一种邻苯二胺基桥联多芳氧基稀土-镁杂金属配合物及其制备方法与应用
一种邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物及其制备方法与应用
技术领域
1.本发明属于聚合物制备技术领域,尤其是指一种邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物及其制备方法与应用。
背景技术:
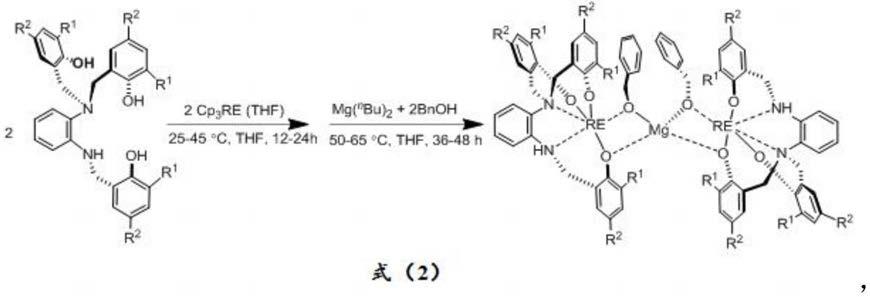
2.二氧化碳作为一种廉价的、无毒的且可再生的c1资源,将其转化为有用的化学品具有长远的战略意义。将二氧化碳作为原料不仅可以满足化学产品的绿色要求,也可以减少对化石资源的依赖。因此,对二氧化碳的转化越来越受到全社会的关注。二氧化碳转化的两个主要策略包括:(1)将co2选择性还原为co、ch4、meoh、烯烃和其他化合物;(2)将co2转化为高附加值的产品,例如环状碳酸酯、聚碳酸酯和噁唑烷酮,同时保持碳的氧化态为+4价。在这些方法中,二氧化碳与环氧化物共聚合成聚碳酸酯便是其中一种典型的例子。聚碳酸酯具有高的拉伸强度、热稳定性以及生物降解性,在食品包装、光学介质和粘合剂中得到了广泛的应用。在二氧化碳与环氧化物生成聚碳酸酯方面,二氧化碳具有较强的热力学稳定性,在参与反应时通常需要高温高压等比较苛刻的反应条件,因此在比较温和的条件下将二氧化碳转为相应的化学品依然存在很大的挑战。
3.早在1969年,日本化学家inoue首次报道了使用znet2/h2o体系非均相催化二氧化碳与环氧化物生成聚碳酸酯。这一体系在十分严苛的条件下,催化活性也很差,并且所得到聚合物的分子量较低(参见:inoue,s.;koinuma,h.;tsuruta,t.j.polym.sci.part b:polym.lett.1969,7,287.)。但是这一反应的发现开辟了co2与环氧化合物为原料生成聚碳酸酯的新领域,为以后的研究开创了先河,在此之后,研究者们致力于对共聚催化剂的开发,目的为了提高催化活性和选择性。
4.在过去的几十年里,报道了大量的包括过渡金属如钴、铬、镍、铁、锌等和主族元素铝、硼等均相的和非均相的金属配合物催化二氧化碳与环氧化物生成聚碳酸酯的研究,其中也有许多突出的工作。
5.2003年,coates报道了一种醋酸桥联的β二亚胺型的双锌金属配合物,在催化合成聚碳酸酯方面表现出很高的催化活性。经过一系列的探究发现,这种松散的双核结构的催化活性优于相应的单核金属配合物(参见:moore,d.r.;cheng,m.;lobkovsky,e.b.;coates,g.w.j.am.chem.soc.2003,125,11911)。
6.2015年,williams课题组首次报道了使用大环辅助配体稳定的锌镁杂核金属配合催化环氧环己烷与二氧化碳共聚,通过比较发现杂核金属配合物比相应的同核双金属配合物表现出更高的催化活性。报道的这种杂核金属配合物在0.01mol%的催化剂用量下,tof值达到660h
‑1(参见:garden,j.a.;saini,p.k.;williams,c.k.j.am.chem.soc.2016,137,15078.)。同年,姚英明等人使用邻苯二胺基桥联多芳氧基配体稳定的稀土
‑
锌杂核金属配合物催化环氧环己烷与二氧化碳共聚。在7bar二氧化碳压力下,表现出较好的催化活性。并且这是首例报道的含稀土的杂核金属配合物催化环氧环己烷与二氧化碳共聚的研究(参
见:qin,j.;xu,b.;zhang,y.;yuan,d.and yao,y.green chem.2016,18,4270.)。
7.2020年,williams课题组将金属钴与镁进行结合形成的杂核双金属配合物,应用于二氧化碳与环氧烷共聚时,表现出十分优异的催化活性,在140℃,20个二氧化碳压力下,得到的tof>12000h
‑1。(参见:deacy,a.c.;kilpatrick,a.f.r.;regoutz,a.;williams,c.k.nat.chem.2020,12,372.)。
技术实现要素:
