Cas12蛋白、含有Cas12蛋白的基因编辑系统及应用
cas12蛋白、含有cas12蛋白的基因编辑系统及应用
技术领域
1.本技术属于基因编辑技术领域,具体涉及cas12蛋白、含有该cas12蛋白的基因编辑系统及其相关应用。
背景技术:
2.crispr/cas系统是细菌和古细菌为抵御外源病毒或质粒入侵而进化的一种获得性免疫系统。在crispr/cas12a和crispr/cas12j系统中,crrna(crispr
‑
derived rna)和cas12蛋白形成复合体后,识别靶位点的pam(protospacer adjacent motif)序列。在识别后,crrna会与靶向dna序列形成互补结构,cas蛋白行使切割dna的功能,使dna发生断裂损伤。crispr/cas12b系统还含有tracrrna(trans
‑
activating rna),它和crrna以及cas12b共同形成复合物发挥功能。tracrrna和crrna通过连接序列可以融合成为单链单链向导rna(single guide rna,sgrna)。当dna发生断裂损伤后,细胞内的两种主要dna损伤修复机制负责修复:非同源末端连接(non
‑
homologous end
‑
joining,nhej)和同源重组(homologous recombination,hr)。nhej修复的结果会引起碱基的缺失或插入,可以进行基因敲除;在提供同源模板的情况下,利用hr修复可以进行基因的定点插入和碱基的精确替换。
3.除了基础科研外,crispr/cas12基因编辑系统还具有广泛的临床应用前景。利用crispr/cas12基因编辑系统做基因治疗时,需要把cas和单链向导rna导入到体内。目前做基因治疗最有效的表达载体是腺相关病毒(aav)。但是aav病毒包装的dna一般不超过4.5kb。spcas9因为pam序列简单(识别ngg)和活性高而得到广泛应用。但是spcas9蛋白有1368个氨基酸,加上sgrna和启动子,无法有效地包装到aav病毒中,限制了其在临床中的应用。为了克服这个问题,几个分子量小的cas9被发明出来,包括sacas9(pam序列为nngrrt);stlcas9(pam序列为nnagaw);nmcas9(pam序列为nnnngatt);nme2cas9(pam序列为nnnncc);cjcas9(pam序列为nnnnryac)。但是这些cas9或者容易脱靶(即非靶向位点切割),或者pam序列复杂,或者编辑活性低,难以广泛应用。
4.因此,寻找编辑活性高、特异性高、pam序列简单的小型crispr/cas系统是解决上述问题的希望所在。
技术实现要素:
5.针对上述问题,本发明人进行了反复研究,发现一系列cas12蛋白以及与之相对应的单链向导rna,两者能构成有效地进行基因编辑的crispa/cas12基因编辑系统,由此完成了本发明。
6.因此,在第一方面,本发明提供了一种缀合物,所述缀合物包含:
7.a)cas12蛋白,所述cas12蛋白为分别具有seq id no:1至seq id no:6所示氨基酸序列的cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白或chcas12b蛋白,或者为具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%序列同一性并且保留其生物
学活性的氨基酸序列;以及
8.b)修饰部分。
9.在第二方面,本发明提供了一种融合蛋白,所述融合蛋白包含:
10.a)cas12蛋白,所述cas12蛋白为分别具有seq id no:1至seq id no:6所示氨基酸序列的cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白或chcas12b蛋白,或者为具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%序列同一性并且保留其生物学活性的氨基酸序列;
11.b)另外的蛋白或多肽;以及
12.c)任选的用于连接所述cas12蛋白或其同源物与所述另外的蛋白或多肽的接头。
13.在第三方面,本发明提供了一种单链向导rna,所述单链向导rna包含crispr重复序列,所述crispr重复序列具有seq id no:15至seq id no:18中任一个所示的核酸序列,或者具有与seq id no:15至seq id no:18中任一项所示的核酸序列至少90%序列同一性且保留其生物学活性的核酸序列,或者具有基于seq id no:15至seq id no:18中任一项所述的核酸序列改造得到的保留其生物学活性的核酸序列。
14.在第四方面,本发明提供了一种分离的核酸分子,所述分离的核酸分子包含编码以下的核酸序列:
15.a)cas12蛋白,所述cas12蛋白为分别具有seq id no:1至seq id no:6所示氨基酸序列的cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白或chcas12b蛋白,或者为具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%序列同一性并且保留其生物学活性的氨基酸序列;
16.b)本发明第一方面的缀合物;或者
17.c)本发明第三方面的融合蛋白。
18.在第五方面,本发明提供了一种分离的核酸分子,所述分离的核酸分子包含编码本发明第三方面的单链向导rna的核酸序列。
19.在第六方面,本发明提供了一种载体,所述载体包含编码以下的核酸序列:
20.a)cas12蛋白,所述cas12蛋白为分别具有seq id no:1至seqid no:6所示氨基酸序列的cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白或chcas12b蛋白,或者为具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%序列同一性并且保留其生物学活性的氨基酸序列;
21.b)本发明第一方面的缀合物;或者
22.本发明第二方面的融合蛋白。
23.在第七方面,本发明提供了一种载体,所述载体包含编码本发明第三方面的单链向导rna的核酸序列。
24.在第八方面,本发明提供了一种crispr/cas12基因编辑系统,其包含:
25.a)蛋白组分,其包含:
26.1)cas12蛋白,所述cas12蛋白为分别具有seq id no:1至seq id no:6所示氨基酸
序列的cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白或chcas12b蛋白,或者为具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%序列同一性并且保留其生物学活性的氨基酸序列;
27.2)本发明第一方面的缀合物;或者
28.3)本发明第二方面的融合蛋白;
29.b)核酸组分,其包含:
30.本发明第三方面的单链向导rna。
31.在第九方面,本发明提供了一种细胞,所述细胞包含:本发明第六方面的分离的核酸分子、或者本发明第七方面的载体。
32.在第十方面,本发明提供了一种对细胞内或体外环境中的靶序列进行基因编辑的方法,所述方法包括:使cas12蛋白、本发明第一方面的缀合物或本发明第二方面的融合蛋白与本发明第三方面的单链向导rna、使本发明第六方面和第七方面的载体、或使本发明第八方面的crispr/cas12基因编辑系统与细胞内或体外环境中的靶序列相接触,其中,所述cas12蛋白为分别具有seq id no:1至seq id no:6所示氨基酸序列的cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白或chcas12b蛋白,或者为具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%序列同一性并且保留其生物学活性的氨基酸序列,所述靶序列位于原间隔邻近序列(pam)的5’端,并且,对于所述cas12j
‑
8蛋白、所述mb4cas12a蛋白、所述mlcas12a蛋白、所述mocas12a蛋白、所述bgcas12a蛋白、和所述chcas12b蛋白、或者它们的同源物、缀合物或融合蛋白,所述pam分别具有序列5
’‑
ttn、5
’‑
yyn、5
’‑
yyn、5
’‑
yyn、5
’‑
yyn和5
’‑
ttn。
33.在第十一方面,本发明提供了一种试剂盒,所述试剂盒包括:cas12蛋白、本发明第一方面的缀合物或本发明第二方面的融合蛋白与本发明第三方面的单链向导rna,本发明第四方面和第五方面的分离的核酸分子,本发明第六方面和第七方面的载体,或者本发明第八方面的crispr/cas12基因编辑系统;以及如何对细胞内或体外环境中的靶序列进行基因编辑的说明书;其中,所述cas12蛋白为分别具有seq id no:1至seq id no:6所示氨基酸序列的cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白或chcas12b蛋白,或者为具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%序列同一性并且保留其生物学活性的氨基酸序列。
34.本课题组开发了可在真核细胞环境高效进行基因编辑的cas12j
‑
8编辑工具。该cas12j
‑
8蛋白具有较少数量的氨基酸,特别是具有目前可用于真核基因编辑器中最少数量的氨基酸,因此可有效地包装到表达载体例如腺相关病毒载体中。并且,该蛋白具有特异性高、pam简单的特性,而且蛋白分子量小可轻易被腺相关病毒等载体工具包装,非常适合后期作为基因治疗工具的开发。
35.此外,cas12j
‑
8蛋白的pam为ttn,pam简单,编辑范围广泛。而且,经过我们的实验证明,cas12j
‑
8蛋白在随机位点的编辑效率较fncas12a蛋白具有显著性优势,在真核环境下的基因编辑能力强。相较于同系列的cas12j
‑
2蛋白,cas12j
‑
8具有极为显著的编辑优势,
随机位点上的编辑能力显著高于cas12j
‑
2,更适合进行基因编辑的开发和应用研究。
36.本发明的cas12a蛋白及cas12b蛋白现有的其他cas12a蛋白及cas12b蛋白具有较高的编辑活性,特异性较高,且具有较为简单的pam序列,同时本发明的cas12a蛋白及cas12b蛋白的pam为yyn,拓展了cas12a蛋白及cas12b蛋白的领域,及增加了cas12a蛋白和cas12b蛋白的应用范围。
附图说明
37.图1示出crispr/cas12j
‑
8基因编辑系统对两个靶位点进行基因编辑后的编辑效率结果的示意图;
38.图2示出crispr/chcas12b基因编辑系统对两个靶位点进行基因编辑后的编辑效率结果的示意图;
39.图3示出crispr/mb4cas12a基因编辑系统对两个靶位点进行基因编辑后的编辑效率结果的示意图;
40.图4示出crispr/mocas12a基因编辑系统对两个靶位点进行基因编辑后的编辑效率结果的示意图;
41.图5示出crispr/bgcas12a基因编辑系统对两个靶位点进行基因编辑后的编辑效率结果的示意图;
42.图6示出crispr/micas12a基因编辑系统对两个靶位点进行基因编辑后的编辑效率结果的示意图;
43.图7和图8示出crispr/cas12j
‑
8基因编辑系统在gfp报告系统hek293t细胞系中的特异性检测结果的示意图;
44.图9示出crispr/chcas12b基因编辑系统在gfp报告系统hek293t细胞系中的特异性检测结果的示意图;
45.图10示出crispr/mb4cas12a基因编辑系统在gfp报告系统hek293t细胞系中的特异性检测结果的示意图;
46.图11示出crispr/mocas12a基因编辑系统在gfp报告系统hek293t细胞系中的特异性检测结果的示意图;
47.图12示出crispr/bgcas12a基因编辑系统在gfp报告系统hek293t细胞系中的特异性检测结果的示意图;
48.图13示出crispr/micas12a基因编辑系统在gfp报告系统hek293t细胞系中的特异性检测结果的示意图;
49.图14示出cas12j
‑
8abe碱基编辑器对各内源位点靶位点进行编辑的结果图。
50.图15示出利用gfp报告细胞系文库检测crispr/cas系统对靶基因进行编辑的示意图。
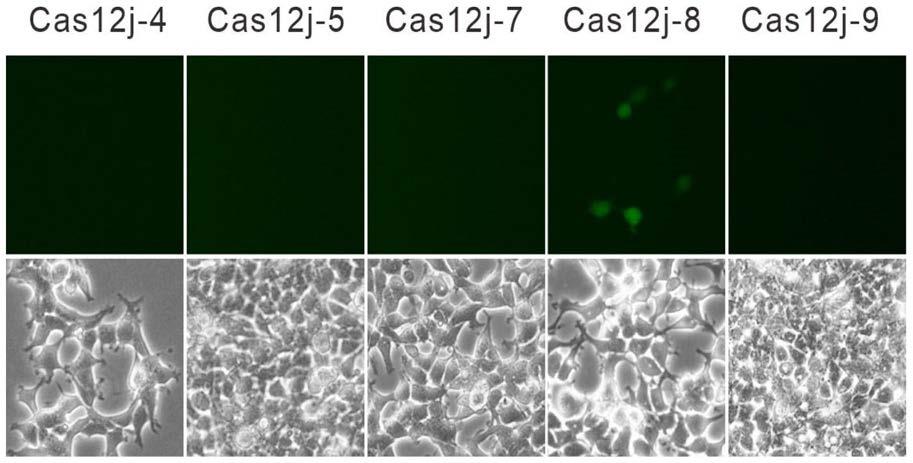
51.图16示出使用几个crispr/cas12j基因编辑系统对gfp报告细胞系进行处理后的细胞照片,其中上图为荧光图像,下图为普通显微图像。
具体实施方式
52.下面将进一步对本发明进行详细的描述。应理解,上文的发明内容部分以及下文
的详细描述仅为具体阐释本发明之目的,无意于以任何方式对本发明进行限制。本发明的保护范围由随附的权利要求书确定。在不背离本发明的精神和主旨的情况下,本领域技术人与可以对各具体实施方式进行更改。
53.定义
54.除非另有说明,否则本技术中使用的科学和技术名词具有本领域技术人员所通常理解的含义。为了更好地理解本发明,下面提供相关术语的定义和解释。
55.本文中使用的术语“cas12蛋白”、“cas12”和“cas”在本技术中可互换使用,指包括cas12蛋白或其功能活性片段在内的rna指导的核酸酶。cas12蛋白是crispr/cas12基因组编辑系统的蛋白组分,能在单链向导rna(grna)的指导下靶向并切割dna靶序列,形成dna双链断裂(dsb)。dna双链断裂能够激活细胞内固有的修复机制非同源末端连接(non
‑
homologousendjoining,nhej)和同源重组(homologous recombination,hr),由此对细胞中的dna损伤进行修复。在修复过程中,对该特定的dna序列进行定点编辑。
56.本文中使用的术语“单链向导rna”、“sgrna(single guided rna)”在本技术中可互换使用并且具有本领域技术人员通常理解的含义。一般而言,单链向导rna或者sgrna可以包含crispr重复序列(repeat sequence)和向导序列(guide sequence),向导序列在本文中也称为向导rna(guide rna或grna)。在内源性crispr系统背景下,向导序列也称为间隔序列(spacer)。在某些情况下,向导序列是与靶序列具有足够相似性从而与所述靶序列杂交并引导crispr/cas12复合物与所述靶序列的特异性结合的任何多核苷酸序列。在某些实施方案中,当最佳比对时,向导序列与其相应靶序列之间的互补程度为至少50%、至少60%、至少70%、至少80%、至少90%、至少95%、或至少99%。确定最佳比对在本领域的普通技术人员的能力范围内。例如,存在公开和可商购的比对算法和程序,诸如但不限于clustalw、matlab中的史密斯
‑
沃特曼算法(smith
‑
waterman)、bowtie、geneious、biopython以及seqman。
57.本文中所使用的术语“crispr/cas12复合物”是指单链向导rna(single guide rna)或成熟crrna与cas12蛋白结合所形成的复合体,其包含与靶序列杂交并由此使cas12蛋白与所述靶序列结合的向导序列。该复合体能够识别并切割能与该单链向导rna或成熟crrna杂交的多核苷酸。
58.因此,在形成crispr/cas12复合物的情况下,“靶序列”是指被设计为具有靶向性的向导序列所靶向的多核苷酸,例如与该向导序列具有互补性的序列,其中靶序列与向导序列之间的杂交将促进cas12发挥其活性,例如切割靶序列的活性。完全互补性不是必需的,只要存在足够互补性以引起杂交并且促进cas12发挥其活性即可。靶序列可以包括任何多核苷酸,如dna或rna。在某些情况下,所述靶序列位于细胞的细胞核或细胞质中。在某些情况下,该靶序列可位于真核细胞的一个细胞器例如线粒体或叶绿体内。
59.在本文中使用的术语“靶序列”或“靶多核苷酸”可以是对细胞(例如,真核细胞)而言任何内源或外源的多核苷酸。例如,该靶多核苷酸可以是一种存在于真核细胞的细胞核中的多核苷酸。该靶多核苷酸可以是一个编码基因产物(例如,蛋白质)的序列或一个非编码序列(例如,调节多核苷酸或无用dna)。在某些情况下,该靶序列应该与原间隔序列临近基序(pam)相关。对pam的精确序列和长度要求根据使用的cas蛋白而不同,但是pam典型地是临近原间隔序列(靶序列)的2
