一种卟啉马来酰亚胺衍生物共价功能化二硫化钼纳米片非线性杂化材料及其制备与应用
1.本发明属于有机
‑
无机功能复合材料和军工强激光防护材料技术领域,涉及一种卟啉马来酰亚胺衍生物共价功能化二硫化钼纳米片非线性杂化材料及其制备与应用。
背景技术:
2.自从2004年novoselov等人发明石墨烯以来,二维材料成为了功能材料领域上的重要组成部分,由于强的量子限域效应和层内刚性共价结构网络,二维材料展现出优异的光、电、热、力性能,在光催化、电催化、超级电容器、线性光学和非线性光学等领域都具有潜在应用。其中,层状过渡金属二硫化物材料表现出厚度依赖的带隙,晶型多样等特性,弥补了石墨烯零带隙,晶型单一的缺憾,现已成为二维材料领域的一颗新星。最近的研究表明,二硫化钼纳米片在近红外激光领域表现出了一定的非线性响应,有望应用于激光锁模,光信号处理,光限幅等领域上,然而,二硫化钼在常规有机溶剂中分散性较差,限制了基于二硫化钼纳米片的器件制备,同时,二硫化钼的非线性光学响应相对较弱,因此对二硫化钼纳米片进行修饰改进,以提升其非线性响应及在常规溶剂中分散性已成为二维二硫化钼材料的研究热点之一。
3.在对氧化石墨烯的研究发现,传统非线性活性分子卟啉的共价功能化往往能显著提升其在有机溶剂中的分散性,同时由于两者的协同效应,基于氧化石墨烯的共价杂化产物具有更优异的反饱和吸收性能。而对于二硫化钼的共价官能化研究较少。目前,已有研究报道硫辛酸衍生物可以与二硫化钼纳米片上硫缺陷处的钼原子配位,但这种修饰方式受二硫化钼本身缺陷程度影响较大,修饰程度往往较低,另外,硫辛酸具有较长的饱和碳链,不利于电子的离域,因而此类杂化材料在近红外飞秒激光领域响应较弱,迄今为止,针对二硫化钼纳米片的新型共价修饰杂化材料的制备及其在近红外飞秒激光下非线性行为尚有待进一步研究。
技术实现要素:
4.本发明的目的就是为了提供一种卟啉马来酰亚胺衍生物共价功能化二硫化钼纳米片非线性杂化材料及其制备与应用。
5.本发明的目的可以通过以下技术方案来实现:
6.本发明的技术方案之一提供了一种卟啉马来酰亚胺衍生物共价功能化二硫化钼纳米片非线性杂化材料,其由二硫化钼纳米片,以及通过卟啉共价键连到二硫化钼纳米片上的卟啉的马来酰亚胺衍生物por组成,所述卟啉的马来酰亚胺衍生物 por的化学结构式为:
[0007][0008]
本发明的技术方案之二提供了一种卟啉马来酰亚胺衍生物共价功能化二硫化钼纳米片非线性杂化材料的制备方法,包括以下步骤:
[0009]
(1)取二硫化钼块体粉末浸没于溶剂中,搅拌,所得悬浊液离心,洗涤,干燥,得到干燥二硫化钼粉末;
[0010]
(2)再取干燥二硫化钼粉末加入异丙醇与水的混合液中,超声分散,离心,干燥,得到少层二硫化钼粉末;
[0011]
(3)取吡咯与苯甲醛自聚反应制得四苯基卟啉,再将所得四苯基卟啉中一个 meso位取代苯基硝化制得5
‑
(4
‑
硝基苯基)
‑
10,15,20
‑
三苯基卟啉,继续还原制得 5
‑
(4
‑
氨基苯基)
‑
10,15,20
‑
三苯基卟啉;
[0012]
(4)取5
‑
(4
‑
氨基苯基)
‑
10,15,20
‑
三苯基卟啉与马来酸酐发生缩合反应,制得卟啉的马来酰亚胺衍生物por;
[0013]
(5)将卟啉的马来酰亚胺衍生物por超声分散于乙腈溶液中,加入少层二硫化钼粉末,搅拌反应,过滤,洗涤,干燥,得到目的产物卟啉马来酰亚胺衍生物共价功能化二硫化钼纳米片mos2‑
