一种钠-葡萄糖协同转运蛋白2抑制剂的制备方法与流程
1.本公开属于医药领域,涉及一种钠-葡萄糖协同转运蛋白2(sglt-2)抑制剂的制备方法。
背景技术:
2.近年来对糖尿病发病机制的深入研究,为ii型糖尿病的治疗提供了越来越多的途径。而钠-葡萄糖协同转运蛋白2(sodium-glucose transporter 2,sglt-2)抑制剂的发现,为糖尿病的治疗提供了又一新的思路。sglt-2的功能是负责转运葡萄糖。sglt-2抑制剂的治疗机制是通过选择性抑制sglt-2的活性来降低血糖。选择sglt-2作为靶点,一方面是因为它对葡萄糖的绝对重吸收作用,另一方面是因为它仅表达于肾脏。由于sglt-2抑制剂不介入葡萄糖代谢,因此该治疗方法是一种新颖的血糖控制方法。目前的研究还发现,sglt-2的作用机制不依赖于β细胞的功能异常或胰岛素抵抗的程度,其效果也不会随着β细胞功能的衰竭或严重胰岛素抵抗而下降。因此,有理由认为sglt-2抑制剂用于当前ii型糖尿病的治疗具有良好的前景。
3.wo2012019496公开了一种新的sglt-2抑制剂,其化合物结构如式i’所示,
[0004][0005]
wo2016050134公开了该化合物的l-脯氨酸复合物,结构如式i所示,
[0006][0007]
wo2012019496公开了式i’化合物的制备方法,合成工艺较为繁琐,需要对四氢吡喃环的四个羟基分别进行叔丁基二甲基硅(tbs)和苄基(bn)保护,并在反应过程中依次脱去,延长了工艺路线。同时,该工艺路线中存在需要柱层析纯化的步骤,不利于大规模生产,苄基保护的脱除需要利用昂贵的钯试剂,成本较高。
[0008]
cn107686496a公开了采用“一锅法”构建五元杂环的方法,合成效率有所提高,但仍未能有效解决上述问题。
技术实现要素:
[0009]
为了克服现有技术的不足,本公开的目的在于提供一种新的sglt-2抑制剂的制备
方法。
[0010]
本公开一方面提供了新的反应中间体,如式a所示化合物:
[0011][0012]
其中,r1、r2独立地选自h、-ch2oh、-ch2otms或-ch(=o),
[0013]
r3、r4、r5独立地选自h或tms,且当r1为-ch2oh而r2为h时,r3、r4、r5中至少有一个为tms。
[0014]
在一些实施方案中,式a所示化合物具体选自以下化合物:
[0015][0016]
本公开还提供了新的反应中间体,如式vi所示化合物:
[0017][0018]
r6、r7、r8、r9独立地选自h或q,q为
[0019]
其中,r
10
独立地选自氢、氘、卤素、硝基、氰基、苯基、任选的被卤素取代的c
3-c6环烷基、任选的被卤素取代的c
1-c6烷基以及任选的被卤素取代的c
1-c6烷氧基,
[0020]
n为选自1-5的整数。
[0021]
在一些实施方案中,r6为q,r7、r8、r9独立地选自h或q;优选r6、r7、r8、r9为q;
[0022]r10
可以独立地选自氢、卤素、硝基、氰基、任选的被卤素取代的c
3-c6环烷基、任选的被卤素取代的c
1-c3烷基以及任选的被卤素取代的c
1-c3烷氧基;
[0023]
在一些实施方案中,r
10
可以独立地选自氢、卤素、硝基、氰基以及c
1-c3烷氧基。
[0024]
在一些实施方案中,q可具体选自以下结构:
[0025]025][0026]
在一些实施方案中,q具体选自在一些实施方案中,q具体选自在一些实施方案中,q具体选自优选优选
更优选为
[0027]
本公开还提供了式iii所示化合物的制备方法,包括式ii所示化合物在碱性物质存在的条件下制备得到式iii所示化合物的步骤,
[0028][0029]