8.为解决上述技术问题,本发明提供了一种邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物及其制备方法与应用。该方法具有高产率、反应条件温和等优点,在常温常压下有很高的催化效率,并且所得到的聚碳酸酯链节单元>99%。
9.一种邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物,所述金属配合物结构式如下所示:
10.其中,re选自la、nd、sm、eu、gd、y;
11.r1、r2独立的为氢、叔丁基、甲基、异丙基、甲氧基、枯基和卤素基团中的一种或多种。
12.一种邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物的制备方法,包括以下制备方法:
13.将邻苯二胺基桥联多芳氧基配体前体与三茂基稀土金属配合物反应12
‑
24小时后,接着与原位生成的苄氧基镁混合反应,在50
‑
65℃下反应36
‑
48小时,得到所述邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物,反应式如下:
14.15.re选自la、nd、sm、eu、gd、y;
16.r1、r2独立的为氢、叔丁基、甲基、异丙基、甲氧基、枯基和卤素基团中的一种或多种。
17.在本发明的一个实施例中,所述苄氧基镁与与稀土金属化合物的摩尔比为1
‑
1.3:2。
18.邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物在催化制备脂肪族聚碳酸酯中的应用。
19.在本发明的一个实施例中,应用包括以下步骤:在二氧化碳气氛中,以环氧环己烷为原料,在邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物催化作用下,在有机溶剂中发生共聚反应,得到所述脂肪族聚碳酸酯。所述有机溶剂为甲苯、四氢呋喃、乙腈、二氯甲烷、氯仿、乙醚,乙酸乙酯。进一步的有机溶剂为甲苯。
20.在本发明的一个实施例中,所述邻苯二胺基桥联多芳氧基稀土
‑
镁杂金属配合物与环氧环己烷的摩尔比为1:1000
‑
2000。
21.在本发明的一个实施例中,所述共聚反应温度为30
‑
90℃,所述二氧化碳气氛的压力为1bar
‑
20bar。进一步地,在20bar的二氧化碳压力下,当稀土中心金属为钕的杂金属配合物催化环氧环己烷与二氧化碳共聚,反应温度升高至50℃,反应的催化活性增加。进一步地,为了使反应条件温和,将二氧化碳的压力降低至7个大气压,催化活性略微降低,进一步地升高反应温度,反应活性增加,90℃下,在9个小时内,可以使环氧环己烷完全转化为聚碳酸酯,缩短反应时间。优选地,温度为90℃时,在7bar的二氧化碳压力下反应3个小时,可以达到81%的收率。进一步地,在30℃,1bar下,反应48小时,得到45%的收率。
22.进一步地,当稀土中心金属为钐的杂金属配合物催化环氧环己烷与二氧化碳共聚,30℃,20bar的二氧化碳压力下反应6小时可以达到98%的收率;进一步地,将压力降低至7个大气压,反应12小时,环氧环己烷可以得到完全的转化;升高温度至90℃,反应活性降低。进一步地,在30℃下,将压力降低至1个大气压,反应36小时,可以达到83%的收率。
23.在本发明的一个实施例中,所述共聚反应需要加入终止剂,所述终止剂为含盐酸的乙醇溶液。
24.在本发明的一个实施例中,所述的环氧环己烷和有机溶剂的体积比例为1:0.5
‑
2。进一步地,所述的环氧环己烷和甲苯的体积比例为1:0.5
‑
2,当不加入甲苯作为反应溶剂时,催化活性和聚合物的选择性降低,当v
环氧环己烷
:v
甲苯
=1:1,展现出最优的催化活性。
25.本发明利用上述催化剂催化环氧环己烷与二氧化碳共聚,其原理如下:
26.在聚合的引发阶段,首先可能是二氧化碳插入mg
‑
obn,形成新的碳酸盐,新形成的末端碳酸盐进攻被稀土金属中心配位活化的环氧环己烷,形成醇盐。后面,二氧化碳与环氧环己烷交替插入形成聚碳酸酯,直至反应被终止。通过1h nmr对聚合后的粗产物分析,可以证实得到的是完全交替的共聚物,没有聚醚等副产物的生成。