‑
5个碱基序列。本领域技术人员能够鉴定与给定的cas蛋白
一起使用的pam序列。
60.本文中使用的术语“多核苷酸”、“核酸序列”、“核苷酸序列”或“核酸片段”可互换使用并且是单链或双链rna或dna聚合物,任选地可含有合成的、非天然的或改变的核苷酸碱基。核苷酸通过如下它们的单个字母名称来指代:“a”为腺苷或脱氧腺苷(分别对应rna或dna),“c”表示胞苷或脱氧胞苷,“g”表示鸟苷或脱氧鸟苷,“u”表示尿苷,“t”表示脱氧胸苷,“r”表示嘌呤(a或g),“y”表示嘧啶(c或t),“k”表示g或t,“h”表示a或c或t,“i”表示肌苷,并且“n”表示任何核苷酸。
61.本文中使用的术语“多肽”、“肽”、和“蛋白(质)”在本技术中可互换使用,指氨基酸残基的聚合物。该术语适用于其中一个或多个氨基酸残基是相应的天然存在的氨基酸的人工化学类似物的氨基酸聚合物,并且适用于天然存在的氨基酸聚合物。术语“多肽”、“肽”、“氨基酸序列”和“蛋白质”还可包括修饰形式,包括但不限于糖基化、脂质连接、硫酸盐化、谷氨酸残基的γ羧化、羟化和adp
‑
核糖基化。
62.本文中使用的术语序列“同一性”或者“同源性”具有本领域公认的含义,并且可以利用公开的技术计算两个核酸或多肽分子或区域之间序列同一性的百分比。可以沿着多核苷酸或多肽的全长或者沿着该分子的区域测量序列同一性。(参见,例如computational molecular biology,lesk,a.m.,ed.,oxford university press,new york,1988;biocomputing:informatics and genome projects,smith,d.w.,ed.,academic press,new york,1993;computer analysis of sequence data,part i,griffin,a.m.,and griffin,h.g.,eds.,humana press,new jersey,1994;sequence analysis in molecular biology,von heinje,g,academic press,1987;and sequence analysis primer,gribskov,m.and devereux,j.,eds.,m stockton press,new york,1991)。虽然存在许多测量两个多核苷酸或多肽之间的同一性的方法,但是术语“同一性”是技术人员公知的在肽或蛋白中适合于保守型氨基酸置换的,并且一般可以进行而不改变所得分子的生物活性。通常,本领域技术人员认识到多肽的非必需区中的单个氨基酸置换基本上不改变生物活性(参见例如watson et al.,molecular biology of the gene,4th edition,1987,the benjamin/cummings pub.co.,p.224)。
63.本文中所使用的术语“载体”是指可将多聚核苷酸插入其中的一种核酸运载工具。当载体能使插入的多核苷酸编码的蛋白获得表达时,或者当载体能使得插入的多核苷酸发生转录(例如转录生成mrna或功能rna)时,载体称为表达载体。载体可以通过转化、转导或者转染而被导入宿主细胞,使其携带的遗传物质元件在宿主细胞中获得表达。载体是本领域技术人员公知的,包括但不限于:质粒载体、病毒载体等。载体还可以含有多种调控表达的调控序列。“调控序列”和“调控元件”在本文中可互换使用,指位于编码序列的上游(5
′
非编码序列)、中间或下游(3
′
非编码序列)、并且影响相关编码序列的转录、rna加工或稳定性或者翻译的核苷酸序列。调控序列可以包括但不限于启动子序列、转录起始序列、增强子序列、选择元件及报告基因等。所述调控序列可以是不同来源的,也可以是相同来源但以不同于通常天然存在的方式排列的。另外,载体还可含有复制起始位点。
64.本文中使用的术语“启动子”指能够控制另一核酸片段转录的核酸片段。在本发明的一些实施方案中,启动子是能够控制细胞中基因转录的启动子,无论其是否来源于所述细胞。启动子可以是组成型启动子或组织特异性启动子或发育调控启动子或诱导型启动
子。
65.本文中使用的术语“组成型启动子”指一般将引起基因在多数细胞类型中在多数情况下表达的启动子。“组织特异性启动子”和“组织优选启动子”可互换使用,并且指主要但非必须专一地在一种组织或器官中表达,而且也可在一种特定细胞或细胞型中表达的启动子。“发育调控启动子”指其活性由发育事件决定的启动子。“诱导型启动子”响应内源性或外源性刺激(环境、激素、化学信号等)而选择性表达可操纵连接的dna序列。
66.将核酸分子(例如质粒、线性核酸片段、rna等)或蛋白质“导入”生物体是指用所述核酸或蛋白质转化生物体细胞,使得所述核酸或蛋白质在细胞中能够发挥功能。本发明所用的“转化”包括稳定转化和瞬时转化。
67.本文中使用的术语“稳定转化”指将外源核苷酸序列导入基因组中,导致外源基因稳定遗传。一旦稳定转化,外源核酸序列稳定地整合进所述生物体和其任何连续世代的基因组中。
68.本文中使用的术语“瞬时转化”指将核酸分子或蛋白质导入细胞中,执行功能而没有外源基因稳定遗传。瞬时转化中,外源核酸序列不整合进基因组中。
69.本文中使用的术语“互补性”是指一个核酸序列与另一个核酸序列借助于传统的沃森
‑
克里克或其他非传统类型形成一个或多个氢键的能力。互补百分比表示一个核酸分子中可与另一个核酸序列形成氢键(例如,沃森
‑
克里克碱基配对)的残基的百分比(例如,10个之中有5、6、7、8、9、10个互补,则互补百分比为50%、60%、70%、80%、90%和100%)。“完全互补”表示一个核酸序列的所有连续残基与另一个核酸序列中的相同数目的连续残基均形成氢键。如本文使用的“基本上互补”是指在一个具有8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50个或更多个核苷酸的区域上至少为60%、65%、70%、75%、80%、85%、90%、95%、97%、98%、99%或100%的互补程度,或者是指在严格条件下杂交的两个核酸。
70.本文中使用的与杂交相关的术语“严格条件”是指与靶序列具有互补性的一个核酸主要地与该靶序列杂交并且基本上不杂交到非靶序列上的条件。严格条件通常是序列依赖性的,并且取决于许多因素。一般而言,该序列越长,则该序列特异性地杂交到其靶序列上的温度就越高。严格条件的非限制性实例描述于蒂森(tijssen)(1993)的《生物化学和分子生物学中的实验室技术
‑
核酸探针杂交》(laboratory techniques in biochemistry and molecular biology
‑
hybridization with nucleic acid probes),第1部分,第二章,“杂交原理概述和核酸探针分析策略”(“overview of principles of hybridization andthe strategy of nucleic acid probe assay”),爱思唯尔(elsevier),纽约。
71.本文中使用的术语“杂交”是指其中一个或多个多核苷酸反应形成一种复合物的反应,该复合物经由这些核苷酸残基之间的碱基的氢键键合而稳定化。氢键键合可以借助于沃森
‑
克里克碱基配对、hoogstein结合或以任何其他序列特异性方式而发生。该复合物可包含形成一个双链体的两条链、形成多链复合物的三条或多条链、单个自我杂交链、或这些的任何组合。杂交反应可以构成一个更广泛的过程(如pcr的开始、或经由一种酶的多核苷酸的切割)中的一个步骤。能够与一个给定序列杂交的序列被称为该给定序列的“互补物”。
72.衍生化蛋白
73.可以对cas12蛋白进行衍生化,例如将其连接至另外的分子(例如另外的蛋白或多肽)。通常,蛋白的衍生化(例如标记)不会不利影响该蛋白的期望活性(例如,起与单链向导rna结合的活性、核酸内切酶活性、在向导rna引导下与靶序列特定位点结合并切割的活性)。因此,在本发明中,可以将cas12蛋白功能性连接(通过化学偶合、基因融合、非共价连接或其它方式)于一个或多个其它分子部分,例如另外的蛋白或多肽、可检测标记、药用试剂等。
74.特别地,可以将cas12蛋白连接其他功能性单元。例如,可以将其与核定位信号(nls)序列连接,以提高本发明的蛋白进入细胞核的能力。例如,可以将其与靶向部分连接,以使得cas12蛋白具有靶向性。例如,可以将其与可检测标记连接,以便于对cas12蛋白进行检测。例如,可以将其与表位标签连接,以便于对cas12蛋白的表达、检测、示踪和/或纯化。
75.因此,在第一方面,本发明提供了一种缀合物,所述缀合物包含:
76.a)cas12蛋白,所述cas12蛋白为:
77.1)具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白,
78.具有seq id no:2所示氨基酸序列的mb4cas12a蛋白,
79.具有seq id no:3所示氨基酸序列的mlcas12a蛋白,
80.具有seq id no:4所示氨基酸序列的mocas12a蛋白,
81.具有seq id no:5所示氨基酸序列的bgcas12a蛋白,或
82.具有seq id no:6所示氨基酸序列的chcas12b蛋白,
83.或者为
84.2)具有seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%、至少81%、至少82%、至少83%、至少84%、至少85%、至少86%、至少87%、至少88%、至少89%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.1%、至少99.2%、至少99.3%、至少99.4%、至少99.5%、至少99.6%、至少99.7%、至少99.8%、至少99.9%、至少99.95%、至少99.99%、至少99.999%、至少100%、或者80%
‑
100%中任一百分比的序列同一性并且保留其生物学活性的氨基酸序列的同源物;
85.b)修饰部分;以及
86.c)任选的用于连接所述cas12蛋白与所述修饰部分的接头。
87.在本发明中,所谓cas12蛋白的“生物学活性”是指该蛋白与单链向导rna结合的活性、核酸内切酶活性(包括单链切割活性和双链切割活性)、和/或在向导rna(grna)引导下与靶序列特定位点结合并切割的活性,但不限于此。
88.可以理解,除了cas12蛋白自身外,还可以使cas12蛋白与其他物质例如其他蛋白或者可标记标签等结合从而赋予其他的功能性。
89.因此,在一个实施方案中,所述修饰部分可以为另外的蛋白或多肽、可检测标记或其组合。
90.在一个进一步的实施方案中,所述另外的蛋白或多肽选自表位标签、报告蛋白或核定位信号(nls)序列、胞嘧啶脱氨酶(cbe)、腺嘌呤脱氨酶(abe)、胞嘧啶甲基化酶dnmt3a和mq1、胞嘧啶去甲基化酶tet1、转录激活蛋白vp64、p65和rta、转录抑制蛋白krab、组蛋白乙酰化酶p300、组蛋白去乙酰化酶lsd1、和内切酶foki中的一种或者多种。
91.表位标签是本领域技术人员熟知的,其实例包括但不限于his、v5、flag、ha、myc、vsv
‑
g、trx等,并且本领域技术人员已知如何根据期望目的(例如,纯化、检测或示踪)选择合适的表位标签。
92.报告蛋白是本领域技术人员熟知的,其实例包括但不限于gst、hrp、cat、gfp、hcred、dsred、cfp、yfp、bfp等。
93.可检测标记是本领域技术人员熟知的,其实例包括荧光染料,例如异硫氰酸荧光素(fitc)或dapi。
94.本发明的cas12蛋白可以通过接头与所述修饰部分偶联、缀合或融合,也可以不通过接头而直接地与所述修饰部分连接。接头是本领域熟知的,其实例可以包括但不限于包含1
‑
50个氨基酸(如glu或ser)或氨基酸衍生物(如ahx、β
‑
ala、gaba或ava)的接头、或peg等。
95.在第二方面,本发明提供了一种融合蛋白,所述融合蛋白包含:
96.a)cas12蛋白,所述cas12蛋白为:
97.1)具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白,
98.具有seq id no:2所示氨基酸序列的mb4cas12a蛋白,
99.具有seq id no:3所示氨基酸序列的mlcas12a蛋白,
100.具有seq id no:4所示氨基酸序列的mocas12a蛋白,
101.具有seq id no:5所示氨基酸序列的bgcas12a蛋白,
102.或
103.具有seq id no:6所示氨基酸序列的chcas12b蛋白,
104.或者为
105.2)具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%、至少81%、至少82%、至少83%、至少84%、至少85%、至少86%、至少87%、至少88%、至少89%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.1%、至少99.2%、至少99.3%、至少99.4%、至少99.5%、至少99.6%、至少99.7%、至少99.8%、至少99.9%、至少99.95%、至少99.99%、至少99.999%、至少100%、或者80%
‑
100%中任一百分比的序列同一性并且保留其生物学活性的氨基酸序列的同源物;
106.b)另外的蛋白或多肽、以及
107.c)任选的用于连接所述cas12蛋白与所述另外的蛋白或多肽的接头。
108.同本发明第一方面一样,所述另外的蛋白或多肽可以选自表位标签、报告蛋白或核定位信号(nls)序列、胞嘧啶脱氨酶(cbe)、腺嘌呤脱氨酶(abe)、胞嘧啶甲基化酶dnmt3a和mq1、胞嘧啶去甲基化酶tet1、转录激活蛋白vp64、p65和rta、转录抑制蛋白krab、组蛋白乙酰化酶p300、组蛋白去乙酰化酶lsd1、和内切酶foki中的一种或者多种。
109.表位标签是本领域技术人员熟知的,其实例包括但不限于his、v5、flag、ha、myc、vsv
‑
g、trx等,并且本领域技术人员已知如何根据期望目的(例如,纯化、检测或示踪)选择合适的表位标签。报告蛋白是本领域技术人员熟知的,其实例包括但不限于gst、hrp、cat、gfp、hcred、dsred、cfp、yfp、bfp等。
110.报告蛋白是本领域技术人员熟知的,其实例包括但不限于gst、hrp、cat、gfp、
hcred、dsred、cfp、yfp、bfp等。
111.可检测标记是本领域技术人员熟知的,其实例包括荧光染料,例如异硫氰酸荧光素(fitc)或dapi。
112.本发明的cas12蛋白可以通过接头与所述另外的蛋白或多肽偶联、缀合或融合,也可以不通过接头而直接地与所述另外的蛋白或多肽连接。接头是本领域熟知的,其实例包括但不限于包含1
‑
50个氨基酸(如glu或ser)或氨基酸衍生物(如ahx、β
‑
ala、gaba或ava)的接头、或peg等。
113.在一个优选的实施方案中,所述融合蛋白包含:具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、腺嘌呤脱氨酶(abe)、以及任选的连接所述cas12j
‑
8蛋白和所述腺嘌呤脱氨酶(abe)的接头。
114.在一个优选的实施方案中,所述融合蛋白从其n端到c端依次为所述腺嘌呤脱氨酶(abe)、所述接头、以及所述cas12j
‑
8蛋白。
115.在一个更优选的实施方案中,所述融合蛋白的氨基酸序列为seq id no:7所示。
116.本课题组开发了可在真核细胞环境高效进行基因编辑的cas12j
‑
8编辑工具。该cas12j
‑
8蛋白具有较少数量的氨基酸,特别是具有目前可用于真核基因编辑器中最少数量的氨基酸,因此可有效地包装到表达载体例如腺相关病毒载体中。并且,该蛋白具有特异性高、pam简单的特性,而且蛋白分子量小可轻易被腺相关病毒等载体工具包装,非常适合后期作为基因治疗工具的开发。
117.此外,cas12j
‑
8蛋白的pam为ttn,pam简单,编辑范围广泛。而且,经过我们的实验证明,cas12j
‑
