一种蒽环类药物中间体的制备方法与流程
1.本发明属于医药技术领域,具体涉及一种蒽环类药物中间体的制备方法。
背景技术:
2.蒽环类药物(anthracyclines)或蒽环类抗生素(anthracycline antibiotics)是一类来源于波赛链霉菌青灰变种(streptomyces peucetius var.caesius)的化疗药物。蒽环类药物,包括阿霉素、表阿霉素、柔红霉素和阿克拉霉素等广泛地用于治疗血液系统恶性肿瘤和实体肿瘤,如急性白血病、淋巴瘤、乳腺癌、胃癌、软组织肉瘤和卵巢癌等。蒽环类药物可以与其他化疗药物及分子靶向药物联合应用,以蒽环类药物为基础的联合治疗通常是一线治疗的标准方案,具有抗瘤谱广,抗肿瘤作用强,疗效确切等优势。
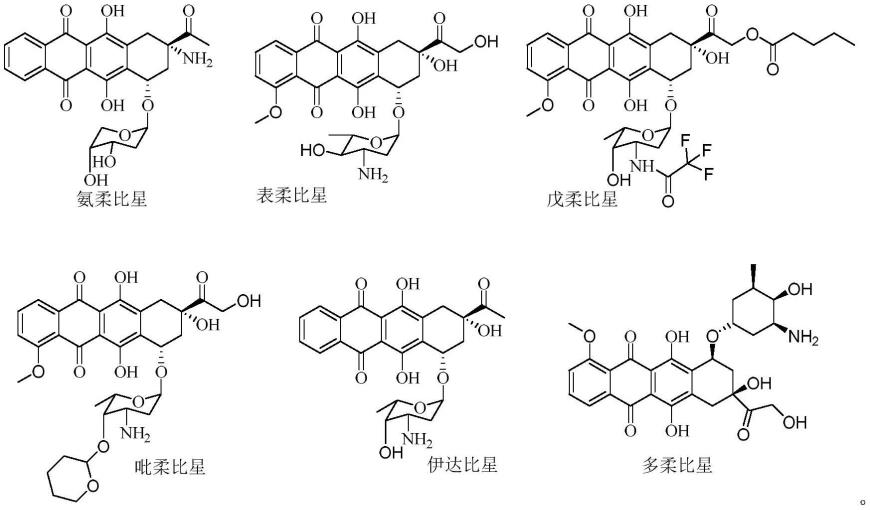
3.常见的蒽环类药物的结构式如下:
[0004][0005]
上述药物合成中,通常涉及对侧链酮羰基的保护反应,如文献j.org.chem.,1987,52,4477-4485报道的盐酸氨柔比星的合成路线:其中间体7与乙二醇反应对羰基进行保护,生成缩酮衍生物(r)-(-)-9-乙酰胺基-9-[1,1-(亚乙基二氧基)乙基]-6,11-二羟基-7,8,9,10-四氢-并四苯-5,12-二酮(1),具体路线如下:
[0006][0007]
现有技术中对上述蒽环类药物合成中侧链羰基的保护常采用乙二醇、1,3-丙二醇或者新戊二醇等作为保护试剂,高温回流反应,不仅容易导致副反应的发生,且对机器设备要求较高,增加能耗和生产成本。
[0008]
鉴于目前上述蒽环类药物侧链羰基保护中间体合成中存在的诸多问题,本技术发明人经过不断探索和实验,研究寻找到一种操作简便、生产周期较短、反应能耗低、收率更高、更适合工业化生产的工艺路线。
技术实现要素:
[0009]
针对目前蒽环类药物侧链羰基保护中间体(i)制备工艺中存在的技术问题,本发明提供了一种新的酮羰基保护制备该中间体化合物的方法。该方法反应条件温和,操作过程简便,反应周期短,所制得的目标产品具有较高的纯度、收率。
[0010]
本发明的具体技术方案如下:
[0011]
一种蒽环类药物中间体(i)的制备方法,具体步骤如下:
[0012]
室温,将sm、丁酮乙二醇缩酮(med)、乙二醇、催化剂加入干燥的反应溶剂中,控温搅拌反应,制得目标化合物i,反应路线如下:
[0013][0014]
其中r1=h,r2=ohornhcoch3,r3=ohorh或;
[0015][0016]
优选地,所述催化剂选自对甲苯磺酸、樟脑磺酸、α-萘磺酸、β-萘磺酸中的一种或其组合,优选β-萘磺酸;其中樟脑磺酸可为1r-(-)-10-樟脑磺酸、1s-(+)-10-樟脑磺酸中的单一异构体或其混合物或消旋物。
[0017]
优选地,所述反应溶剂选自苯、甲苯、二甲苯、二氯甲烷、氯仿中的一种或其组合,优选二氯甲烷。
[0018]
优选地,所述sm与med的投料摩尔比为1:1.2~1.8,优选1:1.4。
[0019]
优选地,所述sm与乙二醇的投料摩尔比为1:0.02~0.15,优选1:0.06。
[0020]
优选地,所述sm与催化剂的投料摩尔比为1:0.01~0.05,优选1:0.03。
[0021]
优选地,所述控温温度为10~50℃,优选20~25℃。
[0022]
优选地,所述sm为氨柔比星合成中间体,其结构如下:
[0023][0024]
优选地,所述sm为表柔比星、多柔比星、吡柔比星、戊柔比星合成中间体中的一种,其结构如下:
[0025][0026]
优选地,所述sm为伊达比星合成中间体或其他具有如下结构的蒽环类药物合成中间体,具体结构如下:
[0027][0028]
在一优选方案中需进行后处理操作,具体步骤为:反应结束后,向反应液中加入饱和碳酸氢钠溶液,分液取有机层,依次经纯化水、饱和食盐水洗涤,合并有机相,减压浓缩,干燥,制得目标化合物。
[0029]
本发明中所述的干燥的反应溶剂,是反应溶剂经本领域常规干燥方法获得,如分子筛除水,精馏等常规手段。
[0030]
所述后处理中的干燥为本领域常规干燥手段。
[0031]
本发明的有益效果:
[0032]
本发明提供了一种新的制备蒽环类药物中间体(i)的方法,以丁酮乙二醇缩酮代替现有技术中常用的乙二醇作为原料进行反应,室温下几乎可以定量得到相关中间体,并可显著降低反应温度以及分水器的使用,简化反应操作,减少能耗。同时在含有氰基的相关中间体的制备过程中可有效抑制氰基水解,提高产品收率及纯度。本发明所述的制备工艺相较于现有技术,制得的产品收率及纯度均较高,更适合工业化生产。
具体实施方式
[0033]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0034]
实施例1
[0035]
室温,将sm-1(39.30g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(1.24g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品42.62g,收率97.5%,纯度99.33%。
[0036]
实施例2
[0037]
室温,将sm-1(39.31g,0.10mol)、丁酮乙二醇缩酮(13.94g,0.12mol)、乙二醇(0.37g,0.02mol)、樟脑磺酸(0.70g,0.003mol)加入干燥的二甲苯(300ml)中,控温25~30℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品41.74g,收率95.5%,纯度99.19%。
[0038]
实施例3
[0039]
室温,将sm-1(39.31g,0.10mol)、丁酮乙二醇缩酮(12.78g,0.11mol)、乙二醇(0.37g,0.02mol)、1s-(+)-10-樟脑磺酸(0.70g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温30~35℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用乙酸乙酯(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品39.65g,收率90.7%,纯度98.23%。
[0040]
实施例4
[0041]
室温,将sm-1(39.30g,0.10mol)、丁酮乙二醇缩酮(20.91g,0.18mol)、乙二醇(0.37g,0.02mol)、1r-(-)-10-樟脑磺酸(0.70g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温15~20℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用甲基叔丁基醚(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品42.40g,收率97.0%,纯度99.04%。
