一种2-苯基苯并噻唑衍生物的制备方法与流程
一种2
‑
苯基苯并噻唑衍生物的制备方法
技术领域
1.本技术属于有机化学合成技术领域,特别是涉及一种2
‑
苯基苯并噻唑衍生物的制备方法。
背景技术:
[0002]2‑
苯基苯并噻唑的分子式为c13h9ns,不溶于水。2
‑
苯基苯并噻唑衍生物由于本身特殊的结构,使得其具有多方面的应用。在其结构的2位引入不同的活性基团,会在不同的领域都有不同的应用和更优异的性能,由于近年来科学家们对有机电至发光材料的不断研究和开发,2位引入各种取代基需要越来越高的纯度和收率,而且随着oled有机发光材料的不断突破和市场化,对其中间体材料有了更大的需求,因此需要不停的创新和对工艺不断的进行优化和改进。
[0003]
现有的2
‑
苯基苯并噻唑衍生物选用原材料污染大且价格昂贵,反应步骤长反应复杂,产率低,后处理复杂。
技术实现要素:
[0004]
1.要解决的技术问题
[0005]
基于现有的2
‑
苯基苯并噻唑衍生物原材料污染大且价格昂贵,反应步骤长反应复杂,产率低,后处理复杂的问题,本技术提供了一种2
‑
苯基苯并噻唑衍生物的制备方法。
[0006]
2.技术方案
[0007]
为了达到上述的目的,本技术提供了一种2
‑
苯基苯并噻唑衍生物的制备方法,以取代芳基羧酸和邻氨基苯硫酚为原料发生反应得到2
‑
苯基苯并噻唑衍生物。
[0008]
本技术提供的另一种实施方式为:在脱水缩合剂存在下邻氨基苯硫酚和取代芳基羧酸通过两次脱水缩合得到2
‑
苯基苯并噻唑衍生物。
[0009]
本技术提供的另一种实施方式为:所述方法包括如下步骤:在反应容器中加入脱水缩合剂和溶剂,升温至80℃~100℃开启搅拌,然后加入取代芳基羧酸和邻氨基苯硫酚,在150℃~180℃下搅拌1~5小时,降温至100℃以下,导入水中淬灭,搅拌1~5小时后抽滤,得固体后在用乙酸乙酯溶解后过硅胶柱,浓缩后得到2
‑
苯基苯并噻唑衍生物w1。
[0010]
本技术提供的另一种实施方式为:所述脱水缩合剂为多聚磷酸。
[0011]
本技术提供的另一种实施方式为:所述溶剂为磷酸。
[0012]
本技术提供的另一种实施方式为:所述抽滤采用抽滤漏斗,用所述抽滤漏斗抽滤后得到的是固体粗品需要进一步纯化。
[0013]
本技术提供的另一种实施方式为:所述取代芳基羧酸为苯甲酸,对溴苯甲酸,间溴苯甲酸,对氨基苯甲酸,间氨基苯甲酸,间硝基苯甲酸,2
‑
萘甲酸或者4
‑
联苯甲酸。
[0014]
本技术提供的另一种实施方式为:所述2
‑
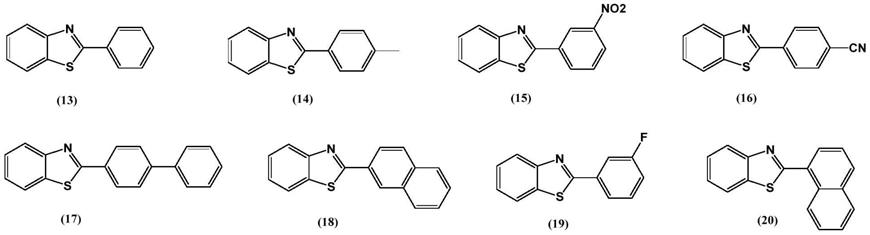
苯基苯并噻唑衍生物具体结构式如下:
[0015][0016]
本技术还提供一种2
‑
苯基苯并噻唑衍生物的应用,将所述的2
‑
苯基苯并噻唑衍生物应用于合成药物,有机电至发光材料,高分子材料或者有机柔性材料。
[0017]
3.有益效果
[0018]
与现有技术相比,本技术提供的2
‑
苯基苯并噻唑衍生物及其制备方法的有益效果在于:
[0019]
本技术提供的2
‑
苯基苯并噻唑衍生物制备方法,原材料便宜易得且没有太多危险性,采用常规反应釜即可,合成操作过程简便容易操作,相对于其它方案反应步骤少相应的能耗更少,后处理更加简单。
[0020]
本技术提供的2
‑
苯基苯并噻唑衍生物制备方法,在简便的后处理之后能得到高纯度产品,产率高所以成本与其它方法比较更加的低廉,没有产生废溶剂,产生的废水少,原子利用率高所以更加的绿色环保。
附图说明
[0021]
图1为本技术的2
‑
苯基苯并噻唑衍生物合成路线示意图。
具体实施方式
[0022]
在下文中,将参考附图对本技术的具体实施例进行详细地描述,依照这些详细的描述,所属领域技术人员能够清楚地理解本技术,并能够实施本技术。在不违背本技术原理的情况下,各个不同的实施例中的特征可以进行组合以获得新的实施方式,或者替代某些实施例中的某些特征,获得其它优选的实施实施方式。
[0023]