27.本发明的上述技术方案相比现有技术具有以下优点:
28.1)本发明使用的催化剂结构明确,产率高,分离提纯简单。
29.2)本发明选用的催化剂活性高,催化剂用量小,催化剂用量为0.1
‑
0.05mol%。共聚产率高。在较低的催化剂用量下,所得到聚合物的提纯十分方便。
30.3)本发明的制备方法中,原料价格廉价,反应条件温和,可以在常温常压下反应。
在适当的温度和压力下,可以以较短的时间达到不错的收率。反应操作和后处理过程简单。
31.4)本发明所使用的杂金属配合物催化剂结构明确、产率较高,催化效率高,并且在催化聚合过程中没有聚醚和环状碳酸酯副产物的生成,制得的聚碳酸酯的分子量较高,并且具有较窄分子量分布。尤其将金属镁与稀土金属结合得到的杂金属配合物的催化活性比相应的单金属配合物的催化活性高出一个数量级,并且不需要加入季铵盐或有机碱等作为助催化剂。展现出高活性的原因是不同金属在催化过程中起到不同的作用,环氧化物配位到稀土金属中心被活化,镁中心在催化过程中稳定聚合物链的碳酸根。活性聚合物链在两种金属之间穿梭,杂金属之间存在协同催化,提高了杂金属化合物的催化性能。
附图说明
32.为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中
33.图1是本发明实施例1所制备的配合物llamglal的核磁氢谱图。
34.图2是本发明实施例1所制备的配合物llamglal的核磁碳谱图。
35.图3是本发明实施例3所制备的配合物lsmmgsml的核磁氢谱图。
36.图4是本发明实施例3所制备的配合物lsmmgsml的核磁碳谱图。
37.图5是本发明实施例6所制备的配合物lymgyl的核磁氢谱图。
38.图6是本发明实施例6所制备的配合物lymgyl的核磁碳谱图。
39.图7是本发明实施例8所制备的环氧环己烷与二氧化碳共聚得到的聚碳酸酯的核磁氢谱图。
40.图8是本发明实施例8所制备的环氧环己烷与二氧化碳共聚得到的聚碳酸酯的核磁碳谱图。
41.图9是本发明中脂肪族聚碳酸酯反应示意图。
具体实施方式
42.下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
43.实施例1
44.邻苯二胺基桥联多芳氧基稀土
‑
镁杂核金属配合物llamglal的制备:
45.(1)将1.52克lh3(2.00毫摩尔)溶于四氢呋喃,加入到含有0.812克lacp3(thf)(2.00毫摩尔)的四氢呋喃溶液中,室温搅拌反应12小时,体系为淡黄色透明溶液,记为混合物1;次日,称取正丁基镁溶液(1.0毫摩尔/毫升,1.0毫升)滴加到苄醇溶液(1.0毫摩尔/毫升,2.0毫升)中,室温搅拌3小时,得到混合物2。将混合物1转至混合物2中,在50℃下搅拌三天。
46.(2)除去溶剂,加入0.5毫升四氢呋喃和3毫升正己烷,离心。清液转移,室温下放置两日,析出黄色晶体,将析出的晶体在真空下抽干封存(1.24克,产率61%)。核磁氢谱(400mhz,thf
‑
d8)δ7.73(d,j=8.1hz,2h,ar
–
h),7.28
‑
7.03(m,14h,ar
‑
h),6.94(d,j=5.8hz,6h,ar
–
h),6.83(s,2h,ar
–
h),6.75(d,j=2.5hz,2h,ar
‑
h),6.59(s,2h,ar
–
h),6.33(s,2h,ar
‑
h),4.53(d,j=11.5hz,6h,phch2o,nch2ar),4.10(d,j=2.3hz,1h,nch2ar),3.89
(d,j=3.8hz,2h,nch2ar),3.75(s,2h,nch2ar),3.39(s,1h,nch2ar),3.22(s,2h,nch2ar),2.98(s,2h,nch2ar),1.45(s,18h,c(ch3)3),1.31
‑
1.22(m,36h,c(ch3)3),1.18(s,18h,c(ch3)3),1.09
‑
0.95(m,36h,c(ch3)3)(见图1)。核磁碳谱(101mhz,thf