在一些实施方案中,反应溶剂选自水、c1~c6醇类、丙酮、二氯甲烷、氯仿、乙酸乙酯、四氢呋喃、二噁烷、乙腈、n,n-二甲基甲酰胺、n,n-二甲亚砜中的一种或多种,优选水、甲醇、乙醇、丙酮、二氯甲烷中的一种或多种,更优选甲醇;
[0030]
所述碱性物质选自li2co3、na2co3、ba(oh)2、k3po4、cs2co3、k2co3、kf、csf、bu4nf、lioh、naoh、koh、三乙胺、吡啶、dipea、dabco、naor、kor中的一种或多种,其中r独立地选自c1~c6烷基,其中,碱性物质优选li2co3、na2co3、k2co3、cs2co3中的一种或两种,更优选na2co3;
[0031]
式ii所示化合物与碱性物质的摩尔比为1:0.1-5,优选为1:0.15-2,更优选为1:0.2-0.5;
[0032]
在一些实施方案中,该反应还包括加入酸性物质淬灭的步骤,所述酸性物质优选为醋酸。
[0033]
本公开还提供了一种如式i’所示化合物或其药学上可接受的氨基酸复合物的制备方法,包括根据本公开的方法制备式iii所示化合物的步骤,
[0034][0035]
在一些实施方案中,式i’化合物药学上可接受的氨基酸复合物具体选自如式i所示的l-脯氨酸复合物;优选l-脯氨酸一水合物。
[0036][0037]
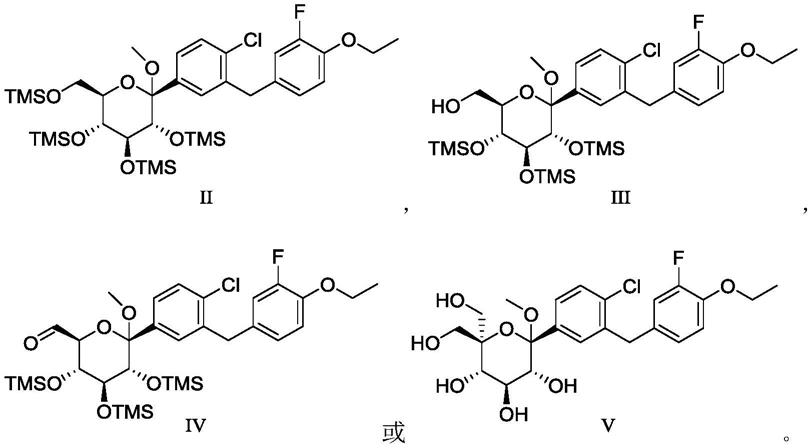
进一步地,本公开所述式i’所示化合物或其药学上可接受的氨基酸复合物的制备方法,或i所示化合物的制备方法,还可包括由式a所示化合物制备式ii所示化合物的步骤,
[0038][0039]
进一步地,所述方法还可包括由式iii所示化合物经氧化反应制备式iv所示化合物的步骤,
[0040][0041]
在一些实施方案中,氧化反应所用试剂包含三氧化硫-吡啶;
[0042]
进一步地,所述方法还可包括由式iv所示化合物制备式v所示化合物的步骤,
[0043][0044]
进一步地,所述方案还可包括由式v所示化合物制备式i’所示化合物的步骤,
[0045][0046]
进一步地,所述方法还包括将式i’所示化合物转化为本公开所述式vi化合物,纯化式vi化合物,再将其转化为式i'所式化合物的步骤,
[0047][0048]
本公开还提供一种纯化式i’所示化合物的方法,所述方法包括如下步骤:将式i’所示化合物转化为本公开所述式vi化合物,纯化式vi化合物,再将其转化为式i'所式化合物。
[0049][0050]
本公开还提供了一种如式i’所示化合物或其药学上可接受的氨基酸复合物的制备方法,所述方法包括如下步骤:将式i’所示化合物转化为本公开所述式vi化合物,纯化式vi化合物,再将其转化为式i'所示化合物,
[0051][0052]
在一些实施例中,所述纯化优选包括重结晶和/或打浆;
[0053]
进一步地,所述纯化的溶剂为乙腈/异丙醇的混合溶剂,配比为乙腈:异丙醇=1:0.5-2,优选乙腈:异丙醇=1:1-1.5;更优选乙腈:异丙醇=1:1.5。
[0054]
本公开还提供了一种如式i’所示化合物或其药学上可接受的氨基酸复合物的制备方法,包括由本公开所述式vi化合物制备式i’所示化合物的步骤,
[0055][0056]
在一些实施方案中,式i’化合物药学上可接受的氨基酸复合物具体选自如式i所示的l-脯氨酸复合物;优选l-脯氨酸一水合物。
[0057]
本公开还提供一种由式b所示化合物制备得到式ii所示化合物的方法,包括以下步骤:
[0058][0059]
本公开整体上还提供了一种式i’及式i所示化合物的制备方法:
[0060][0061]
为了进一步提高产品的纯度,在一些实施方案中,式i’及式i所示化合物的制备方法中还包括对式i’化合物进行衍生化得到式vi化合物,vi化合物经纯化后重新得到式i’所示化合物的步骤:
[0062][0063]
本公开的技术方案具备以下有益的效果:
[0064]
(1)在合成式i’化合物的过程中仅需要使用并脱除tms一种保护基,缩短了工艺步骤,降低了生产成本;(2)由式ii化合物选择性脱除单个tms得到式iii化合物的反应特异性高,所示式iii化合物纯度高,可直接进行下一步反应;(3)衍生化的式vi化合物可通过重结晶和/或打浆进行纯化,后续步骤所得终产品纯度上升,可满足药品的纯度要求;(4)该制备方法全程无需进行柱层析,适合大规模生产。
[0065]
除非有相反陈述,在说明书和权利要求书中使用的术语具有下述含义。
[0066]“任选地”或“任选”是指意味着随后所描述的事件或环境可以但不必发生,该说明包括该事件或环境发生或不发生的场合。例如“任选的被卤素取代的c
1-c6烷基”是指卤素可
以但不必须存在,该说明包括烷基被卤素取代的情形和烷基不被卤素取代的情形。
[0067]“a和/或b”意味着以下任一情形:a;b;a和b,且“a和b”中,a与b的前后顺序不受限制。例如“包括重结晶和/或打浆”意味着以下任一情形:“包括重结晶”;“包括打浆”;“包括重结晶和打浆”,且当为“包括重结晶和打浆”时,重结晶和打浆的前后顺序不受限制。
[0068]
本发明所述化合物的化学结构中,键表示未指定构型,即如果化学结构中存在手性异构体,键可以为或者同时包含或者同时包含两种构型。
[0069]
本公开所述化合物或其可药用盐、或其异构体的任何同位素标记的衍生物都被本公开所覆盖。能够被同位素标记的原子包括但不限于氢、碳、氮、氧、磷、氟、氯、碘等。它们可分别被同位素2h(d)、3h、
11
c、
13
c、
14
c、
15
n、
18
f、
31
p、
32
p、
35
s、
36
cl和
125
i等代替。除另有说明,当一个位置被特别地指定为氘(d)时,该位置应理解为具有大于氘的天然丰度(其为0.015%)至少3000倍的丰度的氘(即,至少45%的氘掺入)。
[0070]“卤素”是指氟,氯,溴和碘。
[0071]“烷基”指饱和的脂族烃基团,包括1至20个碳原子的直链和支链基团。优选含有1至10个碳原子的烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基、叔丁基或戊基等。更优选的是含有1至6个碳原子的低级烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基或叔丁基、戊基、庚基等。
[0072]“环烷基”指饱和或部分不饱和单环或多环环状烃取代基,环烷基环包含3至20个碳原子,优选包含3至6个碳原子。单环环烷基的非限制性实例包括环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基等;多环环烷基包括螺环、稠环和桥环的环烷基。
[0073]“烷氧基”指-o-(烷基)和-o-(环烷基),其中烷基和环烷基的定义如上所述。非限制性实施例包含甲氧基、乙氧基、丙氧基、丁氧基、环丙氧基、环丁氧基、环戊氧基、环己氧基等。
[0074]“tms”指三甲基硅基,“tms-cl”指三甲基氯硅烷。
[0075]“pnb”指对硝基苯甲酰基,“pnb-cl”指对硝基苯甲酰氯。
具体实施方式
[0076]
以下将结合具体实例详细地解释本发明,使得本专业技术人员更全面地理解本发明。具体实例仅用于说明本发明的技术方案,并不以任何方式限定本发明。
[0077]
本公开中实施例中未注明具体条件的实验方法,通常按照常规条件,或按照原料或商品制造厂商所建议的条件。未注明具体来源的试剂,为市场购买的常规试剂。
