可视化CRISPR/Cas9基因编辑系统及使用方法
可视化crispr/cas9基因编辑系统及使用方法
技术领域
1.本发明涉及基因工程技术领域,尤其涉及一种可视化crispr/cas9基因编辑系统及使用方法。
背景技术:
2.目前,已经开发了多个经典的适用于植物基因编辑的crispr/cas9系统。由于crispr/cas9系统具有操作简单、高效和可控的特性,已被广泛用于作物遗传育种改良。但是在作物中利用crispr/cas9系统进行基因编辑育种,仍然存在前期阳性转基因植株筛选工作量大而繁琐、后期筛选非转基因突变体周期长而困难等问题。尤其是在筛选不含转基因突变体的过程中,主要通过自交或者回交来剔除携带cas9基因元件的外源t
‑
dna区,利用该方式获得不含转基因的基因编辑植株,一般需要种植2代以上才能获得稳定突变单株,时间跨度往往在2年以上甚至更久。另外,研究人员也尝试了利用基因枪和纳米颗粒将cas9蛋白和grna直接导入植物细胞中或利用农杆菌介导的转基因瞬时表达cas9蛋白和grna等方法,来避免cas9基因等原件整合至基因组中,从而获得非转基因的基因编辑植株。但上述策略发生基因编辑的效率较低,且需要使用高通量的方法来鉴定编辑植株,成本非常高。此外,也有使用除草剂来杀死转基因植株,从而筛选非转基因植株的方法报道,但该方法对环境不友好,且不能避免转基因花粉的漂移,可能存在转基因安全风险。
3.crispr/cas9基因编辑系统简单操作,尤其是刘耀光课题利用bsai内切酶开发的标准化生物砖组装技术,极大简化了crispr/cas9基因编辑载体构建,加速了crispr/cas9基因编辑系统在作物遗传改良上的应用。但在引入dsred和zmaa1表达框后,bsai等iis型限制性内切酶不能使用,使构建载体的难度加大。
技术实现要素:
4.本发明要解决的技术问题是,面对急需解决的问题,寻求一种可视化筛选阳性转基因植株及快速剔除转基因成分的crispr/cas9基因编辑系统及使用方法。利用胚乳荧光蛋白标记携带外源cas9基因t
‑
dna区的转基因种子,以筛选不含转基因的突变体种子。同时,利用花粉失活技术使携带cas9基因的花粉失活,增加无转基因成分种子的比例,且避免因转基因花粉漂移带来的生物安全隐患。
5.为了实现上述目的,本发明提供了一种可视化crispr/cas9基因编辑系统,包括基因编辑元件、外源t
‑
dna可视化追踪元件和转基因花粉失活元件;所述基因编辑元件、外源t
‑
dna可视化追踪元件和转基因花粉失活元件构建到表达载体的同一个t
‑
dna区中,使三个基因连锁遗传;
6.所述转基因花粉失活元件为玉米α
‑
淀粉酶基因zmaa1的表达框,由花粉特异性启动子pg47驱动zmaa1基因,由in2
‑
1终止子终止;
7.所述花粉特异性启动子pg47的dna序列如seq id no.12所示;
8.所述zmaa1基因的dna序列如seq id no.13所示;
9.所述in2
‑
1终止子的dna序列如seq id no.14所示。
10.上述的可视化crispr/cas9基因编辑系统,进一步的,所述基因编辑元件为crispr/cas9表达结构,crispr/cas9表达结构包括cas9基因表达框和grna表达框。
11.上述的可视化crispr/cas9基因编辑系统,进一步的,所述cas9基因表达框由ubi启动子驱动cas9基因,由nos终止子终止;
12.所述ubi启动子的dna序列如seq id no.1所示;
13.所述cas9基因的dna序列如seq id no.2所示;
14.所述nos终止子的dna序列如seq id no.3所示。
15.上述的可视化crispr/cas9基因编辑系统,进一步的,所述grna表达框包括小rna启动子和guide
‑
rna,所述小rna启动子和guide
‑
rna之间用1.8kb的间隔序列隔开;
16.所述小rna启动子为osu3、osu6a、osu6b、osu6c中的一种;
17.所述osu3的dna序列如seq id no.4所示;
18.所述osu6a的dna序列如seq id no.5所示;
19.所述osu6b的dna序列如seq id no.6所示;
20.所述osu6c的dna序列如seq id no.7所示;
21.所述guide
‑
rna序列如seq id no.8所示;
22.所述间隔序列的dna序列seq id no.15所示。