‑
d8)δ162.7,161.9,145.6,134.6,134.0,128.2,126.7,126.6,126.3,125.9,125.1,123.7,122.7(ar
‑
c),63.2,61.0,58.3(phch2),34.8,34.6,33.5,33.4(c(ch3)3),31.6,31.5,31.3,31.2,29.6(c(ch3)3)(见图2)。元素分析anal.calcd for c
116
h
156
n4o8la2mg:c,68.41;h,7.72;n,2.75;found:c,67.85;h,7.90;n,2.73。
47.实施例2
48.邻苯二胺基桥联多芳氧基稀土
‑
镁杂核金属配合物lndmgndl的制备:
49.(1)将1.52克lh3(2.00毫摩尔)溶于四氢呋喃,加入到含有0.823克ndcp3(thf)(2.00毫摩尔)的四氢呋喃溶液中,室温搅拌反应12小时,体系为蓝色透明溶液,记为混合物1;次日,称取正丁基镁溶液(1.0毫摩尔/毫升,1.0毫升)滴加到苄醇溶液(1.0毫摩尔/毫升,2.0毫升)中,室温搅拌3小时,得到混合物2。将混合物1转至混合物2中,在50℃下搅拌三天。
50.(2)除去溶剂,加入0.5毫升四氢呋喃和3毫升正己烷,离心。清液转移,室温下放置两日,析出蓝色晶体,将析出的晶体在真空下抽干封存(1.33克,产率65%)。元素分析anal.calcd for c
116
h
156
n4o8nd2mg:c,68.05;h,7.68;n,2.74.found:c,67.46;h,7.79;n,2.72。
51.实施例3
52.邻苯二胺基桥联多芳氧基稀土
‑
镁杂核金属配合物lsmmgsml的制备:
53.(1)将1.52克lh3(2.00毫摩尔)溶于四氢呋喃,加入到含有0.835克smcp3(thf)(2.00毫摩尔)的四氢呋喃溶液中,室温搅拌反应12小时,体系为黄色透明溶液,记为混合物1;次日,称取正丁基镁溶液(1.0毫摩尔/毫升,1.0毫升)滴加到苄醇溶液(1.0毫摩尔/毫升,2.0毫升)中,室温搅拌3小时,得到混合物2。将混合物1转至混合物2中,在50℃下搅拌三天。
54.(2)除去溶剂,加入0.5毫升四氢呋喃和3毫升正己烷,离心。清液转移,室温下放置两日,析出淡黄色晶体,将析出的晶体在真空下抽干封存(1.32克,产率63%)。核磁氢谱(400mhz,thf
‑
d8)δ12.93(s,2h,nch2ar),9.91(s,2h,ar
–
h),7.83(d,j=2.6hz,2h,ar
–
h),7.52(s,2h,ar
–
h),7.32
–
6.88(m,14h,ar
–
h),6.33(t,j=7.5hz,2h,ar
–
h),6.01(d,j=2.3hz,2h,ar
–
h),5.89(s,2h,ar
–
h),5.69(t,j=6.7hz,2h,ar
–
h),5.31(s,2h,ar
–
h),4.68(s,2h,phch2o ornch2ar),4.28(s,2h,phch2o ornch2ar),4.12(s,4h,phch2o or nch2ar),3.27(s,18h,c(ch3)3),2.70(s,18h,c(ch3)3),2.55(s,18h,c(ch3)3),1.33(s,18h,c(ch3)3),1.22(s,18h,c(ch3)3),0.82(s,18h,c(ch3)3),
‑
1.72(s,2h,nch2ar),
‑
2.65(s,2h,nch2ar),
‑
4.57(s,2h,nch2ar)(见图3)。核磁碳谱(101mhz,thf
‑
d8)δ171.4,168.9,166.1,139.1,135.2,134.8,134.7,134.2,134.0,133.8,128.1,127.7,126.5,126.2,125.9,125.2,124.4,123.7,123.5,123.3,122.5,122.4,121.4(ar
‑
