2.0,聚合物tm=123.2℃。
[0216]
实施例37.利用配合物co-2及mmao联合催化加压下的乙烯聚合:
[0217]
基本同实施例35g),区别在于:主催化剂为co-2。聚合活性:1.72
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=10.0kg
·
mol-1
、分子量分布mw/mn=2.2,聚合物tm=129.4℃。
[0218]
实施例38.利用配合物co-3及mmao联合催化加压下的乙烯聚合:
[0219]
基本同实施例35g),区别在于:主催化剂为co-3。聚合活性:0.18
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=166.6kg
·
mol-1
、分子量分布mw/mn=2.7,聚合物tm=132.7℃。
[0220]
实施例39.利用配合物co-4及mmao联合催化加压下的乙烯聚合:
[0221]
基本同实施例35g),区别在于:主催化剂为co-4。聚合活性:3.98
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=2.9kg
·
mol-1
、分子量分布mw/mn=1.7,聚合物tm=124.7℃。
[0222]
实施例40.利用配合物co-5及mmao联合催化加压下的乙烯聚合:
[0223]
基本同实施例35g),区别在于:主催化剂为co-5。聚合活性:1.20
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=9.9kg
·
mol-1
、分子量分布mw/mn=2.3,聚合物tm=129.5℃。
[0224]
实施例41.利用配合物co-6及mmao联合催化加压下的乙烯聚合:
[0225]
基本同实施例35g),区别在于:主催化剂为co-6。聚合活性:4.06
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=83.9kg
·
mol-1
、分子量分布mw/mn=3.4,聚合物tm=133.4℃。
[0226]
实施例42.利用配合物co-7及mmao联合催化加压下的乙烯聚合:
[0227]
基本同实施例35g),区别在于:主催化剂为co-7。聚合活性:11.06
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=2.1kg
·
mol-1
、分子量分布mw/mn=2.7,聚合物tm=122.7℃。
[0228]
实施例43.利用配合物co-8及mmao联合催化加压下的乙烯聚合:
[0229]
基本同实施例35g),区别在于:主催化剂为co-8。聚合活性:2.80
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=73.6kg
·
mol-1
、分子量分布mw/mn=5.7,聚合物tm=131.9℃。
[0230]
实施例44.利用配合物co-9及mmao联合催化加压下的乙烯聚合:
[0231]
基本同实施例35g),区别在于:主催化剂为co-9。聚合活性:3.72
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=141.3kg
·
mol-1
、分子量分布mw/mn=5.8,聚合物tm=134.2℃。
[0232]
实施例45.利用配合物co-10及mmao联合催化加压下的乙烯聚合:
[0233]
基本同实施例35g),区别在于:主催化剂为co-10。聚合活性:4.28
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=318.7kg
·
mol-1
、分子量分布mw/mn=3.9,聚合物tm=134.8℃。
[0234]
实施例46.利用配合物co-11及mmao联合催化加压下的乙烯聚合:
[0235]
基本同实施例35g),区别在于:主催化剂为co-11。聚合活性:5.16
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=1.2kg
·
mol-1
、分子量分布mw/mn=2.4,聚合物tm=122.1℃。
[0236]
实施例47.利用配合物co-12及mmao联合催化加压下的乙烯聚合:
[0237]
基本同实施例35g),区别在于:主催化剂为co-12。聚合活性:4.05
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=66.8kg
·
mol-1
、分子量分布mw/mn=7.5,聚合物tm=131.6℃。
[0238]
实施例48.利用配合物co-13及mmao联合催化加压下的乙烯聚合:
[0239]
基本同实施例35g),区别在于:主催化剂为co-13。聚合活性:20.10
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=1.7kg
·
mol-1
、分子量分布mw/mn=3.0,聚合物tm=122.4℃。
[0240]
取所得聚合物10mg,溶于3ml氘代1,1,2,2-四氯乙烷,在100℃条件下,测试该聚合物的1h数据,如图4所示。信号累积64次。
[0241]
取所得聚合物60mg,溶于3ml氘代1,1,2,2-四氯乙烷,在100℃条件下,测试该聚合
物的
13
c数据,如图4所示。信号累积3000次。
[0242]
实施例49.利用配合物co-14及mmao联合催化加压下的乙烯聚合:
[0243]
基本同实施例35g),区别在于:主催化剂为co-14。聚合活性:2.80
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=63.6kg
·
mol-1
、分子量分布mw/mn=6.5,聚合物tm=132.4℃。
[0244]
实施例50.利用配合物co-15及mmao联合催化加压下的乙烯聚合:
[0245]
基本同实施例35g),区别在于:主催化剂为co-15。聚合活性:3.77
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=127.9kg
·
mol-1
、分子量分布mw/mn=5.2,聚合物tm=133.8℃。
[0246]
实施例51.利用配合物co-16及mmao联合催化加压下的乙烯聚合:
[0247]
基本同实施例35g),区别在于:主催化剂为co-16。聚合活性:4.89
×
106g(pe)(mol co)-1
h-1
,聚合物分子量mw=315.1kg
·
mol-1
、分子量分布mw/mn=4.0,聚合物tm=134.9℃。
[0248]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1