8蛋白在随机位点的编辑效率较fncas12a蛋白具有显著性优势,在真核环境下的基因编辑能力强。相较于同系列的cas12j
‑
2蛋白,cas12j
‑
8蛋白具有极为显著的编辑优势,随机位点上的编辑能力显著高于cas12j
‑
2蛋白,更适合进行基因编辑的开发和应用研究。
118.本发明的cas12a蛋白及cas12b蛋白相较于现有的其他cas12a蛋白及cas12b蛋白具有较高的编辑活性,特异性较高,且具有较为简单的pam序列,同时cas12a蛋白及cas12b蛋白的pam为yyn,拓展了cas12a蛋白及cas12b蛋白的领域,及增加了cas12a蛋白和cas12b蛋白的应用范围。
119.单链向导rna
120.在第三方面,本发明提供了一种单链向导rna,所述单链向导rna包括crispr重复序列,所述crispr重复序列具有:
121.a)针对cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白的seq id no:15所示的核酸序列,
122.针对mb4cas12a蛋白、m1cas12a蛋白和mocas12a蛋白、其同源物、缀合物或融合蛋白的seq id no:16所示的核酸序列,
123.针对bgcas12a蛋白、其同源物、缀合物或融合蛋白的seq id no:17所示的核酸序列,或
124.针对chcas12b蛋白、其同源物、缀合物或融合蛋白的seq id no:18所示的核酸序列;
125.或者
126.b)与seq id no:15至seq id no:18中任一个所示的核酸序列至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.9%或者至少100%的序列同一性且保留其生物学活性的核酸序列;或者
127.c)基于seq id no:15至seq id no:18中任一个所述的核酸序列改造得到的且保留其生物学活性的核酸序列。
128.在一个实施方案中,所述改造可以为碱基磷酸化、碱基硫化、碱基甲基化、碱基羟基化、序列的缩短和序列的加长中的一种或者多种。
129.在一个进一步的实施方案中,所述序列的缩短和所述序列加长包括相对于基础序列存在一个、两个、三个、四个、五个、六个、七个、八个、九个或者十个碱基的缺失或者添加。
130.在又一个实施方案中,所述单链向导rna可以在所述crispr重复序列的3’端进一步包括crispr间隔序列,所述crispr间隔序列为长度为20、21、22、23、24、25、26、27、28、29、30个核苷酸(优选24个核苷酸)且能够与靶序列互补配对的序列。
131.在一个优选的实施方案中,所述crispr间隔序列为长度为24个核苷酸且能够与靶序列互补配对的序列。
132.在一个进一步的实施方案中,所述单链向导rna在所述间隔序列的3’端进一步包括终止子。作为示例,所述终止子可以为多个如至少六个(例如七个或者八个)u构成的终止子。
133.所述单链向导rna能够与上述的cas12蛋白、缀合物或者融合蛋白结合而形成复合物,该复合物可以识别相应的pam并由此与靶序列结合,进而实现对靶序列的剪切或者说基因编辑。
134.编码核酸以及载体
135.在第四方面,本发明提供了一种分离的核酸分子,所述分离的核酸分子包含编码以下的核酸序列:
136.a)cas12蛋白,所述cas12蛋白为:
137.1)具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白,
138.具有seq id no:2所示氨基酸序列的mb4cas12a蛋白,
139.具有seq id no:3所示氨基酸序列的mlcas12a蛋白,
140.具有seq id no:4所示氨基酸序列的mocas12a蛋白,
141.具有seq id no:5所示氨基酸序列的bgcas12a蛋白,
142.或
143.具有seq id no:6所示氨基酸序列的chcas12b蛋白,
144.或者为
145.2)具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%、至少81%、至少82%、至少83%、至少84%、至少85%、至少86%、至少87%、至少88%、至少89%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.1%、至少99.2%、至少99.3%、至少99.4%、至少99.5%、至少99.6%、至少99.7%、至少99.8%、至少99.9%、至少99.95%、至少99.99%、至少99.999%、至少100%、或者80%
‑
100%中任一百分比的序列同一性并且保留其生物学活性的氨基酸序列的同源物;
146.b)本发明第一方面的缀合物;或者
147.c)本发明第二方面的融合蛋白。
148.在一个实施方案中,所述分离的核酸分子包含seq id no:8、seq id no:9、seq id no:10、seq id no:11、seq id no:12、seq id no:13中任一个所示的核酸序列或其简并序列。
149.在一个实施方案中,所述分离的核酸分子包含编码seq id no:7所示融合蛋白的核酸序列。
150.在一个优选的实施方案中,所述分离的核酸分子包含seq id no:14所示的核酸序列或其简并序列。
151.在一个进一步的实施方案中,所述分离的核酸分子还编码本发明第三方面的与所述cas12蛋白对应的单链向导rna。
152.作为一个示例,所述分离的核酸分子包含编码具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白(例如seq id no:7所示的融合蛋白)的核酸序列,例如seq id no:8、或seq id no:14所示的核酸序列,并且包含编码针对该cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白的包含seq id no:15所示crispr重复序列、包含与seq id no:15具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:15改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:19所示的核酸序列。
153.作为一个示例,所述分离的核酸分子包含编码具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列,例如seq id no:9、seq id no:10或seq id no:11所示的核酸序列,并且包含编码针对该cas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:16所示crispr重复序列、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:20所示的核酸序列。
154.作为一个示例,所述分离的核酸分子包含编码具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列,例如seq id no:12所示的核酸序列,并且包含编码针对该bgcas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:17所示crispr重复序列、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:21所示的核酸序列。
155.作为一个示例,所述分离的核酸分子包含编码具有seq id no:6所示氨基酸序列的chcas12b蛋白、其同源物、缀合物或融合蛋白的核酸序列,例如seq id no:13所示的核酸序列,并且包含编码针对该chcas12b蛋白、其同源物、缀合物或融合蛋白的包含seq id no:18所示crispr重复序列、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:22所示的核酸序列。
156.在第五方面,本发明提供了一种分离的核酸分子,所述分离的核酸分子编码本发明第三方面的单链向导rna。
157.在一个实施方案中,所述分离的核酸分子包含seq id no:19、seq id no:20、seq id no:21、和seq id no:22中任一个所示的核酸序列或其简并序列。
158.在一个优选的实施方案中,所述分离的核酸分子还包含编码crispr间隔序列的核酸序列。
159.在利用本领域已知的某些工具例如表达载体将本发明的分离的核酸分子转染到相应的细胞中后,本发明的分离的核酸分子可以表达出本发明上文所述的cas12蛋白、其缀合物或融合蛋白、和/或上文所述的单链向导rna,并在此行使相应的功能,例如进行基因编辑。
160.另外,本发明的分离的核酸分子可以单独地/分别地表达cas12蛋白、其缀合物或融合蛋白、以及单链向导rna,也可以一体地表达所述的表达产物,选择何种表达方式根据具体情况而定。
161.再者,所述表达产物具有上文记载的相应作用和/或功能,为简洁起见在此不再赘述。
162.在第六方面,本发明提供了一种载体,其包含编码以下的核酸序列:
163.a)cas12蛋白,所述cas12蛋白为:
164.1)具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白,
165.具有seq id no:2所示氨基酸序列的mb4cas12a蛋白,
166.具有seq id no:3所示氨基酸序列的mlcas12a蛋白,
167.具有seq id no:4所示氨基酸序列的mocas12a蛋白,
168.具有seq id no:5所示氨基酸序列的bgcas12a蛋白,
169.或
170.具有seq id no:6所示氨基酸序列的chcas12b蛋白,
171.或者为
172.2)具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%、至少81%、至少82%、至少83%、至少84%、至少85%、至少86%、至少87%、至少88%、至少89%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.1%、至少99.2%、至少99.3%、至少99.4%、至少99.5%、至少99.6%、至少99.7%、至少99.8%、至少99.9%、至少99.95%、至少99.99%、至少99.999%、至少100%、或者80%
‑
100%中任一百分比的序列同一性并且保留其生物学活性的氨基酸序列的同源物;
173.b)本发明第一方面的缀合物;或者
174.c)本发明第二方面的融合蛋白。
175.在一个实施方案中,所述载体包含seq id no:8、seq id no:9、seq id no:10、seq id no:11、seq id no:12、seq id no:13中任一个所示的核酸序列或其简并序列。
176.在一个实施方案中,所述载体包含编码seq id no:7所示融合蛋白的核酸序列。
177.在一个优选的实施方案中,所述载体包含seq id no:14所示的核酸序列或其简并序列。
178.所述载体可以为表达载体,例如质粒载体例如puc19载体、附着体载体、paav2_itr载体、逆转录病毒载体、慢病毒载体、腺病毒载体或腺相关病毒载体。
179.在又一个实施方案中,所述载体进一步包含编码本发明第三方面的与所述cas12蛋白对应的单链向导rna的核酸序列。
180.作为一个示例,所述载体包含编码具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白(例如seq id no:7所示的融合蛋白)的核酸序列,例如seq id no:8或seq id no:14所示的核酸序列,并且包含编码针对该cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白的包含seq id no:15所示crispr重复序列、包含与seq id no:15具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq td no:15改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:19所示的核酸序列。
181.作为一个示例,所述载体包含编码具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列,例如seq id no:9、seq id no:10或seq id no:11所示的核酸序列,并且包含编码针对该cas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:16所示crispr重复序列、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:20所示的核酸序列。
182.作为一个示例,所述载体包含编码具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列,例如seq id no:12所示的核酸序列,并且包含编码针对该bgcas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:17所示crispr重复序列、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:21所示的核酸序列。
183.作为一个示例,所述载体包含编码具有seq id no:6所示氨基酸序列的chcas12b蛋白、其同源物、缀合物或融合蛋白的核酸序列,例如seq id no:13所示的核酸序列,并且包含编码针对该chcas12b蛋白、其同源物、缀合物或融合蛋白的包含seq id no:18所示crispr重复序列、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列,例如seq id no:22所示的核酸序列。
184.在第七方面,本发明提供了一种载体,所述载体包含编码本发明第三方面的单链向导rna的核酸分子。
185.在一个实施方案中,所述载体包含seq id no:19、seq id no:20、seq id no:21和seq id no:22中任一个所示的核酸序列或其简并序列。
186.在一个优选的实施方案中,所述载体还包含编码crispr间隔序列的核酸序列。
187.根据上文的记载可知,在将本发明的载体转染到细胞中后,在载体中克隆的核酸序列可以被表达为cas12蛋白、其缀合物或融合蛋白、和/或上文所述的单链向导rna,并在此行使相应的功能,例如进行基因编辑。
188.另外,可以将多种载体例如两种载体转染到细胞中,其中一种载体表达所述cas12蛋白、其缀合物或融合蛋白,而另一种载体表达单链向导rna。随后,表达出来的cas12蛋白、其缀合物或融合蛋白与表达出来的单链向导rna复合形成复合物,并在此行使相应的功能,
例如进行基因编辑。
189.当然,也可以将编码所述cas12蛋白、其缀合物或融合蛋白的核酸序列以及编码所述单链向导rna的核酸序列克隆到一个载体中,使得该载体转染到细胞内后表达所述cas12蛋白、其缀合物或融合蛋白以及所述单链向导rna两者,并在此行使相应的功能,例如进行基因编辑。
190.crispr/cas12基因编辑系统
191.在第八方面,本发明提供了一种crispr/cas12基因编辑系统,其包含:
192.a)蛋白组分,其包含:
193.1)cas12蛋白,所述cas12蛋白为:
194.1.1)具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白,
195.具有seq id no:2所示氨基酸序列的mb4cas12a蛋白,
196.具有seq id no:3所示氨基酸序列的mlcas12a蛋白,
197.具有seq id no:4所示氨基酸序列的mocas12a蛋白,
198.具有seq id no:5所示氨基酸序列的bgcas12a蛋白,或
199.具有seq id no:6所示氨基酸序列的chcas12b蛋白,
200.或者为