c),70.7,64.3,53.0(phch2),36.9,36.8,36.7,33.7,33.7,33.1(c(ch3)3),31.7,31.6,31.4,31.3,30.9,30.8,30.8,22.6,13.5(c(ch3)3)(见图4)。元素分析anal.calcd for c
116
h
156
n4o8sm2mg:c,67.65;h,7.63;n,2.72;found:c,67.28;h,7.78;n,2.79。
55.实施例4
56.邻苯二胺基桥联多芳氧基稀土
‑
镁杂核金属配合物leumgeul的制备:
57.(1)将1.52克lh3(2.00毫摩尔)溶于四氢呋喃,加入到含有0.839克eucp3(thf)(2.00毫摩尔)的四氢呋喃溶液中,室温搅拌反应12小时,体系为红色透明溶液,记为混合物1;次日,称取正丁基镁溶液(1.0毫摩尔/毫升,1.0毫升)滴加到苄醇溶液(1.0毫摩尔/毫升,2.0毫升)中,室温搅拌3小时,得到混合物2。将混合物1转至混合物2中,在50℃下搅拌三天。
58.(2)除去溶剂,加入0.5毫升四氢呋喃和3毫升正己烷,离心。清液转移,室温下放置两日,析出红色晶体,将析出的晶体在真空下抽干封存(1.41克,产率67%)。元素分析anal.calcd for c
116
h
156
n4o8eu2mg:c,67.54;h,7.62;n,2.72;found:c,67.02;h,7.77;n,2.81。
59.实施例5
60.邻苯二胺基桥联多芳氧基稀土
‑
镁杂核金属配合物lgdmggdl的制备:
61.(1)将1.52克lh3(2.00毫摩尔)溶于四氢呋喃,加入到含有0.839克gdcp3(thf)(2.00毫摩尔)的四氢呋喃溶液中,室温搅拌反应12小时,体系为无色透明溶液,记为混合物1;次日,称取正丁基镁溶液(1毫摩尔/毫升,1毫升)滴加到苄醇溶液(1毫摩尔/毫升,2毫升)中,室温搅拌3小时,得到混合物2。将混合物1转至混合物2中,在50℃下搅拌三天。
62.(2)除去溶剂,加入0.5毫升四氢呋喃和3毫升正己烷,离心。清液转移,室温下放置两日,析出无色晶体,将析出的晶体在真空下抽干封存(1.24克,产率60%)。元素分析anal.calcd for c
116
h
156
n4o8gd2mg:c,67.20;h,7.58;n,2.70;found:c,66.65;h,7.49;n,2.83。
63.实施例6
64.邻苯二胺桥联多芳氧基稀土
‑
镁杂核金属配合物lymgyl的制备:
65.(1)将1.52克lh3(2.00毫摩尔)溶于四氢呋喃,加入到含有0.712克ycp3(thf)(2.00毫摩尔)的四氢呋喃溶液中,室温搅拌反应12小时,体系为淡黄色透明溶液,记为混合物1;次日,称取正丁基镁溶液(1.0毫摩尔/毫升,1.0毫升)滴加到苄醇溶液(1.0毫摩尔/毫升,2.0毫升)中,室温搅拌3小时,得到混合物2。将混合物1转至混合物2中,在50℃下搅拌三天。
66.(2)除去溶剂,加入0.5毫升四氢呋喃和3毫升正己烷,离心。清液转移,室温下放置两日,析出黄色晶体,将析出的晶体在真空下抽干封存(1.22克,产率63%)。核磁氢谱(400mhz,thf
‑
d8)δ7.71(s,2h,ar
‑
h),7.59(s,1h,ar
‑
h),7.34
‑
7.11(m,19h,ar
‑
h),7.04
‑
6.99(m,3h,ar
‑
h),6.97(s,1h,ar
‑
h),6.92(s,1h,ar
‑
h),6.87(s,1h,ar
‑
h),6.71(s,1h,ar
‑
h),6.50
‑
6.44(m,1h,ar
‑
h),4.64
‑
4.36(m,6h,phch2o,nch2ar),4.17(s,1h,nch2ar),4.03(d,j=12.4hz,2h,phch2o,nch2ar),3.89(s,2h,nch2ar),3.74(s,3h,nch2ar),3.49(s,2h,nch2ar),1.51(s,18h,c(ch3)3),1.27(s,18h,c(ch3)3),1.22(s,36h,c(ch3)3),1.10(s,18h,c(ch3)3),1.03(s,18h,c(ch3)3)(见图5)。核磁碳谱(101mhz,thf