23.上述的可视化crispr/cas9基因编辑系统,进一步的,所述外源t
‑
dna可视化追踪元件为红色荧光蛋白基因dsred的表达框,由胚乳特异性启动子ltp驱动dsred基因,由pinii终止子终止;
24.所述胚乳特异性启动子ltp的dna序列如seq id no.9所示;
25.所述dsred基因的dna序列如seq id no.10所示;
26.所述pinii终止子的dna序列如seq id no.11所示。
27.上述的可视化crispr/cas9基因编辑系统,进一步的,在转基因花粉失活元件和外源t
‑
dna可视化追踪元件之间引入酶促组装入口序列;所述酶促组装入口序列的dna序列如seq id no.16所示。
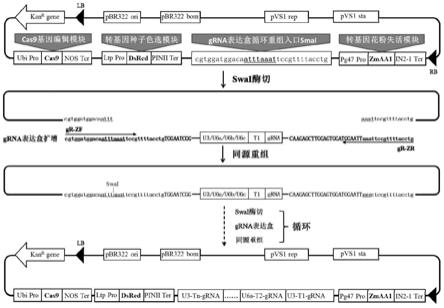
28.基于一个总的技术构思,本发明还提供了一种上述可视化crispr/cas9基因编辑系统的使用方法,包括以下步骤:
29.s1、根据待剔除基因的靶位点设计接头引物序列,合成靶位点重组接头t1
‑
adapter和t2
‑
adapter;根据小rna启动子和guide
‑
rna设计引物grna
‑
f、osu3
‑
r和osu6
‑
r,分别扩增对应的u3
‑
grna表达框前体和u6
‑
grna表达框前体;
30.s2、将u3
‑
grna表达框前体和u6
‑
grna表达框前体分别与靶位点重组接头t1
‑
adapter和t2
‑
adapter进行酶促组装得到酶促组装产物u3
‑
t1
‑
grna和u6
‑
t2
‑
grna;
31.s3、将所述酶促组装产物u3
‑
t1
‑
grna和u6
‑
t2
‑
grna转化至大肠杆菌感受态细胞后,得到含有靶位点完整grna表达框的转化子pu3
‑
t1
‑
grna和pu6
‑
t2
‑
grna;
32.s4、利用重组引物gr
‑
zf和gr
‑
zr扩增所述含有靶位点完整grna表达框的转化子pu3
‑
t1
‑
grna和pu6
‑
t2
‑
grna得到含有重组引物和完整grna表达框的扩增产物u3
‑
t1
‑
grna表达框、u6
‑
t1
‑
grna表达框;
33.s5、将所述u3
‑
t1
‑
grna表达框和c9dz载体进行酶促组装得到酶促组装产物一;
34.s6、将所述酶促组装产物一转化至大肠杆菌感受态细胞后,筛选含有单个靶位点的基因编辑载体的转化子;
35.s7、将所述转化子与所述u6
‑
t2
‑
grna表达框进行酶促组装得到酶促组装产物二;
36.s8、将所述酶促组装产物二导入水稻中,获得靶位点发生突变的基因编辑植株;
37.s9、从靶位点发生突变的基因编辑植株后代中,根据胚乳是否含有荧光,筛选无红色荧光的种子,即为剔除了转基因成分且靶位点发生突变的目的植株。
38.上述的使用方法,进一步的,所述grna
‑
f的序列如seq id no.23所示;
39.所述osu3
‑
r的序列如seq id no.24所示;
40.所述osu6
‑
r的序列如seq id no.25所示。
41.上述的使用方法,进一步的,所述重组引物gr
‑
zf的dna序列如seq id no.26所示;
42.所述重组引物gr
‑
zr的dna序列如seq id no.27所示。
43.上述的使用方法,进一步的,所述载体为c9dz载体。
44.上述的使用方法,进一步的,所述待编辑基因为水稻穗颈伸长基因oseui1,编辑所述水稻穗颈伸长基因oseui1并可视化剔除转基因成分的方法为:
45.(1)根据oseui1的靶位点设计接头引物序列,合成靶位点重组接头eui1
‑
u3
‑
t1
‑
adapter和eui
‑
u6
‑
t2
‑
adapter;
46.(2)将u3
‑
grna表达框前体和u6
‑
grna表达框前体分别与靶位点重组接头eui1
‑
u3
‑
t1
‑
adapter和eui
‑
u6
‑
t2
‑
adapter进行酶促组装得到酶促组装产物eui1
‑
u3
‑
t1
‑
grna和eui1
‑
u6
‑
t2
‑
grna;
47.(3)将所述酶促组装产物eui1
‑
u3
‑
t1
‑
grna和eui1
‑
u6
‑
t2
‑
grna转化至大肠杆菌感受态细胞后,得到含有靶位点完整grna表达框的转化子peui1
‑
u3
‑
t1
‑
grna和peui1
‑
u6
‑
t2
‑