201.1.2)具有与seq id no:1、seq id no:2、seq id no:3、seq td no:4、seq td no:5和seq id no:6中任一个所示的氨基酸序列至少80%、至少81%、至少82%、至少83%、至少84%、至少85%、至少86%、至少87%、至少88%、至少89%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.1%、至少99.2%、至少99.3%、至少99.4%、至少99.5%、至少99.6%、至少99.7%、至少99.8%、至少99.9%、至少99.95%、至少99.99%、至少99.999%、至少100%、或者80%
‑
100%中任一百分比的序列同一性并且保留其生物学活性的氨基酸序列的同源物;
202.2)本发明第一方面的缀合物;或者
203.3)本发明第二方面的融合蛋白;以及
204.b)核酸组分,其包含:本发明第三方面的与a)中的蛋白组分对应的单链向导rna;
205.并且,所述蛋白组分和所述核酸组分相互结合形成复合物。
206.作为一个示例,所述蛋白组分包含具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白,所述核酸组分包含单链向导rna,所述单链向导rna为包含seq id no:15所示crispr重复序列的单链向导rna、包含与seq id no:15具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:15改造得到的且保留其生物学活性的改造序列的单链向导rna。
207.作为一个示例,所述蛋白组分包含具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其同源物、缀合物或融合蛋白,所述核酸组分包含单链向导rna,所述单链向导rna为包含seq id no:16所示crispr重复序列的单链向导rna、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna。
208.作为一个示例,所述蛋白组分包含具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其同源物、缀合物或融合蛋白,所述核酸组分包含单链向导rna,所述单链向导rna为
包含seq id no:17所示crispr重复序列的单链向导rna、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna。
209.作为一个示例,所述蛋白组分包含具有seq id no:6所示氨基酸序列的chcas12b蛋白、其同源物、缀合物或融合蛋白,所述核酸组分包含单链向导rna,所述单链向导rna为包含seq id no:18所示crispr重复序列的单链向导rna、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna。
210.上文中,针对单链向导rna提及的表述“至少90%序列同一性”可以为例如至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.9%或者至少100%的序列同一性。
211.本发明的crispr/cas12基因编辑系统可以由本文所述的cas12蛋白、其同源物、或者它们的缀合物或融合蛋白与本文所述的单链向导rna直接地构成,也可以本文所述的载体表达得到的表达产物构成。
212.本发明的crispr/cas12基因编辑系统通过其中包含的cas12蛋白和单链向导rna共同作用而实现对靶序列的识别、定位、切割和基因编辑。
213.本发明crispr/cas12基因编辑系统能够精确定位靶序列。所谓“精确定位”有两层含义:第一层含义是指本发明的crispr/cas12基因编辑系统自身能够识别并结合靶序列,第二层含义是指本发明的crispr/cas12基因编辑系统能够将与所述cas12蛋白融合的其他蛋白或特异性识别所述sgrna的蛋白带至靶序列的位置。
214.本发明的crispr/cas12基因编辑系统对非靶序列具有低容忍度。在本文中,所谓“具有低容忍度”是指本发明的crispr/cas12基因编辑系统基本上不能或者完全不能识别并结合非靶序列,或基本上不能或者完全不能将与所述cas12蛋白融合的其他蛋白或特异性识别所述sgrna的蛋白带至非靶序列的位置。
215.本发明的crispr/cas12因编辑系统,由于其中含有的cas12蛋白所识别的靶序列上的pam序列更简单,由此可以靶向基因组中更多的dna序列。
216.细胞
217.在第九方面,本发明提供了一种细胞,所述细胞包含:本发明第四发明和第五方面的分离的核酸分子、或者本发明第六方面和第七方面的载体。
218.作为一个示例,所述细胞可以为原核细胞或者真核细胞。对于所述真核细胞,作为示例,其可以为植物细胞或者动物细胞。对于所述动物细胞,作为示例,其可以为哺乳动物细胞例如人类细胞。
219.方法
220.在第十方面,本发明提供了一种对细胞内或体外环境中的靶序列进行基因编辑的方法,所述方法包括使以下(1)至(4)中任一项与细胞内或体外环境中的靶序列相接触:
221.(1)cas12蛋白、本发明第一方面的缀合物或者本发明第二方面的融合蛋白,和本发明第三方面的与所述cas12蛋白对应的单链向导rna,
222.其中,所述cas12蛋白为:
223.1)具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白,
224.具有seq id no:2所示氨基酸序列的mb4cas12a蛋白,
225.具有seq id no:3所示氨基酸序列的mlcas12a蛋白,
226.具有seq id no:4所示氨基酸序列的mocas12a蛋白,
227.具有seq id no:5所示氨基酸序列的bgcas12a蛋白,或
228.具有seq id no:6所示氨基酸序列的chcas12b蛋白,
229.或者为
230.2)具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%、至少81%、至少82%、至少83%、至少84%、至少85%、至少86%、至少87%、至少88%、至少89%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.1%、至少99.2%、至少99.3%、至少99.4%、至少99.5%、至少99.6%、至少99.7%、至少99.8%、至少99.9%、至少99.95%、至少99.99%、至少99.999%、至少100%、或者80%
‑
100%中任一百分比的序列同一性并且保留其生物学活性的氨基酸序列的同源物;
231.(2)本发明第六方面和第七方面的载体;
232.(3)本发明第六方面的载体;以及
233.(4)本发明第八方面的crispr/cas12基因编辑系统;
234.其中,在与靶序列接触后,所述cas12蛋白、其同源物、缀合物或融合蛋白识别各自的原间隔邻近序列(pam),所述pam位于靶序列的5’端,并且,对于所述cas12j
‑
8蛋白、所述mb4cas12a蛋白、所述mlcas12a蛋白、所述mocas12a蛋白、所述bgcas12a蛋白、和所述chcas12b蛋白、或它们各自的同源物、缀合物或融合蛋白,所述pam分别为5
’‑
ttn、5
’‑
yyn、5
’‑
yyn、5
’‑
yyn、5
’‑
yyn和5
’‑
ttn。
235.对于上文所述的第(1)项:
236.作为一个示例,具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白,以及包含seq id no:15所示crispr重复序列、包含与seq id no:15具有至少90%序列同一性的同源序列、或包含基于seq id no:15改造得到的且保留其生物学活性的改造序列的单链向导rna;
237.作为一个示例,具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其同源物、缀合物或融合蛋白,以及包含seq id no:16所示crispr重复序列、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna;
238.作为一个示例,具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其同源物、它们的缀合物或融合蛋白的核酸序列,以及包含seq id no:17所示crispr重复序列、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna;
239.作为一个示例,具有seq id no:6所示氨基酸序列的chcas12b蛋白、其同源物、缀合物或融合蛋白,以及包含seq id no:18所示crispr重复序列、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna。
240.对于上文中的第(2)项:
241.作为一个示例,包含编码具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白(例如seq id no:7所示的融合蛋白)的核酸序列(例如seq id no:8或seq id no:14所示的核酸序列)的载体,以及包含编码针对该cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白的包含seq id no:15所示crispr重复序列、包含与seq id no:15具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:15改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:19所示的核酸序列)的载体;
242.作为一个示例,包含编码具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:9、seq id no:10或seq id no:11所示的核酸序列)的载体,以及包含编码针对该mb4cas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:16所示crispr重复序列、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:20所示的核酸序列)的载体;
243.作为一个示例,包含编码具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:12所示的核酸序列)的载体,以及包含编码针对该bgcas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:17所示crispr重复序列、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:21所示的核酸序列)的载体;
244.作为一个示例,包含编码具有seq id no:6所示氨基酸序列的chcas12b蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:13所示的核酸序列)的载体,以及包含编码针对该chcas12b蛋白、其同源物、缀合物或融合蛋白的包含seq id no:18所示crispr重复序列、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:22所示的核酸序列)的载体。
245.在一个实施方案中,所述细胞为原核细胞或者真核细胞,所述真核细胞为例如植物细胞或动物细胞,所述动物细胞为例如哺乳动物细胞如人类细胞。
246.在一个实施方案中,所述基因编辑包括对靶序列的基因敲除、定点碱基的改变、定点插入、基因转录水平的调控、dna甲基化调控、dna乙酰化修饰、组蛋白乙酰化修饰、单碱基转换以及染色质成像追踪中的一种或者多种。
247.进一步地,在一个实施方案中,所述单碱基转换包括碱基腺嘌呤到鸟嘌呤的转换、胞嘧啶到胸腺嘧啶的转换或胞嘧啶到尿嘧啶的转换。
248.在一个实施方案中,在所述方法中,所述单链向导rna的crispr间隔序列与所述靶序列形成完全碱基互补配对结构,而与非靶序列形成不完全碱基互补配对的结构。
249.在本文中,所述不完全碱基互补配对结构是指其中包括一部分碱基互补配对和一部分非碱基互补配对的结构,所述非碱基互补配对包括例如碱基错配(mismatch)和/或碱基凸出(bulge)等。
250.在一个实施方案中,所述不完全碱基互补配对结构包括一个或者多个例如两个或
者更多个碱基错配。
251.由此,本发明的cas12蛋白可以对所述靶序列上的靶位点进行切割,并且在cas12蛋白的切割作用下,靶序列发生双链断裂。进一步地,当所述方法在细胞内进行时,切割后的靶序列可以通过细胞内的非同源末端连接修复或同源重组修复途径进行修复,从而实现对靶序列的基因编辑。
252.本发明的crispr/cas12基因编辑系统以及采用该基因编辑系统的基因编辑方法,经实验发现其具有40%
‑
70%(对于cas12j
‑
8蛋白)、12%
‑
56%(对于chcas12b蛋白)以及10%
‑
20%(对于其他各cas12a蛋白)的编辑效率。另外,对于crispr/cas12j
‑
8基因编辑系统,前14bp的向导rna的mismatch具有接近0%的容错率。因此,该基因编辑系统可以高特异性地编辑靶基因,具有编辑效率高、脱靶率低的特点,可广泛应用于细胞中或者体外环境中的基因编辑。
253.试剂盒
254.在第十一方面,本发明提供了一种试剂盒,所述试剂盒用于对细胞内或者体外环境中的靶序列进行基因编辑,包括:
255.a)选自以下1)至6)中的任一项:
256.1)cas12蛋白或其同源物、本发明第一方面的缀合物、或者本发明第二方面的融合蛋白,和本发明第三方面的与所述cas12蛋白对应的单链向导rna,
257.其中,所述cas12蛋白为:
258.1.1)具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白,
259.具有seq id no:2所示氨基酸序列的mb4cas12a蛋白,
260.具有seq id no:3所示氨基酸序列的mlcas12a蛋白,
261.具有seq id no:4所示氨基酸序列的mocas12a蛋白,
262.具有seq id no:5所示氨基酸序列的bgcas12a蛋白,
263.或
264.具有seq id no:6所示氨基酸序列的chcas12b蛋白,
265.或者为
266.1.2)具有与seq id no:1、seq id no:2、seq id no:3、seq id no:4、seq id no:5和seq id no:6中任一个所示的氨基酸序列至少80%、至少81%、至少82%、至少83%、至少84%、至少85%、至少86%、至少87%、至少88%、至少89%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.1%、至少99.2%、至少99.3%、至少99.4%、至少99.5%、至少99.6%、至少99.7%、至少99.8%、至少99.9%、至少99.95%、至少99.99%、至少99.999%、至少100%、或者80%
‑
100%中任一百分比的序列同一性并且保留其生物学活性的氨基酸序列的同源物;
267.2)本发明第四和第五方面的分离的核酸分子;
268.3)本发明第五方面的分离的核酸分子;
269.4)本发明第六和第七方面的载体;
270.5)本发明第六方面的载体;或者
271.6)本发明第八方面的crispr/cas12基因编辑系统;
272.以及
273.b)如何对细胞内或体外环境中的靶序列进行基因编辑的说明书。
274.对于上文中的第1)项:
275.作为一个示例,具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白,以及包含seq id no:15所示crispr重复序列的单链向导rna、包含与seq id no:15具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:15改造得到的且保留其生物学活性的改造序列的单链向导rna;