[0078]
化合物的结构是通过核磁共振(nmr)或/和质谱(ms)来确定的。nmr位移(δ)以10-6
(ppm)的单位给出。nmr的测定是用bruker avance-400核磁仪,测定溶剂为氘代二甲基亚砜(dmso-d6),氘代氯仿(cdcl3),氘代甲醇(cd3od),内标为四甲基硅烷(tms)。化合物的光学异构体(异构体)空间构型可进一步通过测量单晶参数的方式确认。
[0079]
hplc的测定使用waters acquity ultra high performance lc、shimadzu lc-20a systems、shimadzu lc-2010ht series或安捷伦agilent 1200lc高压液相色谱仪(acquity uplc beh c18 1.7um 2.1x50mm色谱柱、ultimate xb-c183.0*150mm色谱柱或xtimate c18 2.1*30mm色谱柱)。
[0080]
ms的测定用waters sqd2质谱仪,以正/负离子模式扫描,质量扫描范围为100~1200。
[0081]
手性hplc分析测定使用chiralpak ic-3 100
×
4.6mm i.d.,3um、chiralpak ad-3 150
×
4.6mm i.d.,3um、chiralpak ad-3 50
×
4.6mm i.d.,3um、chiralpak as-3 150
×
4.6mm i.d.,3um、chiralpak as-3 100
×
4.6mm i.d.,3μm、chiralcel od-3 150
×
4.6mm i.d.,3um、chiralcel od-3 100
×
4.6mm i.d.,3μm、chiralcel oj-h 150
×
4.6mm i.d.,5um、chiralcel oj-3 150
×
4.6mm i.d.,3um色谱柱;
[0082]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.15mm~0.2mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。
[0083]
快速柱纯化系统使用combiflash rf150(teledyne isco)或者isolara one(biotage)。
[0084]
正向柱层析一般使用烟台黄海硅胶100~200目、200~300目或300~400目硅胶为载体,或者使用常州三泰预填预填超纯正相硅胶柱(40-63μm,60,12g,,25g,40g,80g或其他规格)。
[0085]
反相柱层析一般使用常州三泰预填超纯c18硅胶柱(20-45μm,40g,80g,120g,220g或其他规格)。
[0086]
高压柱纯化系统使用waters autop,配合使用waters xbridge beh c18 obd prep column,5μm,19mm x 150mm或者atlantis t3obd prep column,5μm,19mm x 150mm。
[0087]
手性制备柱使用daicel chiralpak ic(250mm*30mm,10um)或phenomenex-amylose-1(250mm*30mm,5um)。
[0088]
本公开中的已知的起始原料可以采用或按照本领域已知的方法来合成,或可购买自上海泰坦科技,abcr gmbh&co.kg,acros organics,aldrich chemical company,韶远化学科技(accela chembio inc)、达瑞化学品等公司。
[0089]