por。
[0014]
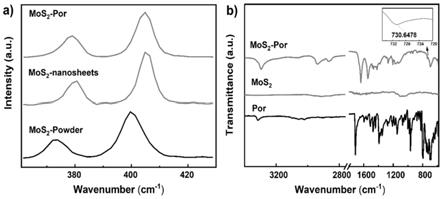
进一步的,步骤(1)中,所述溶剂为水。通过将二硫化钼块体粉末先浸没于溶剂中,然后干燥,可以使得mos2块体层间距稍稍变大,有利于后续剥离效率的提升。
[0015]
进一步的,步骤(2)中,所述异丙醇与水的体积比为(2
‑
4):(6
‑
8),优选为3:7。在上述比例下,其具有与二硫化钼匹配的表面能,其剥离效果最佳,同时少层二硫化钼分散液维持稳定不聚沉时间更久。在超声过程中,需间隔振荡二硫化钼纳米片分散液,并更换超声清洗液并添加冰块以维持低温,防止温度升高带来的氧化。
[0016]
进一步的,步骤(3)中,自聚反应所用反应溶剂为丙酸,且苯甲醛与吡咯的摩尔比为1:(1.5~2.5),优选为1:2。优选的,反应时,先将苯甲醛溶解在丙酸中,当反应体系稳定回流后,缓慢添加吡咯的丙酸溶液。
[0017]
进一步的,步骤(3)中,硝化所用试剂为质量分数为65%
‑
68%的浓硝酸和亚硝酸钠,两者的添加量之比为6ml:(180~220)mg,优选为6ml:200mg,硝化过程的体系温度维持在0℃。通过薄层色谱法追踪反应进度,当新的主要点生成时,立刻加入氨水终止反应,以防多硝化的发生。
[0018]
进一步的,步骤(3)中,还原过程中采用质量分数为35%
‑
37%浓盐酸作为反应溶剂,并在惰性气体保护气氛下进行(为反应提供还原性环境),所用还原剂为 sncl2·
2h2o(还原剂用量相对于硝基卟啉可过量添加以提升还原产率),且还原过程中体系温度为60~80℃,优选为70℃。另外,需要注意的是,由于盐酸的高挥发性,反应溶剂占反应瓶体积不超过1/5,同时选取体积更大的球形冷凝管以防止反应液溢出。
[0019]
进一步的,步骤(4)中,缩合反应过程具体为:
[0020]
取5
‑
(4
‑
氨基苯基)
‑
10,15,20
‑
三苯基卟啉溶于四氢呋喃中,加入马来酸酐,室温搅拌,然后除去四氢呋喃,再加入乙酸酐,继续加入乙酸钠,升温搅拌,冷却、分离、洗涤,得到卟啉的马来酰亚胺衍生物por。
[0021]
更进一步的,5
‑
(4
‑
氨基苯基)
‑
10,15,20
‑
三苯基卟啉、马来酸酐、乙酸钠的质量比为(500~700):108:(450~550)。另外,优选的,升温搅拌的温度为140~150℃。
[0022]
进一步的,步骤(5)中,卟啉的马来酰亚胺衍生物por、少层二硫化钼粉末的质量比为(50~70):10。为提高修饰程度,卟啉的马来酰亚胺衍生物por过量加入,同时二硫化钼纳米片可以分批加入,同时反应过程中,间断对反应体系进行超声,使反应体系分散均匀的同时,避免了二硫化钼纳米片的聚沉。
[0023]
进一步的,步骤(5)中,反应体系温度控制为50~70℃,优选为60℃,且少层二硫化钼粉末分批次加入反应体系中。
[0024]
本发明的技术方案之三提供了一种卟啉马来酰亚胺衍生物共价功能化二硫化钼纳米片非线性杂化材料在近红外飞秒激光领域中的应用。该杂化材料由于卟啉的表面修饰,在氯仿中分散性显著提升。共轭连接促进了卟啉与二硫化钼之间的相互作用,杂化材料的荧光显著猝灭。在飞秒近红外激光领域,将二硫化钼纳米片本身的饱和吸收转变为反饱和吸收,对调控二维材料的非线性响应具有很强的借鉴意义。