276.作为一个示例,具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其具有与seq id no:2、seq id no:3或seq id no:4具有至少80%序列同一性的氨基酸序列的同源物、它们的缀合物或融合蛋白,以及包含seq id no:16所示crispr重复序列的单链向导rna、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna;
277.作为一个示例,具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其具有与seq id no:5具有至少80%序列同一性的氨基酸序列的同源物、它们的缀合物或融合蛋白,以及包含seq id no:17所示crispr重复序列的单链向导rna、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna;
278.作为一个示例,具有seq id no:6所示氨基酸序列的chcas12b蛋白、其具有与seq id no:6具有至少80%序列同一性的氨基酸序列的同源物、它们的缀合物或融合蛋白,以及包含seq id no:18所示crispr重复序列的单链向导rna、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列的单链向导rna、或者包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna。
279.对于上文中的第2)项:
280.作为一个示例,包含编码具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白(例如seq id no:7所示的融合蛋白)核酸序列(例如seq id no:8或seq id no:14所示的核酸序列)的分离的核酸分子,以及包含编码针对该cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白的包含seq id no:15所示crispr重复序列、包含与seq id no:15具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:15改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:19所示的核酸序列)的分离的核酸分子;
281.作为一个示例,包含编码具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列(seq id no:9、seq id no:10或seq id no:11所示的核酸序列)的分离的核酸分子,以及包含包含编码针对该cas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:16所示crispr重复序列、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:20所示的核酸序列)的分离的核酸分子;
282.作为一个示例,包含编码具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:12所示的核酸序列)的分离的核酸分
子,以及包含编码针对该bgcas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:17所示crispr重复序列、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:21所示的核酸序列)的分离的核酸分子;
283.作为一个示例,包含编码具有seq id no:6所示氨基酸序列的chcas12b蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:13所示的核酸序列)的分离的核酸分子,以及包含编码针对该chcas12b蛋白、其同源物、缀合物或融合蛋白的包含seq id no:18所示crispr重复序列、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:22所示的核酸序列)的分离的核酸分子。
284.对于上文中的第4)项:
285.作为一个示例,包含编码具有seq id no:1所示氨基酸序列的cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白(例如seq id no:7所示的融合蛋白)的核酸序列(例如seq id no:8或seq id no:14所示的核酸序列)的载体,以及包含编码针对该cas12j
‑
8蛋白、其同源物、缀合物或融合蛋白的包含seq id no:15所示crispr重复序列、包含与seq id no:15具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:15改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:19所示的核酸序列)的载体;
286.作为一个示例,包含编码具有seq id no:2、seq id no:3或seq id no:4所示氨基酸序列的cas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:9、seq id no:10或seq id no:11所示的核酸序列)的载体,以及包含编码针对该cas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:16所示crispr重复序列、包含与seq id no:16具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:16改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:20所示的核酸序列)的载体;
287.作为一个示例,包含编码具有seq id no:5所示氨基酸序列的bgcas12a蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:12所示的核酸序列)的载体,以及包含编码针对该bgcas12a蛋白、其同源物、缀合物或融合蛋白的包含seq id no:17所示crispr重复序列、包含与seq id no:17具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:17改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:21所示的核酸序列)的载体;
288.作为一个示例,包含编码具有seq id no:6所示氨基酸序列的chcas12b蛋白、其同源物、缀合物或融合蛋白的核酸序列(例如seq id no:13所示的核酸序列)的载体,以及包含编码针对该chcas12b蛋白、其同源物、缀合物或融合蛋白的包含seq id no:18所示crispr重复序列、包含与seq id no:18具有至少90%序列同一性且保留其生物学活性的同源序列、或包含基于seq id no:18改造得到的且保留其生物学活性的改造序列的单链向导rna的核酸序列(例如seq id no:22所示的核酸序列)的载体。
289.当然,本领域技术人员可以理解,本发明试剂盒中还可以包含其他有助于进行基因编辑的试剂。
290.对本发明涉及序列的简单描述
291.seq id no:1:cas12j
‑
8蛋白序列
292.seq id no:2:mb4cas12a蛋白序列
293.seq id no:3:mlcas12a蛋白序列
294.seq id no:4:mocas12a蛋白序列
295.seq id no:5:bgcas12a蛋白序列
296.seq id no:6:chcas12b蛋白序列
297.seq id no:7:包含cas12j
‑
8蛋白的融合蛋白
298.seq id no:8:cas12j
‑
8蛋白的编码序列
299.seq id no:9:mb4cas12a蛋白的编码序列
300.seq id no:10:mlcas12a蛋白的编码序列
301.seq id no:11:mocas12a蛋白的编码序列
302.seq id no:12:bgcas12a蛋白的编码序列
303.seq id no:13:chcas12b蛋白的编码序列
304.seq id no:14:包含cas12j
‑
8蛋白的融合蛋白编码序列
305.seq id no:15:与cas12j
‑
8蛋白联用的crispr重复序列
306.seq id no:16:与mb4cas12a、mlcas12a和mocas12a蛋白联用的crispr重复序列
307.seq id no:17:与bgcas12a蛋白联用的crispr重复序列
308.seq id no:18:与chcas12b蛋白联用的crispr重复序列
309.seq id no:19:与cas12j
‑
8蛋白相关的单链向导rna的crispr重复序列的dna序列
310.seq id no:20:与mb4cas12a、mlcas12a、和mocas12a蛋白相关的单链向导rna的crispr重复序列的dna序列
311.seq id no:21:与bgcas12a蛋白相关的单链向导rna的crispr重复序列的dna序列
312.seq id no:22:与chcas12b蛋白相关的单链向导rna的crispr重复序列的dna序列
313.seq id no:23:cas12j
‑
4蛋白序列
314.seq id no:24:cas12j
‑
5蛋白序列
315.seq id no:25:cas12j
‑
7蛋白序列
316.seq id no:26:cas12j
‑
9蛋白序列
317.seq id no:27:cas12j
‑
4蛋白的编码序列
318.seq id no:28:cas12j
‑
5蛋白的编码序列
319.seq id no:29:cas12j
‑
7蛋白的编码序列
320.seq id no:30:cas12j
‑
9蛋白的编码序列
321.seq id no:31:与cas12j
‑
4蛋白联用的crispr重复序列的dna序列
322.seq id no:32:与cas12j
‑
5蛋白联用的crispr重复序列的dna序列
323.seq id no:33:与cas12j
‑
7蛋白联用的crispr重复序列的dna序列
324.seq id no:34:与cas12j
‑
9蛋白联用的crispr重复序列的dna序列实施例
325.现参照下列意在举例说明而非限定本发明的实施例来描述本发明。本领域技术人员知晓,在此提供实施例仅出于详细描述本发明之目的,无意于限制本发明所要求保护的范围。
326.除非特别指明,否则基本按照本领域内熟知的以及在各参考文献中描述的常规方法进行实施例中描述的实验和方法。另外,对于实施例中未注明具体条件者,均按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
327.实施例1
328.(1)构建质粒paav2_cas12_itr
329.根据表1中列出的各cas12蛋白的基因检索号,下载其氨基酸序列,其中cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白和chcas12b蛋白的氨基酸序列分别如seq id no:1至seq id no:6所示。
330.表1.cas12蛋白及其ncbi蛋白搜索id和序列编号
331.cas12蛋白名称ncbi蛋白搜索id氨基酸序列cas12j
‑
8无seq id no:1mb4cas12awp_078273923.1seq id no:2mlcas12awp_065256572.1seq id no:3mocas12awp_112744621.1seq id no:4bgcas12aola11341.1seq id no:5chcas12boqb30769seq id no:6
332.将上述各cas12蛋白的编码核酸序列进行密码子优化,获得所述cas12蛋白在人细胞中高表达的基因序列。cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白和chcas12b蛋白的经优化基因序列分别如seq id no:8至seq id no:13所示。
333.将上述获得的seq id no:8至seq id no:13所示的各cas12蛋白高表达的基因序列进行基因合成,并构建至slugcas9骨架质粒(addgene平台,catalog#163793)上,得到质粒paav2_cas12_itr。
334.(2
‑
1)构建质粒cas12j
‑8‑
psk
‑
u6
‑
crrna
335.用bbsi和xhoi限制性内切酶将pbluescriptskii+u6
‑
sgrna(f+e)empty质粒(addgene平台,可以商购,catalog为#74707)进行酶切反应,酶切体系为:1μg质粒psk
‑
bbsi
‑
sasg、5μl 10
×
cutsmart缓冲液(购于neb公司)、1μl bbsi和1μl xhoi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应1小时。
336.然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
337.从琼脂糖凝胶上切下3296bp dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
338.根据cas12j
‑
8蛋白基因组上的repeat序列(其dna序列为seq id no:19),将该repeat序列进行基因合成,并构建于线性化的pbluescriptskii+u6
‑
sgrna(f+e)empty骨架上,得到质粒cas12j
‑8‑
psk
‑
u6
‑
crrna。
339.(2
‑
2)构建质粒psk
‑
bbsi
‑
cas12a
‑
crrna1
340.用bbsi和xhoi限制性内切酶将pbluescriptskii+u6
‑
sgrna(f+e)empty质粒进行酶切反应,酶切体系为:1μg质粒psk
‑
bbsi
‑
sasg、5μl 10
×
cutsmart缓冲液(购于neb公司)、1μl bbsi和1μl xhoi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反
应1小时。
341.然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
342.从琼脂糖凝胶上切下3296bp dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
343.根据cas12a蛋白基因组上的repeat,将截断后的repeat序列(其dna序列分别为seq id no:20和seq id no:21)进行基因合成,并构建于线性化的pbluescriptskii+u6
‑
sgrna(f+e)empty骨架上,得到质粒psk
‑
bbsi
‑
cas12a
‑
crrna1。
344.(2
‑
3)构建质粒hu6
‑
oqb30769_tracr
‑
bsa1
345.用bsai和noti限制性内切酶将px330_sgacta2质粒(addgene平台,catalog为#63712)进行酶切反应,酶切体系为:1μg质粒hu6
‑
sa
‑
tracr
‑
bsai、5μl 10
×
cutsmart缓冲液(购于neb公司)、1μl bsai和1μl noti限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应3小时。
346.然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