实施例中无特殊说明,反应能够均在氩气氛或氮气氛下进行。
[0090]
氩气氛或氮气氛是指反应瓶连接一个约1l容积的氩气或氮气气球。
[0091]
氢气氛是指反应瓶连接一个约1l容积的氢气气球。
[0092]
加压氢化反应使用parr 3916ekx型氢化仪和清蓝ql-500型氢气发生器或hc2-ss型氢化仪。
[0093]
氢化反应通常抽真空,充入氢气,反复操作3次。
[0094]
微波反应使用cem discover-s 908860型微波反应器。
[0095]
实施例中无特殊说明,溶液是指水溶液。
[0096]
实施例中无特殊说明,反应的温度为室温,为20℃~30℃。
[0097]
实施例中的反应进程的监测采用薄层色谱法(tlc)。
[0098]
实施例1
[0099][0100]
将化合物b(30.0g,0.168mol)溶于300ml四氢呋喃中,加入n-甲基吗啉(136.3g,1347.2mmol,8.0eq)加入到1l三口瓶中。冰水浴滴加tms-cl(109.8g,1010.46mmol,6.0eq),加完后升至室温搅拌18h。反应结束后加入正庚烷、饱和碳酸氢钠溶液和水,分液,有机层依次用水、磷酸二氢钠水溶液和饱和食盐水洗,用无水硫酸钠干燥,过滤,减压浓缩,得到84.51g浅色油状物,粗品直接用于下一步。
[0101]
实施例2
[0102][0103]
将化合物d(50.0g,145.5mmol,1.0eq)加入到3l多口瓶中,加入四氢呋喃(240ml)、甲苯(200ml)、正己烷(600ml),搅拌溶清,-65℃以下滴加正丁基锂(95.9ml,152.8mmol,1.05eq,1.6m/l),保温搅拌1h。将化合物c(79.08g,160.1mmol,1.1eq)的甲苯溶液滴加到反应液中,保温搅拌2h。冰浴条件将甲烷磺酸(26.6g,276.4mmol,1.9eq)的甲醇溶液滴加到反应液中,室温搅拌16h。反应结束后加入饱和碳酸氢钠水溶液,分液,有机相用饱和碳酸氢钠水溶液提取,合并水相,再用乙酸乙酯提取,乙酸乙酯相用水洗,合并乙酸乙酯相,用无水硫酸钠干燥,过滤,减压浓缩,得到泡沫状固体57.82g,粗品直接用于下一步。
[0104]
实施例3
[0105][0106]
将化合物a(57.8g,126.51mmol,1.0eq)溶于二氯甲烷(578ml)中,加入咪唑(68.89g,1012.08mmol,8.0eq)。降温至0℃,加入三甲基氯硅烷(82.46g,759.06mmol,6.0eq)后,室温搅拌16h。反应结束后加入饱和碳酸氢钠水溶液和水,分液,有机相用饱和食盐水洗,用无水硫酸钠干燥,过滤,减压浓缩干,得到油状物86.07g,粗品直接用于下一步。
[0107]1h nmr(400mhz,cdcl3)d 7.29-7.45(m,1h),7.29-7.44(m,2h),6.81-6.95(m,3h),3.93-4.16(m,5h),3.82-3.90(m,1h),3.71-3.81(m,1h),3.60-3.68(m,1h),3.46-3.57(m,1h),3.19-3.28(m,1h),2.98-3.16(m,3h),1.37-1.48(m,3h),-0.03-0.25(m,28h),-0.35(s,8h)
[0108]
实施例4
[0109][0110]
将化合物ii(86.07g,115.44mmol,1.0eq)溶于甲醇(396ml)中,降温至-5℃,加入碳酸钠(3.42g,32.32mmol,0.28eq),搅拌4h。反应结束后加入醋酸,搅拌。反应液减压浓缩,加入mtbe和碳酸氢钠水溶液,分液,有机相用饱和食盐水洗,用无水硫酸钠干燥,过滤,减压浓缩,得到油状物84.25g,纯度86.74%,粗品直接用于下一步。