grna;
48.(4)利用重组引物gr
‑
zf和gr
‑
zr扩增含有完整grna表达框的质粒子peui1
‑
u3
‑
t1
‑
grna和peui1
‑
u6
‑
t2
‑
grna;
49.(5)将含有重组引物和完整grna表达框的扩增产物eui1
‑
u3
‑
t1
‑
grna的表达框和c9dz载体进行酶促组装得到酶促组装产物c9dz
‑
eui1
‑
t1;
50.(6)s5、将所述酶促组装产物c9dz
‑
eui1
‑
t1转化至大肠杆菌感受态细胞后,筛选含有单个靶位点的基因编辑载体的转化子pc9dz
‑
eui1
‑
t1;
51.(7)swai酶切pc9dz
‑
eui1
‑
t1后,与含有重组引物和完整grna表达框的扩增产物eui1
‑
u6
‑
t2
‑
grna表达框进行酶促组装得到酶促组装产物pc9dz
‑
eui1
‑
t1
‑
t2;
52.(8)将所述酶促组装产物pc9dz
‑
eui1
‑
t1
‑
t2导入水稻中,获得靶位点发生突变的基因编辑植株;
53.(9)从eui1基因发生突变的编辑植株的后代中,根据胚乳是否含有荧光,筛选无红色荧光的种子,即为剔除了转基因成分且eui1基因突变的目的植株。
54.与现有技术相比,本发明的优点在于:
55.1、本发明提供了一种可视化crispr/cas9基因编辑系统,利用胚乳荧光蛋白标记携带外源cas9基因t
‑
dna区的转基因种子,可以直观筛选不含转基因的基因编辑植株,而不需要通过pcr等分子生物学手段,不但减少了大量的筛选工作,而且节省了时间和成本;花粉失活技术使携带cas9基因的转基因花粉失活,增加了无转基因成分种子的比例,更重要
的是能避免因转基因花粉漂移带来的生物安全隐患。
56.2、本发明提供了一种可视化crispr/cas9基因编辑系统的使用方法,属于循环堆垒酶促组装crispr/cas9基因编辑载体的方法,该方法每次只组装一个grna表达盒,但具有效率好、精度高的优点,只要感受态细胞的容纳能力足够,可在完成上一个grna表达盒组装的基础上,无限组装多个grna表达盒。
附图说明
57.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整的描述。
58.图1本发明实施例1的grna表达盒载体结构图。
59.图2本发明实施例1中可视化剔除转基因成分的crispr/cas9基因编辑载体c9dz的结构及grna表达盒构建技术流程图。
60.图3为本发明实施例2中敲除oseui1基因后株高变高的株系及种子。
61.图4为本发明实施例2中株高增加的无荧光种子发育成植株后的田间表型。
62.图5为本发明实施例2中株高增加的无荧光种子发育成植株后的转基因成分pcr检测。m为takara 100bp dna ladder,1
‑
12为无荧光且株高增加的基因编辑植株,
“‑”
为阴性对照,“+”为阳性对照。
具体实施方式
63.以下结合具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。
64.除非另有定义,下文中所使用的所有专业术语与本领域技术人员通常理解含义相同。本文中所使用的专业术语只是为了描述具体实施例的目的,并不是旨在限制本发明的保护范围。
65.除非另有特别说明,本发明中用到的各种原材料、试剂、仪器和设备等均可通过市场购买得到或者可通过现有方法制备得到。下述实施例中的方法,如无特别说明,均为本领域的常规方法。
66.实施例
67.以下实施例中所采用的材料和仪器均为市售。
68.实施例1
69.一种可视化剔除转基因成分的crispr/cas9基因编辑系统,包括三个基本结构元件,分别为基因编辑元件、外源t
‑
dna可视化追踪元件和转基因花粉失活元件,基因编辑元件、外源t
‑
dna可视化追踪元件和转基因花粉失活元件构建到表达载体的同一个t
‑
dna区中,使三个基因连锁遗传。在转基因花粉失活元件和外源t
‑
dna可视化追踪元件之间引入酶促组装入口序列;酶促组装入口序列的dna序列如seq id no.16所示,具体为:cgtggatggacaatttaaattccgttttacctg。
70.基因编辑元件为crispr/cas9表达结构,crispr/cas9表达结构包括cas9基因表达框和grna表达框;cas9基因表达框由ubi启动子(seq1)驱动cas9基因(seq2),由nos终止子(seq3)终止。ubi启动子(seq1)的核苷酸序列如seq id no.1所示。cas9基因(seq2)的核苷