347.从琼脂糖凝胶上切下2998bp dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
348.根据chcas12b的基因组找出基因组上的repeat及tracr,根据二级结构推断出其rna scaffold序列(其dna序列为seq id no:22),将该序列进行基因合成,并构建于线性化的hu6
‑
sa
‑
tracr
‑
bsai骨架上,得到质粒hu6
‑
oqb30769_tracr
‑
bsal。
349.(3)质粒paav2_cas12
‑
hu6
‑
sgrna_itr载体的构建
350.利用pcr方法线性化(1)中表达cas12蛋白的paav2_cas12_itr质粒和(2)中表达各蛋白对应sgrna的cas12j
‑8‑
psk
‑
u6
‑
crrna、psk
‑
bbsi
‑
cas12a
‑
crrna1和hu6
‑
oqb30769_tracr
‑
bsa1质粒。
351.对于paav2_cas12_itr质粒,引物序列为:
352.atcatgggaaataggccctcaggtacctccccagcatgc;和
353.cgagggggggcccggtacatcatgggaaataggccctc;
354.对于cas12j
‑8‑
psk
‑
u6
‑
crrna、psk
‑
bbsi
‑
cas12a
‑
crrna1和hu6
‑
oqb30769_tracr
‑
bsa1质粒,引物序列为:
355.gagggcctatttcccatgat;和
356.gtaccgggccccccctcg。
357.反应体系如下:
[0358][0359]
pcr运行程序如下:
[0360][0361]
pcr产物在1%琼脂糖凝胶上以120v电压电泳30min,用胶回收试剂盒依据厂家提供的步骤,纯化得到目的dna片段,用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0362]
将线性化paav2_cas12_itr片段与线性化cas12j
‑8‑
psk
‑
u6
‑
crrna、psk
‑
bbsi
‑
cas12a
‑
crrna1和hu6
‑
oqb30769_tracr
‑
bsa1片段对应按照说明书要求比例进行同源重组,所使用的同源重组酶为高保真dna组装预混液(neb),反应体系如下:
[0363][0364]
反应条件如下:
[0365][0366]
将连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0367]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有氨苄青霉素抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0368]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到质粒paav2_cas12
‑
hu6
‑
sgrna_itr,备用。
[0369]
(4)线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr的制备
[0370]
用bbsi限制性内切酶将(3)中制备的各质粒paav2_cas12
‑
hu6
‑
sgrna_itr进行酶切反应,酶切体系为:1μg质粒paav2_cas12
‑
hu6
‑
sgrna_itr、5μl 10
×
cutsmart缓冲液(购于neb公司)、1μl bbsi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应1小时。
[0371]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0372]
从琼脂糖凝胶上切下dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。所述dna片段即为包含以上各cas12蛋白的编码基因的线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr,其大小分别为7135bp(cas12j
‑
8蛋白)、7866bp(mb4cas12a蛋白)、7875bp(mlcas12a蛋白)、7998bp(mocas12a蛋白)、7875bp(bgcas12a)和8606bp(chcas12b)。
[0373]
将回收的线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0374]
(5)质粒paav2_cas12
‑
hu6
‑
sgrna_itr的制备
[0375]
设计各grna,其序列示于如表2。在设计的各grna序列对用的正义链和反义链上分别加上线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr两侧对应的粘性末端序列,并合成两条寡核苷酸单链dna,这两条寡核苷酸单链dna的具体序列也示于下表。
[0376]
[0377]
将寡核苷酸单链dna进行退火得到双链dna。退火反应体系为:1μl 100μm oligo
‑
f、1μl 100μm oligo
‑
r、28μl水。将该退火体系震荡混匀后,放置于pcr仪中运行退火程序,退火程序为:95℃_5min,85℃_1min,75℃_1min,65℃_1min,55℃_1min,45℃_1min,35℃_1min,25℃_1min,4℃保存,降温速率0.3℃/s。退火后,将所得的产物通过dna连接酶(购于neb公司)连接至步骤(2)所得的线性化paav2_cas12
‑
hu6
‑
sgrna_itr质粒。
[0378]
取1μl所得连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0379]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有对应抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0380]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到含有表达目标sgrna序列的质粒paav2_cas12
‑
hu6
‑
sgrna_itr,备用。
[0381]
(6)表达cas蛋白和sgrna的质粒paav2_cas12
‑
hu6
‑
sgrna_itr对hek293t细胞系的转染
[0382]
在第0天,根据转染所需,将含有靶序列的hek293t细胞在6孔板进行铺板,细胞密度约30%左右。
[0383]
第1天,进行转染,转染过程如下:
[0384]
取2μg待转染质粒paav2_cas12
‑
hu6
‑
sgrna_itr加入至100μlopti
‑
mem培养基(购于gibco公司)中,轻轻吹打混匀。
[0385]
将转染试剂脂质体2000(购于invitrogen公司)或聚乙烯亚胺(以下简称pei)(购于polysciences公司)轻弹混匀,吸取5μl2000或pei加入至100μl opti
‑
mem培养基(购于gibco公司)中,轻轻混匀,室温静置5min。
[0386]
将稀释的转染试剂和稀释的质粒进行混合,轻轻吹打混匀,室温静置20min,然后加入到包含待转染细胞hek293t细胞的培养基中,然后将细胞置于37℃、5%co2培养箱中继续培养3天。
[0387]
(7)二代测序文库的制备
[0388]
收集编辑三天后的hek293t细胞,用dna试剂盒(天根生化科技(北京)有限公司,dp304)并依据该dna试剂盒提供的说明书提取基因组dna。
[0389]
进行pcr建库第一轮pcr,用2
×
q5 mastermix进行pcr反应,pcr引物如下所示:
[0390]
表3.二代测序一轮pcr引物列表
[0391][0392]
反应体系如下:
[0393][0394][0395]
pcr运行程序如下:
[0396][0397]
进行测序建库第二轮pcr,用2xq5 mastermix进行pcr反应,pcr引物如下所示:
[0398]
f2引物:
[0399]
aatgatacggcgaccaccgagatctacacnnnnnnnnacactctttccctacacgac;
[0400]
r2引物:
[0401]
caagcagaagacggcatacgagatnnnnnnnngtgactggagttcagacgtgtg。
[0402]
反应体系如下:
[0403][0404]
pcr运行程序如下:
[0405][0406]
将第二轮的pcr产物用胶回收试剂盒依据厂家提供的步骤,纯化330bp、327bp、279bp、239bp、311bp和298bp的dna片段,其中,330bp、327bp分别是a1和a7的大小,279bp和239bp分别是e2和e3位点的大小,311bp和298bp分别是a3和a4位点的大小。由此,二代测序文库制备完毕。
[0407]
(8)二代测序结果的分析
[0408]
将制备好的二代测序文库在高通量测序仪hiseqxten(illumina)上进行双端测序。
[0409]
二代测序计算得到对各自的两个靶位点的编辑效率如图1至图6所示,其中x轴表示靶位点,y轴表示编辑效率(indels%)。从图中可以看出,含有cas12j
‑
8、mb4cas12a、mocas12a、bgcas12a、mlcas12a及chcas12b蛋白的基因编辑系统均可以用于细胞基因编辑,且含有cas12j
‑
8蛋白的基因编辑系统的编辑活性相较于现有的cas12j
‑
2蛋白的基因编辑系统更高。
[0410]
实施例2
[0411]
(1)构建质粒paav2_cas12_itr
[0412]
根据上文表1中列出的各cas12蛋白的基因检索号,下载其氨基酸序列信息,其中cas12j
‑
8蛋白、mb4cas12a蛋白、m1cas12a蛋白、mocas12a蛋白、bgcas12a蛋白和chcas12b蛋白的氨基酸序列分别如seq id no:1至seq id no:6所示。
[0413]
将上述所得的cas12蛋白的编码核酸序列进行密码子优化,获得所述cas蛋白在人细胞中高表达的基因序列。cas12j
‑
8蛋白、mb4cas12a蛋白、mlcas12a蛋白、mocas12a蛋白、bgcas12a蛋白蛋白和chcas12b的基因序列分别如seq id no:8至seq id no:13所示。
[0414]
将上述获得的seq id no:8至seq id no:13所示的各cas蛋白高表达的基因序列进行基因合成,并构建至slugcas9骨架质粒(addgene平台,catalog#163793)上,得到质粒paav2_cas12_itr。
[0415]
(2
‑
1)构建质粒cas12j
‑8‑
psk
‑
u6
‑
crrna
[0416]
用bbsi和xhoi限制性内切酶将pbluescriptskii+u6
‑
sgrna(f+e)empty质粒(addgene平台,可以商购,catalog为#74707)进行酶切反应,酶切体系为:1μg质粒psk
‑
bbsi
‑
sasg、5μl10
×
cutsmart缓冲液(购于neb公司)、1μl bbsi和1μl xhoi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应1小时。
[0417]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0418]
从琼脂糖凝胶上切下3296bp dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
[0419]
根据cas12j
‑
8蛋白基因组上的repeat序列(其dna序列为seq id no:19),将该repeat序列进行基因合成,并构建于线性化的pbluescriptskii+u6
‑
sgrna(f+e)empty骨架上,得到质粒cas12j
‑8‑
psk
‑
u6
‑
crrna。
[0420]
(2
‑
2)构建质粒psk
‑
bbsi
‑
cas12a
‑
crrna1
[0421]
用bbsi和xhoi限制性内切酶将pbluescriptskii+u6
‑
sgrna(f+e)empty质粒进行酶切反应,酶切体系为:1μg质粒psk
‑
bbsi
‑
sasg、5μl 10
×
cutsmart缓冲液(购于neb公司)、1μl bbsi和1μl xhoi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应1小时。
[0422]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0423]
从琼脂糖凝胶上切下3296bp dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
[0424]
根据cas12a蛋白基因组上的repeat,将截断后的repeat序列(其dna序列分别为seq id no:20和seq id no:21)进行基因合成,并构建于线性化的pbluescriptskii+u6
‑
sgrna(f+e)empty骨架上,得到质粒psk
‑
bbsi
‑
cas12a
‑
crrna1。
[0425]
(2
‑
3)构建质粒hu6
‑
oqb30769_tracr
‑
bsa1
[0426]
用bsai和noti限制性内切酶将px330_sgacta2质粒(addgene平台,catalog为#63712)进行酶切反应,酶切体系为:1μg质粒hu6
‑
sa
‑
tracr
‑
bsai、5μl 10
×
cutsmart缓冲液(购于neb公司)、1μl bsai和1μl noti限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应3小时。
[0427]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0428]
从琼脂糖凝胶上切下2998bp dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
[0429]
根据chcas12b的基因组找出基因组上的repeat及tracr,根据二级结构推断出其rna scaffold序列(其dna序列为seq id no:22),将该序列进行基因合成,并构建于线性化的hu6
‑
sa
‑
tracr
‑
bsai骨架上,得到质粒hu6
‑
oqb30769_tracr
‑
bsa1。
[0430]
(3)质粒paav2_cas12
‑
hu6
‑
sgrna_itr载体的构建
[0431]
利用pcr方法线性化(1)中表达cas12蛋白的paav2_cas12_itr质粒和(2)中表达各蛋白对应sgrna的cas12j
‑8‑
psk
‑
u6
‑
crrna、psk
‑
bbsi
‑
cas12a
‑
crrna1和hu6
‑
oqb30769_
tracr
‑
bsa1质粒。
[0432]
对于paav2_cas12_itr质粒,引物序列为:
[0433]
atcatgggaaataggccctcaggtacctccccagcatgc;和
[0434]
cgagggggggcccggtacatcatgggaaataggccctc;
[0435]
对于cas12j
‑8‑
psk
‑
u6
‑
crrna、psk
‑
bbsi
‑
cas12a
‑
crrna1和hu6
‑
oqb30769_tracr
‑
bsa1质粒,引物序列为:
[0436]
gagggcctatttcccatgat;和
[0437]
gtaccgggccccccctcg。
[0438]
反应体系如下:
[0439][0440]
pcr运行程序如下:
[0441][0442]
pcr产物在1%琼脂糖凝胶上以120v电压电泳30min,用胶回收试剂盒依据厂家提供的步骤,纯化得到目的dna片段,用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0443]
将线性化paav2_cas12_itr片段与线性化cas12j