当量指与特定或俗成的数值相当的量;化学专业用语,用作物质相互作用时的质量比值的称谓。
[0024]
参见图1,本技术提供一种2
‑
苯基苯并噻唑衍生物制备方法,以取代芳基羧酸和邻氨基苯硫酚为原料发生反应得到2
‑
苯基苯并噻唑衍生物。在多聚磷酸存在下邻氨基苯硫酚和取代芳基羧酸通过两次脱水缩合得到2
‑
苯基苯并噻唑衍生物。
[0025]
进一步地,所述方法包括如下步骤:在反应容器中加入脱水缩合剂和溶剂,升温至80℃~100℃开启搅拌,然后加入取代芳基羧酸和邻氨基苯硫酚,在150℃~180℃下搅拌1~5小时,降温至100℃以下,导入水中淬灭,搅拌1~5小时后抽滤,得固体后在用乙酸乙酯溶解后过硅胶柱,浓缩后得到2
‑
苯基苯并噻唑衍生物w1。这里的脱水缩合剂和溶剂的比例没有限制。
[0026]
用水淬灭反应的作用:第一点可以破坏多聚磷酸使之变成磷酸失去脱水剂的作用从而使反应停止,第二点用水可以稀释磷酸的浓度使酸变稀便与处理,第三用常温水可以降低反应液温度从而达到降温的效果,第四用水处理可以让反应体系中生成的产品从溶解状态转化成不溶解的固体析出达到分离提纯的效果。
[0027]
产品用抽滤漏斗抽滤后得到的是固体粗品需要进一步纯化,产品在乙酸乙酯中比较好溶解,溶解后变成液体状态,并且乙酸乙酯属于一种的毒性很小的化学物质。
[0028]
下边过硅胶柱就是过层析柱的意思,这是在化学上的一种纯化产品的方法,层析柱里面需要填充介质这种介质大部分选择的是硅胶,因为硅胶便宜且结构稳定不会污染产品或与产品反应,它的原理等同于纯净水的净化,把含杂质的水通过小碎石阻拦杂质得到净化一个道理。
[0029]
进一步地,所述脱水缩合剂为多聚磷酸。
[0030]
进一步地,所述溶剂为磷酸。
[0031]
进一步地,所述取代芳基羧酸为苯甲酸,对溴苯甲酸,间溴苯甲酸,对氨基苯甲酸,间氨基苯甲酸,间硝基苯甲酸,2
‑
萘甲酸,4
‑
联苯甲酸等。
[0032]
本技术还提供一种2
‑
苯基苯并噻唑衍生物,所述衍生物通式为:
[0033][0034]
其中,r为氢,烷基,卤素,氰基,硝基,芳香化合物及其衍生物或者稠环;
[0035]
进一步地,所述烷基为c1~c40,所述芳基为苯、甲苯、联苯或者其他芳基衍生物,所述稠环为萘基,菲或者蒽。
[0036]
进一步地,所述烷基为c1~c5,所述芳基为苯环或者甲苯,所述稠环为萘基。
[0037]
进一步地,选自下述具体结构式:
[0038][0039]
本技术还提供一种2
‑
苯基苯并噻唑衍生物的应用,将所述的2
‑
苯基苯并噻唑衍生物应用于合成药物,有机电至发光材料,高分子材料或者有机柔性材料。
[0040]
向1000ml反应器中加入多聚磷酸,磷酸升温至80℃~100℃开启搅拌,然后加入取代芳基羧酸和邻氨基苯硫酚摩尔比例为1~2:1优选的,所述取代芳基羧酸和邻氨基苯硫酚摩尔比例为1:1,在150℃~180℃搅拌1~5小时,降温至100℃以下,导入水中淬灭,搅拌1小时后抽滤,得固体后在用甲苯溶解后过硅胶柱,浓缩后得到高纯度的2
‑
苯基苯并噻唑衍生物产品。
[0041]
以下实施例用于说明本技术,但不用来限制本技术的范围。本技术中所使用的基本原材料均为常规市售产品。
[0042]
实施例1:
[0043]
向1000ml反应器中加入480ml多聚磷酸,120ml磷酸,升温至80℃,开启搅拌,然后加入(0.384mol,48g)邻氨基苯硫酚搅拌半小时后加入(0.384mol,77g)对溴苯甲酸;升温至150℃,反应1小时后,降温至80℃,加入至1200ml水中淬灭1小时,抽滤漏斗抽滤得到固体粗品,固体粗品用乙酸乙酯溶解后过硅胶柱,浓缩后得到精品产品2
‑
(4
‑
溴苯基)苯并噻唑,hplc=99.2%,102.5g收率92%。
[0044]
实施例2:
[0045]