酸序列如seq id no.2所示。nos终止子(seq3)的核苷酸序列如seq id no.3所示。
71.grna表达框由小rna启动子(小rna启动子可以为osu3(seq4)、osu6a(seq5)、osu6b(seq6)、osu6c(seq7)中的一个)、靶位点和guide
‑
rna(seq8)组成。
72.osu3启动子(seq4)的核苷酸序列如seq id no.4所示。osu6a启动子(seq5)的核苷酸序列如seq id no.5所示。osu6b启动子(seq6)的核苷酸序列如seq id no.6所示。osu6c启动子(seq7)的核苷酸序列如seq id no.7所示。
73.guide
‑
rna(seq8)的核苷酸序列如seq id no.8所示,具体为:
74.gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttcaagagcttggagtggatggaattttcctccg。
75.小rna启动子和guide
‑
rna之间用1.8kb的间隔序列(seq15)隔开,间隔序列的dna序列如seq id no.15所示,具体为:
76.tctgccagcccggctacccggcgaacttcgccggcgccggcggcttcagggacaacgtgaggacgctgctcggcttcgcgcacctggaggccggcgtccacggcgagaccaagtgctggtcgttccagctcgagctgcaccgccacccccccaccgtcgtgaggctcttcgtcgtcgaggaggaggtcgccgcctcgccgcaccgccagtgccacctctgccgccatattggtccgtcgaacaaactacaattaatcaatcaacctttacataggattgatccgatcgatgccatggtgttgtagggtgggggaggcatctgatatgcagcaagaggtatcacttcttgctgccgaggagggaatcggcggcggaagccgacggcctgtgcttcgcgatcaaccacggcggcggcggtggcgcggagaaagcgtcgtcgaaagggacgacgacgacggcctccagcagaggccacctgctacacggcgtcgtgcacctcaacggctacggccacctcgtcgccctccacggcctcgagggcggctccgacttcgtctccggccaccagatcatggacctctgggaccgcatttgctcagccttgcacgtaaggtagtagtagtatacatgtgcgtgtgcatgcatgcaagcaatgcaacgatgtcgggctgcgtgtgagaacatttgcttgggcatggtgtggtgtatgcaaggacggtgagcctggtggacacggcgaggaagggccacatggagctgaggctgctgcacggcgtcgcgtacggcgagacgtggttcgggcggtgggggtacaggtacggccggccgagctacggcgtcgcgctgccgtcgtaccggcagtcgctgcacgtgctcggctccatgccgctctgcgtgctggtgccgcacctgtcgtgcttcagccaggagctccccatggtggtcaccaagtaccaggccatcagcggccacaagctgctcagcctcggcgacctcctccgcttcatgctcgagctgcgcgcccgcctgccggccacctccgtcacggccatggactaccggggcatcatgtcggaggcctcgtgccggtggtcggcgaagcgcgtcgacatggcggcgcgcgccgtcgtggacgcgctccgccgcgccgagccggcggcgcggtgggtcacgcggcaggaggtgcgcgacgcggcgcgcgcctacatcggcgacacgggcctcctcgacttcgtgctcaagtccctcggcaaccacatcgtcggcaactacgtcgtgcgccgcaccatgaacccggtgaccaaggtgctcgagtactgcctcgaggacgtctccagcgtgctcccggcggtcgccgccggcggcggcgtgccggcgcagggcaagatgagggtgaggttccagctcacgcgtgcgcagctcatgagggacctggtgcacctgtaccggcacgtgctcaaggagcccagccaggcgctcaccggcggcgcgttcggcgcgatcccggtggcggtgcggatggtcctggacatcaagcacttcgtcaaagattaccacgaaggacaagccgcggcgagcagcaatggcggtggcggattcgggcatccccacatcaacctgtgctgcacgctgctcgtgagcaacgggagcccggagctagctccaccgtacgagacggtgaccctgccggcgcacgcgacggtgggcgagctgaagtgggaggcgcagagggtgttcagcgagatgtacctcggcctgaggagcttcgcggcggactccgtcgtcggggtcggcgccgac。
77.外源t
‑
dna可视化追踪元件为红色荧光蛋白基因dsred的表达框,由胚乳特异性启动子ltp(seq9)驱动dsred基因(seq10),由pinii终止子(seq11)终止。ltp启动子(seq9)的核苷酸序列如seq id no.9所示。dsred基因(seq10)的核苷酸序列如seq id no.10所示。pinii终止子(seq11)的核苷酸序列如seq id no.11所示。
78.转基因花粉失活元件为玉米α
‑
淀粉酶基因zmaa1的表达框,由花粉特异性启动子pg47(seq12)驱动zmaa1基因(seq13),由in2
‑
1终止子(seq14)终止。pg47启动子(seq12)的核苷酸序列如seq id no.12所示。zmaa1基因(seq13)的核苷酸序列如seq id no.13所示。in2
‑
1终止子(seq14)的核苷酸序列如seq id no.14所示。
79.上述基本结构元件中,grna表达框通过grna表达框前体与靶位点同源重组的方式构建,grna表达框前体构建到pmd18
‑
t上,其中小rna启动子(主要为osu3、osu6a、osu6b、osu6c)与guide
‑
rna之间用1.8kb的间隔序列隔开,方便回收pcr扩增的grna表达框前体并与靶位点进行同源重组(参见附图1)。cas9基因表达框、dsred基因表达框和zmaa1基因的表达框构建到植物表达载体c9dz的同一个t
‑
dna区中,使三个基因连锁遗传(附图2)。在zmaa1基因表达框和dsred基因表达框之间引入33bp酶促组装入口序列cgtggatggacaatttaaattccgttttacctg,该组装入口序列包含一个swai的平末端酶切位点,swai酶切后的线性化载体与grna表达框的正反向引物各有15bp同源,每重组一个grna表达框后又重新生成一个完全一样的组装入口序列,可以用于下一个grna表达框的循环组装(参见附图2)。
80.实施例2
81.一种实施例1的可视化剔除转基因成分的crispr/cas9基因编辑的使用方法,应用的具体对象为水稻穗颈伸长基因oseui1,具体包括以下步骤:
82.(1)利用http://skl.scau.edu.cn/targetdesign/网站targetdesign工具设计oseui1基因的2个靶位点eui1
‑
u3
‑
t1和eui
‑
u6a
‑
t2。根据靶位点设计重组接头引物序列eui1
‑
u3
‑
t1
‑
f和eui1
‑
u3
‑
t1
‑
r、eui1
‑
u6a
‑
t2
‑
f和eui1
‑
u6a
‑
t2
‑
r如下,其中下划线标注的序列为对应的靶位点序列。
83.靶位点向序列如下:
84.eui1
‑
u3
‑
t1(seq17)的核苷酸序列如seq id no.17所示,具体为:gatcgccgggctgtgcatta;
85.eui
‑
u6a
‑
t2(seq18)的核苷酸序列如seq id no.18所示,具体为:caagtacctccagaaaggcc。
86.靶位点重组接头引物序列如下:
87.eui1
‑
u3
‑
t1
‑
f(seq19)的核苷酸序列如seq id no.19所示,具体为:agatgatccgtggcagatcgccgggctgtgcattagttttagagctagaa;