‑8‑
psk
‑
u6
‑
crrna、psk
‑
bbsi
‑
cas12a
‑
crrna1和hu6
‑
oqb30769_tracr
‑
bsa1片段对应按照说明书要求比例进行同源重组,所使用的同源重组酶为高保真dna组装预混液(neb),反应体系如下:
[0444][0445]
反应条件如下:
[0446][0447]
将连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0448]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有氨苄青霉素抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0449]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到质粒paav2_cas12
‑
hu6
‑
sgrna_itr,备用。
[0450]
(4)线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr的制备
[0451]
用bbsi限制性内切酶将(3)中制备的各质粒paav2_cas12
‑
hu6
‑
sgrna_itr进行酶切线性化反应,酶切体系为:1μg质粒paav2_cas12
‑
hu6
‑
sgrna_itr、5μl 10xcutsmart缓冲液(购于neb公司)、1μl bbsi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应1小时。
[0452]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0453]
从琼脂糖凝胶上切下dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)并依据该厂家提供的说明进行回收,最终用超纯水进行洗脱。所述dna片段即为包含以上各cas蛋白的编码基因的线性化质粒paav2_cas12_itr,其大小分别为7135bp(cas12j
‑
8蛋白)、7866bp(mb4cas12a蛋白)、7875bp(mlcas12a蛋白)、7998bp(mocas12a蛋白)、7875bp(bgcas12a)和8606bp(chcas12b)。
[0454]
将回收的线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr用nanodrop
tm lite分光光度计nanodrop(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0455]
(5)质粒paav2_cas12
‑
u6
‑
on target sgrna或paav2_cas12
‑
u6
‑
mismatch sgrna的制备
[0456]
设计各on target grna和mismatch grna的序列,并且其对应的寡核苷酸单链dna如下表4所示,其中mismatch碱基在序列表中显示为带下划线的粗体碱基。
[0457]
将所得的on target grna对应的寡核苷酸单链dna和不同mismatch grna对应的寡核苷酸单链dna分别退火。退火反应体系为:1μl 100μm oligo
‑
f、1μl 100μm oligo
‑
r、28μl水。将该退火体系震荡混匀后,放置于pcr仪中运行退火程序;退火程序如下:95℃_5min,85℃_1min,75℃_1min,65℃_1min,55℃_1min,45℃_1min,35℃_1min,25℃_1min,4℃保
存,降温速率0.3℃/s。退火后,将所得的产物分别通过dna连接酶(购于neb公司)连接至所得的线性化paav2_cas12
‑
hu6
‑
sgrna_itr质粒。
[0458]
取1μl所得连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,37℃培养1h进行大肠杆菌dh5α感受态细胞的活化复苏。
[0459]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有对应抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0460]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即分别得到表达上述on target grna序列的质粒paav2_cas12
‑
hu6
‑
on target grna和表达上述不同mismatch grna序列的质粒paav2_cas12
‑
hu6
‑
mismatch grna,备用。
[0461]
(7)将所得的表达on target grna序列的质粒paav2_cas12
‑
hu6
‑
on target grna和表达mismatch grna序列的质粒paav2_cas12
‑
u6
‑
mismatch grna采用脂质体方式分别转染至含有靶序列(ggatatgttgaagaacaccatgac)的gfp报告系统hek293t细胞系。
[0462]
[0463]
[0464][0465]
所述含有靶序列的gfp报告系统hek293t细胞系是通过下述方式获得的:在起始密码子atg和gfp编码序列之间插入pam序列和特定的靶序列,造成gfp移码突变,然后通过慢病毒感染整合到hek293t细胞中,得到含有靶序列的gfp报告系统hek293t细胞系。当基因编辑系统对靶序列进行切割后,细胞通过自身修复系统会使部分细胞恢复gfp阅读框,产生绿
色荧光,通过流式分析统计gfp阳性细胞比率可以评估基因编辑系统的编辑能力及特异性。
[0466]
上述转染过程包括如下步骤:
[0467]
第0天,根据转染所需,将含有靶序列的gfp报告系统hek293t细胞系在6孔板进行铺板,细胞密度控制在30%。
[0468]
该含有靶序列的gfp报告系统hek293t细胞系中包含cmv
‑
atg
‑
pam
‑
target site
‑
gfp的核苷酸序列,其中,其中pam序列参见图7至图13,靶位点(target site)的序列为ggatatgttgaagaacaccatgac。
[0469]
第1天,进行转染,转染过程如下:
[0470]
分别取2μg待转染质粒paav2_cas12
‑
u6
‑
on target grna或2μg待转染质粒paav2_cas12
‑
u6
‑
mismatch grna加入至100μl opti
‑
mem培养基(购于gibco公司)中,轻轻吹打混匀。
[0471]
将2000(购于invitrogen公司)或pei(购于polysciences公司)轻弹混匀,吸取5μl2000或pei加入至100μl opti
‑
mem培养基中,轻轻混匀,室温静置5min。
[0472]
将经稀释的质粒和经稀释的转染试剂进行混合,轻轻吹打混匀,得到的混合液室温静置20min,然后加入到含有靶序列的gfp报告系统hek293t细胞系的培养基中,并将其置于37℃、5%co2培养箱中继续培养。
[0473]
流式细胞分析技术分析本发明的crispr基因编辑系统对靶序列的编辑效率及脱靶率。
[0474]
具体地,收集在co2培养箱中培养3天后的hek293t细胞系,采用流式细胞仪(bd biosciences facscalibur)对其特异性进行检测,并用flowjo分析软件分析gfp阳性比率并作图。
[0475]
本发明的crispr/cas12基因编辑系统在含有靶序列的gfp报告系统hek293t细胞系中的特异性检测结果示于图7至图13,其中上方横条显示gfp报告系统示意图,在起始密码子atg和gfp编码序列之间插入有特定的pam序列及靶序列,造成gfp移码突变。因此当基因编辑系统对靶序列进行切割后,细胞通过自身修复系统会使部分细胞恢复gfp阅读框,产生绿色荧光。图7至图13中下方的柱状图中的y轴代表gfp阳性细胞百比率(%),x轴代表on
‑
target grna和mismatch grna对应的寡核苷酸单链dna序列。从图7至图13中可以看出,本发明的crispr基因编辑系统在gfp报告系统hek293t细胞系中的靶位点均发生了编辑,且由mismatch grna介导的基因编辑比例显著性低于on
‑
target grna介导的基因编辑比例,由此表明本发明的crispr基因编辑系统的编辑活性高,脱靶率低,特异性高。且在对于crispr/cas12j
‑
8基因编辑系统的研究结果中,在前14bp的单碱基mismatch中并未发现明显错配现象,说明crispr/cas12j
‑
8基因编辑系统对grna与靶序列间的完全配对要求极高,具有较低的容错率和实际应用的较高安全性。
[0476]
实施例3
[0477]
(1)线性化质粒slugabemax的制备
[0478]
以slugabemax质粒(addgene平台,catalog#163798)为模板进行pcr反应,引物序列为:
[0479]
引物1:tctggtggttctcccaagaaga
[0480]
引物2:tgaccccccgctgctgcccc
[0481]
反应体系如下:
[0482][0483][0484]
pcr运行程序如下:
[0485][0486]
pcr产物在1%琼脂糖凝胶上以120v电压电泳30min,用胶回收试剂盒依据厂家提供的步骤,纯化得到4152bp的dna片段,用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0487]
(2)质粒paav2_envtada
‑
cas12j
‑
8itr的制备
[0488]
将线性化slugabemax骨架片段与和公司合成的人源化cas12j
‑
8片段(seq id no:8)按说明书要求比例进行同源重组,所使用的同源重组酶为高保真dna组装预混液(neb),反应体系如下:
[0489][0490]
反应条件如下:
[0491]
[0492]
将连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0493]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有氨苄青霉素抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0494]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到质粒paav2_envtada
‑
cas12j
‑
8_itr,备用。
[0495]
(3)质粒paav2_envtada
‑
dcas12j
‑
8_itr的制备
[0496]
以paav2_envtada
‑
cas12j
‑
8_itr为模板进行环形pcr反应,引物序列为:
[0497]
引物3:caacctggtgaaaaagaacaacttc
[0498]
引物4:gcgatgccgatcacatcgcaca
[0499]
反应体系如下:
[0500][0501]
pcr运行程序如下:
[0502][0503][0504]
pcr产物在1%琼脂糖凝胶上以120v电压电泳30min,用胶回收试剂盒依据厂家提供的步骤,纯化得到6305bp的dna片段,用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,并分别进行t4 pnk处理和t4 dna连接酶处理,反应体系如下:
[0505][0506]
反应条件如下:
[0507][0508]
在反应体系中加入t4 dna连接酶(neb)1μl,震荡混匀后室温孵育2h。
[0509]
将连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0510]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有氨苄青霉素抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0511]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到质粒paav2_envtada
‑
dcas12j
‑
8itr,备用。
[0512]
(5)paav2_envtada
‑
dcas12j
‑
8_itr的线性化制备
[0513]
利用kpn1和not1限制性内切酶(neb)对paav2_envtada
‑
dcas12j
‑
8_itr质粒进行酶切反应,反应体系为:2μg质粒paav2_envtada
‑
dcas12j
‑
8_itr、5μl 10
×
cutsmart缓冲液(购于neb公司)、1μl kpn1限制性内切酶(购于neb公司),1μl not1限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应2小时。
[0514]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0515]
从琼脂糖凝胶上切下dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
[0516]
将回收的线性化片段paav2_envtada
‑
dcas12j
‑
8_itr用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0517]
(6)paav2_envtada
‑
dcas12j
‑8‑
crrna_itr质粒的制备
[0518]
以cas12j
‑8‑
psk
‑
u6
‑
crrna为模板进行pcr反应,引物序列为:
[0519]
引物5:ggaggtaccgatccgacgcgccatctctag
[0520]
引物6:cctgcggccgcgggccccccctcgaaaaaaaaac
[0521]
反应体系如下:
[0522][0523]
pcr运行程序如下:
[0524][0525][0526]
pcr产物在1.5%琼脂糖凝胶上以120v电压电泳30min,用胶回收试剂盒依据厂家提供的步骤,纯化得到394bp的cas12j
‑
8crrna dna片段,用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0527]
将线性化paav2_envtada
‑
dcas12j
‑
8_itr片段与cas12j
‑
8crrna片段按说明书要求比例进行同源重组,所使用的同源重组酶为高保真dna组装预混液(neb),反应体系如下:
[0528][0529]
反应条件如下:
[0530][0531]
将连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0532]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有氨苄青霉素抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0533]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到质粒paav2_envtada
‑
dcas12j
‑8‑
crrna_itr,备用。
[0534]
(7)质粒paav2_envtada
‑
dcas12j
‑8‑
sgrna_itr的制备
[0535]
用bbsi限制性内切酶对paav2_envtada
‑
dcas12j
‑8‑
crrna_itr质粒进行酶切反应,酶切体系为:2μg质粒paav2_envtada
‑
dcas12j
‑8‑
crrna_itr、5μl10
×
cutsmart缓冲液(购于neb公司)、1μl bbsi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应2小时。
[0536]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0537]