向1000ml反应器中加入480ml多聚磷酸,120ml磷酸,升温至80℃,开启搅拌,然后加入(0.384mol,48g)邻氨基苯硫酚搅拌半小时后加入(0.384mol,77g)间溴苯甲酸;升温至160℃,反应2小时后,降温至90℃,加入至1200ml水中淬灭1小时,抽滤漏斗抽滤得到固体粗品,固体粗品用乙酸乙酯溶解后过硅胶柱,浓缩后得到精品产品2
‑
(3
‑
溴苯基)苯并噻唑,hplc=99.3%,91.3g收率85%。
[0046]
实施例3:
[0047]
向1000ml反应器中加入480ml多聚磷酸,120ml磷酸,升温至80℃,开启搅拌,然后
加入(0.384mol,48g)邻氨基苯硫酚搅拌半小时后加入(0.768mol,105.2g)对氨基苯甲酸;升温至170℃,反应3小时后,降温至80℃,加入至1200ml水中淬灭1小时,通过液相检测原料邻氨基苯硫酚以反应完毕但对氨基苯甲酸有大量多余,抽滤漏斗抽滤得到固体粗品,固体粗品用乙酸乙酯溶解后过硅胶柱,浓缩后得到粗品纯度94.3%,再用乙醇重结晶得到产品2
‑
(4
‑
氨基苯基)苯并噻唑,hplc=99.0%,45.1g收率52%。
[0048]
实施例4:
[0049]
向1000ml反应器中加入480ml多聚磷酸,120ml磷酸,升温至100℃,开启搅拌,然后加入(0.384mol,48g)邻氨基苯硫酚搅拌半小时后加入(0.384mol,64.1g)间硝基苯甲酸;升温至180℃,反应1小时后,降温至70℃,加入至1200ml水中淬灭1小时,抽滤漏斗抽滤得到固体粗品,固体粗品用乙酸乙酯溶解后过硅胶柱,浓缩后得到精品产品2
‑
(3
‑
硝基苯基)苯并噻唑,hplc=99.1%,67.8g收率69%。
[0050]
实施例5:
[0051]
向1000ml反应器中加入480ml多聚磷酸,120ml磷酸,升温至90℃,开启搅拌,然后加入(0.384mol,48g)邻氨基苯硫酚搅拌半小时后加入(0.384mol,66.0g)1
‑
萘甲酸;升温至165℃,反应5小时后,降温至85℃,加入至1200ml水中淬灭1小时,抽滤漏斗抽滤得到固体粗品,固体粗品用乙酸乙酯溶解后过硅胶柱,浓缩后得到精品产品2
‑
(1
‑
萘基)苯并噻唑,hplc=99.4%,77.2g收率77%。
[0052]
实施例6:
[0053]
向1000ml反应器中加入480ml多聚磷酸,120ml磷酸,升温至80℃,开启搅拌,然后加入(0.384mol,48g)邻氨基苯硫酚搅拌半小时后加入(0.576mol,80.6g)间氟苯甲酸;升温至150℃,反应2小时后,降温至90℃,液相监测原料邻氨基苯硫酚反应完毕,间氟苯甲酸有少量剩余,加入至1200ml水中淬灭1小时,抽滤漏斗抽滤得到固体粗品,固体粗品用乙酸乙酯溶解后过硅胶柱,浓缩后得到粗品纯度96.2%,再用无水乙醇重结晶得到产品2
‑
(3
‑
溴苯基)苯并噻唑,hplc=99.3%,48.4g收率55%。
[0054]
实施例7:
[0055]
向1000ml反应器中加入480ml多聚磷酸,120ml磷酸,升温至80℃,开启搅拌,然后加入(0.384mol,48g)邻氨基苯硫酚搅拌半小时后加入(0.691mol,136.9g)4
‑
联苯甲酸;升温至160℃,反应3小时后,降温至90℃,液相监测原料邻氨基苯硫酚反应完毕,4
‑
联苯甲酸有大量剩余,加入至1200ml水中淬灭1小时,抽滤漏斗抽滤得到固体粗品,固体粗品用乙酸乙酯溶解后过硅胶柱,浓缩后得到粗品纯度93.8%,再用无水乙醇重结晶得到产品2
‑
(3
‑
溴苯基)苯并噻唑,hplc=99.1%,54.0g收率49%。
[0056]
上述实施例中实施例1为最佳实施例。
[0057]
尽管在上文中参考特定的实施例对本技术进行了描述,但是所属领域技术人员应当理解,在本技术公开的原理和范围内,可以针对本技术公开的配置和细节做出许多修改。本技术的保护范围由所附的权利要求来确定,并且权利要求意在涵盖权利要求中技术特征的等同物文字意义或范围所包含的全部修改。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1