[0042]
实施例5
[0043]
室温,将sm-1(39.30g,0.10mol)、丁酮乙二醇缩酮(22.07g,0.19mol)、乙二醇(0.37g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的氯仿(300ml)中,控温10~15℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品40.26g,收率92.1%,纯度98.04%。
[0044]
实施例6
[0045]
室温,将sm-1(39.30g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.12g,0.01mol)、α-萘磺酸(0.63g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温35~40℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品41.88g,收率95.8%,纯度99.22%。
[0046]
实施例7
[0047]
室温,将sm-1(39.30g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.93g,0.04mol)、对甲苯磺酸(0.52g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温40~45℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相
用氯仿(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品42.49g,收率97.2%,纯度99.14%。
[0048]
实施例8
[0049]
室温,将sm-1(39.32g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.36g,0.02mol)、β-萘磺酸(0.21g,0.001mol)加入干燥的苯(300ml)中,控温45~50℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品42.05g,收率96.2%,纯度98.94%。
[0050]
实施例9
[0051]
室温,将sm-1(39.30g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(1.24g,0.02mol)、β-萘磺酸(1.04g,0.005mol)加入干燥的甲苯(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品42.35g,收率96.9%,纯度99.10%。
[0052]
实施例10
[0053]
室温,将sm-2(40.90g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.37g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品44.05g,收率97.2%,纯度99.31%。esi-hrms(m/z):454.1514[m+h]
+
;1h-nmr(600mhz,dmso-d6)δ:9.02(s,2h),8.06~8.12(m,4h),5.80(s,1h),4.92(t,j=7.9hz,1h),3.95~4.02(m,6h),3.10(s,1h),2.30~2.33(m,1h),1.95~1.98(m,1h),1.92(s,3h),1.33(s,3h);
13
c-nmr(151mhz,dmso-d6)δ:186.38,186.22,170.48,154.26,153.48,146.37,139.84,134.46,133.57,125.69,114.35,110.58,105.74,66.31,65.78,42.59,41.36,23.18,21.26。
[0054]
实施例11
[0055]
室温,将sm-3(38.20g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.37g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品41.55g,收率97.5%,纯度99.16%。
[0056]
实施例12
[0057]
室温,将sm-4(39.81g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.37g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品42.89g,收率97.0%,纯度99.35%。
[0058]
实施例15
[0059]
室温,将sm-5(52.82g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇
(0.36g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品55.29g,收率96.8%,纯度99.02%。
[0060]
实施例18
[0061]
室温,将sm-6(35.20g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.37g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品37.33g,收率98.2%,纯度99.10%。
[0062]
实施例19
[0063]
室温,将sm-7(36.80g,0.10mol)、丁酮乙二醇缩酮(16.26g,0.14mol)、乙二醇(0.37g,0.02mol)、β-萘磺酸(0.62g,0.003mol)加入干燥的二氯甲烷(300ml)中,控温20~25℃反应约1h后,加入饱和碳酸氢钠溶液(150ml)搅拌10~15min后分液取有机相,水相用二氯甲烷(50ml
×
2)萃取,合并有机相,纯化水(140ml
×
3)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,制得目标产品38.58g,收率97.4%,纯度99.41%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1