[0025]
与现有技术相比,本发明具有以下优点:
[0026]
一、反应路线简短,产率较高,容易扩大规模生产
[0027]
二、杂化材料mos2‑
por在氯仿中分散性较修饰前纳米片显著上升。
[0028]
三、杂化材料mos2‑
por的荧光猝灭效率达到了90%以上,远远高于物理吸附的杂化材料,具有更加高效的电子转移和传输效率。
[0029]
四、杂化材料mos2‑
por在近红外飞秒激光下展现了良好的反饱和吸收行为,而原料二硫化钼纳米片仅具有弱的饱和吸收行为,por没有明显的非线性行为。
附图说明
[0030]
图1为卟啉的马来酰亚胺衍生物的合成路线图;
[0031]
图2为本发明所制备的卟啉的马来酰亚胺衍生物共价键连二硫化钼纳米片 mos2‑
por路线图;
[0032]
图3为本发明所制备的mos2‑
por以及原料的拉曼散射和红外吸收图谱;
[0033]
图4为本发明所制备的mos2‑
por的xps谱图;
[0034]
图5为本发明所制备的mos2‑
por的tem图;
[0035]
图6为本发明所制备的mos2和mos2‑
por的氯仿分散液图片(由左到右依次为mos2和mos2‑
por);
[0036]
图7为本发明所制备的mos2‑
por及原料的紫外可见吸收谱图和荧光谱图;
[0037]
图8为本发明所制备的mos2‑
por和mos2纳米片及物理吸附杂化产物的非线性吸收谱图;
[0038]
图9为对比例1和实施例1中的得到产物mos2‑
por的紫外吸收图谱。
具体实施方式
[0039]
下面结合附图和具体实施例对本发明进行详细说明。本实施例以本发明技术方案为前提进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
[0040]
以下实施例中,所用二硫化钼粉末、吡咯和苯甲醛均为安耐吉化学试剂公司的商业化产品,硝酸、丙酮等原料来自于国药试剂公司。所用溶剂来自探索试剂平台。
[0041]
其余若无特别说明的原料试剂或处理技术,则表明其均为本领域的常规市售产品或常规处理技术。
[0042]
实施例1:
[0043]
基于点击反应制备卟啉共价修饰二硫化钼纳米片非线性纳米杂化材料,其合成工艺路线参见图1和图2所示:
[0044]
使用液相剥离法制得少层二硫化钼纳米片,剥离溶剂为异丙醇:水比例为3:7,剥离效果较好,同时缺陷浓度低。取1g mos2块体粉末浸没于水中,持续搅拌5min 后,将mos2悬浊液4200rpm离心5min后,将离心沉淀再次分散于100ml去离子水中,再次离心,离心沉淀置于含干燥剂的真空干燥箱中进行干燥。待干燥完成后称取900mg干燥粉末分散于300ml异丙醇:水比例为3:7(体积比)的混合溶剂中,将其置于200w,40khz的超声波清洗器中进行超声,每隔30min更换超声波清洗器的清洗液并添加冰块,同时振荡mos2分散液,超声总时长6h后,将分散液离心,4200rpm离心30min后,取离心上清液冷冻干燥,获得铅灰色干燥 mos2纳米片粉末100mg。
[0045]
四苯基卟啉的合成使用改进的adler法,首先将吡咯蒸馏获得新制的无色吡咯液体。将5.5ml的苯甲醛加入200ml丙酸中,搅拌,加热至丙酸回流后,缓慢滴加溶有8.8ml吡咯的20ml丙酸溶液,反应40min后,冷却反应液至室温。加入100ml甲醇使产物四苯基卟啉析出,减压抽滤反应液,甲醇洗涤滤饼得到约2.2 g紫色固体卟啉。四苯基卟啉的硝化在冰浴下进行,1g卟啉溶解于180ml二氯甲烷中,加入6ml 65%