88.eui1
‑
u3
‑
t1
‑
r(seq20)的核苷酸序列如seq id no.20所示,具体为:ttctagctctaaaactaatgcacagcccggcgatctgccacggatcatct;
89.eui1
‑
u6a
‑
t2
‑
f(seq21)的核苷酸序列如seq id no.21所示,具体为:ctggcttggctgccgcaagtacctccagaaaggccgttttagagctagaa;
90.eui1
‑
u6a
‑
t2
‑
r(seq22)的核苷酸序列如seq id no.22所示,具体为:ttctagctctaaaacggcctttctggaggtacttgcggcagccaagccag。
91.(2)各取10μl的正向引物f和反向引物r混合均匀后,加热至94℃,静置至室温后,制备成靶位点重组接头eui1
‑
u3
‑
t1
‑
adaptor和eui
‑
u6a
‑
t2
‑
adaptor。
92.(3)利用grna
‑
f/osu3
‑
r和grna
‑
f/osu6a
‑
r分别扩增对应的u3
‑
grna表达框前体和u6a
‑
grna表达框前体。
93.grna
‑
f(seq23)的核苷酸序列如seq id no.23所示,具体为:gttttagagctagaaat
agcaagttaaaataag;
94.osu3
‑
r(seq24)的核苷酸序列如seq id no.24所示,具体为:tgccacggatcatctgcacaactcttttaaac;
95.osu6a
‑
r(seq25)的核苷酸序列如seq id no.25所示,具体为:cggcagccaagccagcaccc。
96.所用pcr反应体系为:kod 2
×
pcr buffer 25ul,2mm dntp 10ul,正向和反向引物各1.5μl,kod fx dna聚合酶1μl,模板1μl约50ng,加入10μl的ddh2o补足至总体积为50μl。
97.pcr反应程序为:
①
98℃预变性3min;
②
35循环(98℃10s,60℃30s,68℃2.5min);
③
最后延伸68℃5min;
④
4℃保存。
98.琼脂糖凝胶电泳回收2.6kb的u3
‑
grna表达框前体和u6a
‑
grna表达框前体。
99.(4)u3
‑
grna表达框前体和u6a
‑
grna表达框前体分别与靶位点重组接头eui1
‑
u3
‑
t1
‑
adaptor和eui
‑
u6a
‑
t2
‑
adaptor进行酶促组装得到酶促组装产物eui1
‑
u3
‑
t1
‑
grna和eui1
‑
u6a
‑
t2
‑
grna。
100.酶促反应体系为:全式金2
×
assembly mix 5ul,grna表达框前体2.0μl(约200ng),靶位点重组接头1.0μl,补足ddh2o至总体积10μl。
101.反应条件:50℃,20min;20℃,2min;之后12℃保存。
102.(5)将酶促组装产物eui1
‑
u3
‑
t1
‑
grna和eui1
‑
u6a
‑
t2
‑
grna通过热激方式转化大肠杆菌感受态细胞得到转化产物。将转化产物涂布于含有卡那霉素抗性的lb平板上过夜培养。第二天挑取单菌落用通用引物m13
‑
f和m13
‑
r进行菌落pcr检测,筛选条带大小为650bp的单菌落并送测序。
103.测序结果显示:peui1
‑
u3
‑
t1
‑
grna和peui1
‑
u6a
‑
t2
‑
grna质粒含有靶位点的完整grna表达框。
104.(6)利用swai酶切c9dz载体,琼脂糖凝胶电泳后回收线性化载体c9dz,酶切体系和酶切条件按照neb公司说明书进行。利用重组引物gr
‑
zf和gr
‑
zr扩增步骤(5)中测序正确的含有完整grna表达框的质粒peui1
‑
u3
‑
t1
‑
grna和peui1
‑
u6a
‑
t2
‑
grna。
105.gr
‑
zf(seq26)的核苷酸序列如seq id no.26所示,具体为:cgtggatggacaatttaaattccgttttacctgtggaatcgg;
106.gr
‑
zr(seq27)的核苷酸序列如seq id no.27所示,具体为:caggtaaaacggaatttaattccatccactccaagctcttg。