[0111]1h nmr(400mhz,cdcl3)d=7.39(d,j=8.8hz,1h),7.34-7.28(m,2h),6.95-6.82(m,3h),4.16-3.95(m,5h),3.86(m,1h),3.79-3.71(m,1h),3.68-3.59(m,1h),3.55-3.46(m,1h),3.22(s,2h),3.16-3.01(m,3h),1.43(t,j=7.0hz,3h),0.31-0.13(m,19h),-0.35(s,8h)
[0112]
实施例5
[0113][0114]
将化合物iii(84.25g,100.13mmol,1.0eq)溶于二氯甲烷(840ml)中,15℃,加入二甲亚砜(337ml),三乙胺(65.5ml),加入三氧化硫吡啶,25℃反应2h。反应液加mtbe稀释,将其倒入5%亚硫酸钠水溶液中,有机相依次用碳酸氢钠水溶液和饱和食盐水洗,用无水硫酸钠干燥,过滤,减压浓缩,得到油状物85.2g,纯度82.4%,粗品直接用于下一步。
[0115]1h nmr(400mhz,cdcl3)d=9.77-8.59(m,1h),7.74-7.68(m,2h),7.32-7.28(m,1h),6.97-6.69(m,3h),4.26-3.75(m,6h),3.74-3.42(m,1h),3.33-3.17(m,1h),3.13-2.79(m,3h),1.48-1.32(m,3h),0.25
‑‑
0.45(m,27h)
[0116]
实施例6
[0117][0118]
将化合物iv(85.2g,100.13mmol,1.0eq)溶于乙醇(850ml)中,20℃,加入多聚甲醛(66.08g,2202.86mmol,22eq),加入20%乙醇钠溶液(187.38g,550.72mmol,5.5eq),先室温搅拌4h,再55℃搅拌2.5h。反应结束后减压浓缩干乙醇,加mtbe,分液,有机相用水洗,合并水相,用mtbe提取,合并有机相,加无水硫酸钠干燥,过滤,减压浓缩,得泡沫状固体58.46g,纯度64.5%,粗品直接用于下一步。
[0119]1h nmr(400mhz,cdcl3)d=7.45-7.30(m,1h),7.26-7.13(m,2h),6.87-6.66(m,
3h),5.75-5.18(m,2h),4.54(d,j=7.8hz,1h),4.10(t,j=9.8hz,1h),4.04-3.91(m,4h),3.86-3.75(m,3h),3.73-3.62(m,4h),3.17(d,j=9.8hz,1h),3.01-2.74(m,3h),1.37-1.28(m,3h)
[0120]
实施例7
[0121][0122]
将化合物v(19.30g,39.64mmol)溶于二氯甲烷(386ml)中,加入浓盐酸(4.8ml),室温搅拌16小时,加入无水硫酸钠,搅拌2h,过滤,滤液减压浓缩至干得泡沫状固体17.73g,纯度72.05%,粗品直接用于下一步。
[0123]
实施例8
[0124][0125]
将化合物i’(17.73g,38.98mmol,1.0eq)溶于二氯甲烷(177ml)中,0-5℃下加入三乙胺(35.50g,350.82mmol,9.0eq),加入pnb-cl(43.40g,233.88mmol,6.0eq),室温反应4小时。反应结束后滴加饱和碳酸氢钠水溶液,分液,有机相再分别用饱和碳酸氢钠水溶液和饱和食盐水洗,硫酸钠干燥,减压浓缩至干得泡沫状固体36.94g,纯度65.9%。
[0126]