从琼脂糖凝胶上切下dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
[0538]
将回收的线性化质粒paav2_envtada
‑
dcas12j
‑8‑
crrna_itr用nanodrop
tm lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0539]
在人类基因组中随机选择满足cas12j
‑
8蛋白pam需求的内源位点靶序列,其对应的寡核苷酸单链dna如下表所示。
[0540][0541]
将寡核苷酸单链dna进行退火得到双链dna。退火反应体系为:1μl 100μm oligo
‑
f、1μl 100μm oligo
‑
r、28μl水。将该退火体系震荡混匀后,放置于pcr仪中运行退火程序,
退火程序为:95℃_5min,85℃_1min,75℃_1min,65℃_1min,55℃_1min,45℃_1min,35℃_1min,25℃_1min,4℃保存,降温速率0.3℃/s。退火后,将所得的产物通过dna连接酶(购于neb公司)连接至线性化paav2_envtada
‑
dcas12j
‑8‑
crrna_itr载体。
[0542]
取1μl所得连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0543]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有对应抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0544]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到含有表达目标sgrna序列的质粒paav2_envtada
‑
dcas12j
‑8‑
crrna
‑
grna_itr,备用。
[0545]
(8)paav2_envtada
‑
dcas12j
‑8‑
crrna
‑
grna_itr质粒对野生型hek293t细胞系的转染
[0546]
将所得的paav2_envtada
‑
dcas12j
‑8‑
crrna
‑
grna_itr质粒采用脂质体方式分别转染至野生型hek293t细胞系。
[0547]
上述转染过程包括如下步骤:
[0548]
第0天,根据转染所需,将hek293t细胞系在6孔板进行铺板,细胞密度控制在30%。
[0549]
第1天,进行转染,转染过程如下:
[0550]
取2μg待转染质粒paav2_envtada
‑
dcas12j
‑8‑
crrna
‑
grna_itr加入至100μlopti
‑
mem培养基(购于gibco公司)中,轻轻吹打混匀。
[0551]
将2000(购于invitrogen公司)或pei(购于polysciences公司)轻弹混匀,吸取5μl2000或pei加入至100μl opti
‑
mem培养基中,轻轻混匀,室温静置5min。
[0552]
将经稀释的质粒和经稀释的转染试剂进行混合,轻轻吹打混匀,得到的混合液室温静置20min,然后加入到备转hek293t细胞的培养基中,并将其置于37℃、5%co2培养箱中继续培养7天。
[0553]
(9)二代测序文库的制备
[0554]
收集编辑七天后的hek293t细胞,用dna试剂盒(天根生化科技(北京)有限公司,dp304)并依据该dna试剂盒提供的说明书提取基因组dna。
[0555]
进行pcr建库第一轮pcr,用2
×
q5 mastermix进行pcr反应,pcr引物如下表所示:
[0556]
表6:针对各内源位点的pcr引物列表
[0557]
[0558]
反应体系如下:
[0559][0560][0561]
pcr运行程序如下:
[0562][0563]
进行pcr建库第二轮pcr,用2
×
q5 mastermix进行pcr反应,pcr引物同上文实施例1中给出的f2引物和r2引物。
[0564]
反应体系如下:
[0565][0566]
pcr运行程序如下:
[0567][0568]
将第二轮的pcr产物用胶回收试剂盒依据厂家提供的步骤,纯化dna片段,由此二代测序文库制备完毕。
[0569]
(10)二代测序结果的分析
[0570]
将制备好的二代测序文库在高通量测序仪hiseqxten(illumina)上进行双端测序。
[0571]
二代测序结果经运算后获得各内源位点靶位点中符合编辑要求的腺嘌呤a的编辑比例,结果示于图14。从该图中可以看出,cas12j
‑
8abe碱基编辑器成功地对这几个内源性位点靶位点进行了细胞单碱基基因编辑,且含有cas12j
‑
8abe碱基编辑器蛋白只有938个氨基酸,可以轻易被aav病毒包装,由此使crispr单碱基编辑器系统在生物体基因治疗上的应用成为了可能。
[0572]
实施例4
[0573]
(1)构建质粒paav2_cas12_itr
[0574]
文末序列表中示出了cas12j
‑
4、cas12j
‑
5、cas12j
‑
7、cas12j
‑
8和cas12j
‑
9蛋白的氨基酸序列(分别如seq id no:23
‑
25、1和26所示)。
[0575]
将各cas12蛋白的编码核酸序列进行密码子优化,获得所述cas12蛋白在人细胞中高表达的基因序列。cas12j
‑
4、cas12j
‑
5、cas12j
‑
7、cas12j
‑
8和cas12j
‑
9蛋白的基因序列分别由seq id no:27
‑
29、8和30所示。
[0576]
将上述获得的seq id no:27
‑
29、8和30所示的各cas12蛋白高表达的基因序列进行基因合成,并分别构建至slugcas9骨架质粒(addgene平台,catalog#163793)上,得到各质粒paav2_cas12_itr。
[0577]
(2)构建质粒cas12j
‑
psk
‑
u6
‑
crrna
[0578]
用bbsi和xhoi限制性内切酶将pbluescriptskii+u6
‑
sgrna(f+e)empty质粒(addgene平台,可以商购,catalog为#74707)进行酶切反应,酶切体系为:1μg质粒psk
‑
bbsi
‑
sasg、5μl10
×
cutsmart缓冲液(购于neb公司)、1μl bbsi和1μl xhoi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应1小时。
[0579]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0580]
从琼脂糖凝胶上切下3296bp dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)依据厂家提供的说明进行回收,最终用超纯水进行洗脱。
[0581]
根据cas12j
‑
4、cas12j
‑
5、cas12j
‑
7、cas12j
‑
8和cas12j
‑
9蛋白基因组上的repeat序列(其dna序列分别由seq id no:31至33、19和34所示),将该repeat序列进行基因合成,并分别构建于线性化的pbluescriptskii+u6
‑
sgrna(f+e)empty骨架上,得到各质粒
cas12j
‑
psk
‑
u6
‑
crrna。
[0582]
(3)质粒paav2_cas12
‑
hu6
‑
sgrna_itr载体的构建
[0583]
利用pcr方法线性化(1)中表达cas12蛋白的paav2_cas12_itr质粒和(2)中表达各蛋白对应sgrna的cas12j
‑
psk
‑
u6
‑
crrna质粒。
[0584]
对于paav2_cas12_itr质粒,引物序列为:
[0585]
atcatgggaaataggccctcaggtacctccccagcatgc;和
[0586]
cgagggggggcccggtacatcatgggaaataggccctc;
[0587]
对于cas12j
‑
psk
‑
u6
‑
crrna质粒,引物序列为:
[0588]
gagggcctatttcccatgat;和
[0589]
gtaccgggccccccctcg。
[0590]
反应体系如下:
[0591][0592][0593]
pcr运行程序如下:
[0594][0595]
pcr产物在1%琼脂糖凝胶上以120v电压电泳30min,用胶回收试剂盒依据厂家提供的步骤,纯化得到目的dna片段,用nanodrop
tm
lite分光光度计(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0596]
将线性化paav2_cas12_itr片段与线性化cas12j
‑
psk
‑
u6
‑
crrna片段对应按照说明书要求比例进行同源重组,所使用的同源重组酶为高保真dna组装预混液(neb),反应体系如下:
[0597][0598]
反应条件如下:
[0599][0600]
将连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0601]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有氨苄青霉素抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0602]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到各质粒paav2_cas12
‑
hu6
‑
sgrna_itr,备用。
[0603]
(4)线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr的制备
[0604]
用bbsi限制性内切酶将(3)中制备的各质粒paav2_cas12
‑
hu6
‑
sgrna_itr进行酶切线性化反应,酶切体系为:1μg质粒paav2_cas12
‑
hu6
‑
sgrna_itr、5μl 10xcutsmart缓冲液(购于neb公司)、1μl bbsi限制性内切酶(购于neb公司),水补足至50μl。使该酶切体系在37℃反应1小时。
[0605]
然后,将酶切产物在1%琼脂糖凝胶上以120v电压电泳30min。
[0606]
从琼脂糖凝胶上切下dna片段,用胶回收试剂盒(天根生化科技(北京)有限公司,dp209)并依据该厂家提供的说明进行回收,最终用超纯水进行洗脱。所述dna片段即为包含以上各cas蛋白的编码基因的线性化质粒paav2_cas12_itr。
[0607]
将回收的线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr用nanodrop
tm lite分光光度计nanodrop(thermo scientific)测定dna浓度,备用或置于
‑
20℃进行长期保存。
[0608]
(5)质粒paav2_cas12
‑
hu6
‑
sgrna_itr的制备
[0609]
设计grna(ggauauguugaagaacaccaugac),并在设计的grna序列用的正义链和反义链上分别加上线性化质粒paav2_cas12
‑
hu6
‑
sgrna_itr两侧对应的粘性末端序列,并合成两条寡核苷酸单链dna,这两条寡核苷酸单链dna的具体序列如下:
[0610]
oligo
‑
f:ggatatgttgaagaacaccatgac
[0611]
oligo
‑
r:gtcatggtgttcttcaacatatcc
[0612]
其中,针对cas12j
‑
4、cas12j
‑
5、cas12j
‑
7、cas12j
‑
8、和cas12j
‑
9的oligo
‑
f的粘性末端分别为cgac、ggac、agac、agac和agac,针对所有cas12蛋白的oligo
‑
r的粘性末端均为aaaa。
[0613]
将寡核苷酸单链dna进行退火得到双链dna。退火反应体系为:1μl 100μm oligo
‑
f、1μl 100μm oligo
‑
r、28μl水。将该退火体系震荡混匀后,放置于pcr仪中运行退火程序,退火程序为:95℃_5min,85℃_1min,75℃_1min,65℃_1min,55℃_1min,45℃_1min,35℃_1min,25℃_1min,4℃保存,降温速率0.3℃/s。退火后,将所得的产物通过dna连接酶(购于neb公司)连接至步骤(2)所得的线性化paav2_cas12
‑
hu6
‑
sgrna_itr质粒。
[0614]
取1μl所得连接产物加到大肠杆菌dh5α感受态细胞(购于上海唯地生物技术有限公司)中,冰上孵育30min,42℃热激1min,冰上孵育2min,加入900μl lb培养基,于37℃培养1小时,以进行大肠杆菌dh5α感受态细胞的活化复苏。
[0615]
将复苏后的大肠杆菌dh5α感受态细胞涂布在含有对应抗性的lb固体平板在37℃培养箱倒置培养,得到的大肠杆菌dh5α单克隆进行sanger测序验证。
[0616]
将测序验证连接正确的大肠杆菌dh5α克隆摇菌,提取质粒,即得到含有表达目标sgrna序列的质粒paav2_cas12
‑
hu6
‑
sgrna_itr,备用。
[0617]
(7)将所得的表达grna序列的质粒paav2_cas12
‑
hu6
‑
sgrna_itr采用脂质体方式分别转染至含有靶序列(ggatatgttgaagaacaccatgac)的gfp报告系统hek293t细胞系文库中。
[0618]
所述含有靶序列的gfp报告系统hek293t细胞系文库是通过下述方式获得的:在起始密码子atg和gfp编码序列之间插入5bp随机序列(作为pam序列)和24bp的protospacer(作为靶序列),造成gfp移码突变而不表达。用cmv启动子启动这个含有插入片段的gfp基因,并构建在慢病毒表达载体上。这段序列由慢病毒介导随机插入到hek293t细胞的基因组中,使之成为稳定的gfp报告细胞系文库。当使用基因编辑系统对靶序列进行切割后,细胞通过自身修复系统会使部分细胞恢复gfp阅读框,产生绿色荧光,通过流式分析统计gfp阳性细胞比率可以评估基因编辑系统的编辑能力及特异性。
[0619]
上述转染过程包括如下步骤:
[0620]
第0天,根据转染所需,将含有靶序列的gfp报告系统hek293t细胞系文库在6孔板进行铺板,细胞密度控制在30%。
[0621]
该含有靶序列的gfp报告系统hek293t细胞系文库中包含cmv
‑
atg
‑
pam
‑
target site
‑
gfp的核苷酸序列,其中,其中pam序列为5bp随机序列,靶位点(target site)的序列为ggatatgttgaagaacaccatgac(图15)。
[0622]
第1天,进行转染,转染过程如下:
[0623]
分别取2μg待转染质粒paav2_cas12
‑
hu6
‑
sgrna_itr加入至100μl opti
‑
mem培养基(购于gibco公司)中,轻轻吹打混匀。
[0624]
将2000(购于invitrogen公司)或pei(购于polysciences公司)轻弹混匀,吸取5μl2000或pei加入至100μl opti
‑
mem培养基中,轻轻混匀,室温静置5min。
[0625]
将经稀释的质粒和经稀释的转染试剂进行混合,轻轻吹打混匀,得到的混合液室温静置20min,然后加入到含有靶序列的gfp报告系统hek293t细胞系文库的培养基中,并将其置于37℃、5%co2培养箱中继续培养。
[0626]
然后,在荧光显微镜下观察各crispr/cas12系统对hek293t细胞系文库中的靶基因进行编辑的情况,结果示于图16。从该图中可以看出,只有crispr/cas12j
‑
8系统组别文
库细胞出绿色荧光,这表明该系统成功地对细胞中的靶基因进行了编辑。但是,其他的任何crispr/cas12j基因编辑系统组别文库细胞均没有发出任何荧光,表明这些系统不能够对靶基因进行有效编辑。
[0627]
[0628]
[0629]
[0630]
[0631]
[0632]
[0633]
[0634]
[0635]
[0636]
[0637]
[0638]
[0639]
[0640]
[0641]
[0642]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1