‑
d8)δ161.2,161.1,160.6,144.8,135.2,135.2,135.1,134.9,134.7,127.8,126.8,126.5,126.3,126.2,126.1,125.9,125.1,124.8,124.1,123.4,123.1,122.8,122.7,122.7(ar
‑
c),64.3(nch2),60.7(phch2o),56.1(nch2),34.8,34.6,34.5,33.5,33.4,33.3(c(ch3)3),31.3,31.2,31.1,29.5,29.4,29.3(c(ch3)3)(见图6)。元素分析anal.calcd for c
116
h
156
n4o8y2mg:c,71.94;h,8.12;n,2.89;found:c,71.16;h,8.19;n,3.06。
67.实施例7
68.0.1mol%nd
‑
mg催化环氧环己烷与二氧化碳反应:
69.在手套箱中,样品瓶中称0.0201g lndmgndl(9.842
×
10
‑3毫摩尔),加入1.0毫升甲苯溶解,再加入1.0毫升环氧环己烷。将样品瓶放在预热到30℃的parr反应釜中,将反应釜密封,充入二氧化碳至20bar,反应12小时,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为86%,聚碳酸酯选择性为99%。在样品瓶中加入2毫升二氯甲烷,再加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。对所得固体产物进行gpc分析,测得共聚物的mn=170.6
×
103g/mol,分子量分布
70.实施例8
71.0.1mol%sm
‑
mg催化环氧环己烷与二氧化碳反应:
72.在手套箱中,样品瓶中称0.0203g lsmmgsml(9.842
×
10
‑3毫摩尔),加入1.0毫升甲苯溶解,再加入1.0毫升环氧环己烷。将样品瓶放在预热到30℃的parr反应釜中,将反应釜密封,充入二氧化碳至20bar,反应12小时,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为99%,聚碳酸酯选择性为99%。在样品瓶中加入2毫升二氯甲烷,再加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。对所得固体产物进行gpc分析,测得共聚物的mn=184.9
×
103g/mol,分子量分布核磁氢谱(400mhz,chloroform
‑
d)δ4.62(s,2h,ch),2.12(s,2h,ch2),1.71(s,2h,ch2),1.36(s,4h,ch2)(见图7)。核磁碳谱(151mhz,chloroform
‑
d)δ153.7(ch),153.0(ch),29.7(ch2),23.0(ch2)(见图8)。
73.实施例9
74.0.1mol%nd
‑
mg催化环氧环己烷与二氧化碳反应:
75.在手套箱中,样品瓶中称0.0201g lndmgndl(9.842
×
10
‑3毫摩尔),加入1.0毫升甲苯溶解,再加入1.0毫升环氧环己烷。将样品瓶放在预热到90℃的parr反应釜中,将反应釜密封,充入二氧化碳至7bar,反应3小时,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为80%,聚碳酸酯选择性为99%,tof值为276h
‑1。在样品瓶中加入2毫升二氯甲烷,再加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。对所得固体产物进行gpc分析,测得共聚物的mn=30.03
×
103g/mol,分子量分布
76.实施例10
77.0.05mol%nd
‑
mg催化环氧环己烷与二氧化碳反应:
78.在手套箱中,样品瓶中称0.0201g lndmgndl(9.842
×
10