在单口烧瓶中加入化合物vi-1粗品(29.00g),再加入乙腈/异丙醇的混合溶剂(乙腈:异丙醇=2:3),加热至回流,自然降温至室温搅拌18小时,过滤,滤饼用乙腈/异丙醇混合溶剂(乙腈:异丙醇=2:3)淋洗,然后真空干燥得到类白色固体15.63g,纯度97.6%。在单口烧瓶中加入上面得到的类白色固体,再加入乙腈/异丙醇的混合溶剂(乙腈:异丙醇=2:3),加热至回流,自然降温至室温搅拌18小时,过滤,滤饼用乙腈/异丙醇混合溶剂乙腈:异丙醇=2:3)淋洗,然后真空干燥得到白色固体14.83g,纯度98.7%,从化合物ii开始五步总收率45.1%。
[0127]1h nmr(400mhz,cdcl3)d=8.34(d,j=8.8hz,2h),8.30-8.21(m,4h),8.17(t,j=9.3hz,4h),8.08(d,j=8.8hz,2h),8.01(d,j=8.8hz,2h),7.94(d,j=8.8hz,2h),7.48-7.41(m,2h),7.35(d,j=8.1hz,1h),6.79-6.70(m,2h),6.66-6.57(m,1h),6.19-6.02(m,2h),5.88(d,j=8.1hz,1h),4.88-4.71(m,2h),4.61(d,j=12.7hz,1h),4.14-3.99(m,3h),3.95(s,2h),1.43(t,j=7.0hz,3h)
[0128]
实施例9
[0129][0130]
将化合物vi-1(14.83g,14.11mmol,1.0eq)溶于thf(74ml)中,加入水(74ml),降温至0-5℃,加入lioh
·
h2o(2.96g,70.55mmol,5.0eq),自然升温至室温反应3小时,反应结束后加入饱和氯化铵中和反应,用乙酸乙酯提取,有机层再用饱和碳酸氢钠以及饱和食盐水(593ml)洗涤,硫酸钠干燥,减压浓缩至干得到泡沫状固体7.03g,纯度99.2%。
[0131]
实施例10
[0132][0133]
将化合物i’(7.03g,理论14.11mmol,1.0eq)置于乙醇(70ml)和正己烷(35ml)中,加入l-脯氨酸(1.79g,15.52mmol,1.1eq)和水(0.7ml),升温至70℃,搅拌30分钟,自然降温至室温搅拌18小时,过滤,滤饼用乙醇/正己烷混合溶剂(乙醇/正己烷=2:1)淋洗,然后真空干燥得到白色固体7.53g,两步收率90.8%,(从化合物vi-1起计算),纯度99.6%,单杂小于0.1%。
[0134]
对比例1
[0135][0136]
将化合物ii(2.0g,2.68mmol,1.0eq)溶于甲醇(9.2ml)中,降温至-5℃,加入碳酸钾(104mg,0.75mmol,0.28eq),搅拌4h,hplc中控,化合物iii占22%,化合物a占48%,大部分tms全部被脱掉。
[0137]
对比例2
[0138][0139]
将化合物i’(1g,2.198mmol,1.0eq)溶于甲苯(10ml)中,0-5℃下加入吡啶(1.39g,17.59mmol,8.0eq),滴加乙酸酐(1.35g,13.19mmol,6.0eq),自然升温至室温反应16小时。反应结束后滴加饱和碳酸氢钠水溶液,分液,有机相再用水洗一遍,硫酸镁干燥,浓缩至干得棕色油状物1.23g,收率89.70%,纯度66.5%。
[0140]
该粗品加入异丙醇,升温至60℃搅拌,降至室温,析出棕色油状物,未能固化,将该溶液旋干。
[0141]
该粗品加乙腈和异丙醇的混合液(乙腈:异丙醇=2:3),加热至回流,自然降温至室温搅拌18小时,无产物析出。
[0142]
由于已根据其特殊的实施方案描述了本发明,某些修饰和等价变化对于精通此领域的技术人员是显而易见的且包括在本发明的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1