107.所用pcr反应体系为:kod 2
×
pcr buffer 25μl,2mm dntp 10μl,正向和反向引物各1.5μl,kod fx dna聚合酶1μl,模板1μl约50ng,加入10μl的ddh2o补足至总体积为50μl。
108.pcr反应程序为:
①
98℃预变性3min;
②
35循环(98℃10s,60℃30s,68℃45s);
③
最后延伸68℃5min;
④
4℃保存。
109.琼脂糖凝胶电泳后回收600bp大小的eui1
‑
u3
‑
t1
‑
grna和eui1
‑
u6a
‑
t2
‑
grna表达框。
110.(7)将线性化载体c9dz和含有重组引物和完整grna表达框的eui1
‑
u3
‑
t1
‑
grna的pcr产物,进行酶促组装得到酶促组装产物c9dz
‑
eui1
‑
t1。
111.酶促反应体系为:全式金2
×
assembly mix 5μl,grna表达框前体2.0μl(约200ng),靶位点重组接头1.0μl,补足ddh2o至总体积10μl。
112.反应条件:50℃,20min;20℃,2min;之后12℃保存。
113.(8)将酶促组装产物c9dz
‑
eui1
‑
t1通过热激方式转化大肠杆菌感受态细胞,转化产物涂布于含有卡那霉素抗性的lb平板上过夜培养。第二天挑取单菌落用引物target
‑
j
‑
f和target
‑
j
‑
r进行菌落pcr检测,筛选条带大小为803bp的单菌落并送测序。
114.pcr反应体系为:kod 2
×
pcr buffer 25ul,2mm dntp 10μl,正向和反向引物各1.5μl,kod fx dna聚合酶1μl,模板1μl约50ng,加入10μl的ddh2o补足至总体积为50μl。
115.pcr反应程序为:
①
98℃预变性3min;
②
35循环(98℃10s,60℃30s,68℃50s);
③
最后延伸68℃5min;
④
4℃保存。
116.测序结果显示:正确的质粒为含有单个靶位点的基因编辑载体pc9dz
‑
eui1
‑
t1。
117.(9)swai酶切pc9dz
‑
eui1
‑
t1载体,与含有重组引物和完整grna表达框的eui1
‑
u6a
‑
t2
‑
grna表达框pcr产物进行酶促组装得到酶促组装产物c9dz
‑
eui1
‑
t1
‑
t2。将酶促组装产物转化大肠杆菌后,用target
‑
j
‑
f和target
‑
j
‑
r引物筛选条带大小为1362bp的单菌落并送测序。
118.pcr反应体系为:kod 2
×
pcr buffer 25μl,2mm dntp 10μl,正向和反向引物各1.5μl,kod fx dna聚合酶1μl,模板1μl约50ng,加入10μl的ddh2o补足至总体积为50μl。
119.pcr反应程序为:
①
98℃预变性3min;
②
35循环(98℃10s,60℃30s,68℃1min);
③
最后延伸68℃5min;
④
4℃保存。
120.测序结果显示:正确的质粒为含有双靶位点的基因编辑载体pc9dz
‑
eui1
‑
t1
‑
t2。
121.(10)将基因编辑载体pc9dz
‑
eui1
‑
t1
‑
t2采用农杆菌介导的方法转化水稻品种中花11。
122.(11)从t0代植株中观察株高,收获株高增加的t0代植株种子,并从株高增加的单株中筛选无荧光的种子(参见附图3)。
123.(12)将株高增加的无荧光种子种下去后,田间表型为株高增加且结出的种子也不发荧光(参见附图4),利用pcr检测植株中的cas9基因和hpt基因,发现不发荧光种子发育成的植株不含外源cas9基因和hpt基因(参见附图5),说明这些植株为无转基因成分且靶位点发生突变的基因编辑植株。
124.以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制。虽然本发明已以较佳实施例揭示如上,然而并非用以限定本发明。任何熟悉本领域的技术人员,在不脱离本发明的精神实质和技术方案的情况下,都可利用上述揭示的方法和技术内容对本发明技术方案做出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同替换、等效变化及修饰,均仍属于本发明技术方案保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1