‑3毫摩尔),加入2.0毫升甲苯溶解,再加入2.0毫升环氧环己烷。将样品瓶放在预热到90℃的parr反应釜中,将反应釜密封,充入二氧化碳至20bar,反应20分钟,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为58%,聚碳酸酯选择性为99%。在样品瓶中加入2毫升二氯甲烷,再加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。对所得固体产物进行gpc分析,测得共聚物的mn=5.58
×
103g/mol,分子量分布
79.实施例11
80.0.1mol%sm
‑
mg催化环氧环己烷与二氧化碳反应:
81.在手套箱中,反应瓶中称0.0203g lsmmgsml(9.842
×
10
‑3毫摩尔),加入1.0毫升甲苯溶解,再加入1.0毫升环氧环己烷。反应瓶连接一个装有二氧化碳的气袋,加热至30℃,反应36小时,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为
83%,聚碳酸酯选择性为99%。在样品瓶中加入2毫升二氯甲烷,加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。对所得固体产物进行gpc分析,测得共聚物的mn=32.57
×
103g/mol,分子量分布
82.实施例12
83.0.05mol%nd
‑
mg催化环氧环己烷与二氧化碳反应:
84.在手套箱中,样品瓶中称0.0201g lndmgndl(9.842
×
10
‑3毫摩尔),加入2.0毫升甲苯溶解,再加入2.0毫升环氧环己烷。将样品瓶放在预热到90℃的parr反应釜中,将反应釜密封,充入二氧化碳至20bar,反应0.3小时,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为58%,聚碳酸酯选择性为99%,tof值为3870h
‑1。在样品瓶中加入2毫升二氯甲烷,再加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。对所得固体产物进行gpc分析,测得共聚物的mn=5.58
×
103g/mol,分子量分布所述tof=产率/(催化剂用量
×
反应时间)。
85.对比例1(和实施例12对比)
86.0.05mol%nd
‑
zn催化环氧环己烷与二氧化碳反应:
87.在手套箱中,样品瓶中称0.0206g lndznndl(9.842
×
10
‑3毫摩尔),加入2.0毫升甲苯溶解,再加入2.0毫升环氧环己烷。将样品瓶放在预热到90℃的parr反应釜中,将反应釜密封,充入二氧化碳至20bar,反应0.3小时,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为14%,聚碳酸酯选择性为99%,tof值为933h
‑1。在样品瓶中加入2毫升二氯甲烷,再加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。因为所得聚合物量太少,没有进行gpc分析。所述tof=产率/(催化剂用量
×
反应时间)。
88.对比例2(和实施例9对比)
89.0.1mol%nd
‑
zn催化环氧环己烷与二氧化碳反应:
90.在手套箱中,样品瓶中称0.0206g lndznndl(9.842
×
10
‑3毫摩尔),加入1.0毫升甲苯溶解,再加入1.0毫升环氧环己烷。将样品瓶放在预热到90℃的parr反应釜中,将反应釜密封,充入二氧化碳至7bar,反应3小时,冰水浴中冷却,放掉多余的二氧化碳。取样,通过核磁氢谱分析,计算转化率为53%,聚碳酸酯选择性为99%,tof值为176h
‑1。在样品瓶中加入2毫升二氯甲烷,再加入10毫升乙醇溶液将聚合物沉降。过滤,置于真空干燥箱抽干,分离得到纯产物。对所得固体产物进行gpc分析,测得共聚物的mn=20.4
×
103g/mol,分子量分布所述tof=产率/(催化剂用量
×
反应时间)。
91.显然,上述实施例仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1