‑
68%的浓硝酸,然后加入200mg亚硝酸钠,约10min后,薄层色谱显示新的主要产物生成,加入氨水中和至中性,二氯甲烷提取,柱层析法分离得到硝基卟啉(即5
‑
(4
‑
硝基苯基)
‑
10,15,20
‑
三苯基卟啉)。氮气气氛下将500mg 硝基卟啉溶于40ml浓盐酸(其浓度为35%~37%),然后加入过量还原剂 sncl2·
2h2o,然后加热至70℃反应5h,然后向反应体系中加入冰水,氨水中和使溶液呈碱性,抽滤,将滤饼重新溶解,旋去溶剂柱色谱分离得到氨基卟啉(即5
‑
(4
‑
氨基苯基)
‑
10,15,20
‑
三苯基卟啉)。
[0046]
将氨基卟啉630mg溶解于四氢呋喃100ml,然后加入马来酸酐108mg,室温下搅拌3h,减压旋去溶剂,然后再加入乙酸酐15ml作为下一步反应溶剂,加入500mg乙酸钠,145℃回流搅拌2h,待反应液冷却至室温,加入200ml去离子水,0℃下静置至固体析出,过滤,甲醇洗涤,柱色谱分离提纯得到产物por。
[0047]
1h
‑
nmr(600mhz,chcl3‑
d,tms,δ/ppm):δ8.90
‑
8.85(m,8h),8.33(d,j=8.2 hz,2h),8.22(d,/j=7.0hz 6h),7.79
‑
7.74(m,11h),7.02(s,2h),
‑
2.79(s,2h).
[0048]
最后将60mg por加入30ml乙腈溶剂下,加热至60℃,将mos2纳米片10mg 在2天内分批加入反应体系,并且每隔3h超声15min以防mos2纳米片聚沉,反应3天后,反应液通过0.22μm的有机系尼龙滤膜,并且用n,n二甲基甲酰胺、四氢呋喃等溶剂清洗,将滤饼重新分
散于n,n二甲基甲酰胺中,再次过滤洗涤,反复过滤
‑
再分散
‑
过滤数次后,物理吸附por已去除,干燥最后得到滤饼,得到卟啉马来酰亚胺共价功能mos2纳米片杂化产物mos2‑
por。
[0049]
图3中,图3a为本发明制备的mos2薄膜和原块体mos2粉末以及杂化产物 mos2‑
por的拉曼图谱。在514.5nm的激光下,块体mos2粉末的e
2g
和a
1g
峰分别位于373.8cm
‑1和399.6cm
‑1,而剥离后的mos2薄膜的e
2g
和a
1g
峰相对块体材料出现显著蓝移,分别位于380.7cm
‑1和404.8cm
‑1。而e
2g
和a
1g
峰的峰位和峰间距与mos2层间的相互作用密切相关,证实了液相剥离的有效性。而经过por修饰后, e
2g
出现一定的红移,位于379.0cm
‑1,而a
1g
峰没有变化。同时e
2g
和a
1g
峰的半峰全宽fwhm相对原薄膜而言显著变大,这可能是mos2层间的范德华力减弱导致的。图3b展示了杂化材料mos2‑
por以及原料mos2薄膜和por的红外图谱,可以发现mos2薄膜上的mo
‑
s振动吸收较弱,而杂化材料mos2‑
por显示出多种por 上的振动吸收峰,特别的是730.89cm
‑1处的新吸收峰可以归属到c
‑
s键的吸收峰,证实了por的成功共价键连。
[0050]
为了进一步表征杂化材料的化学结构,测试得到mos2‑
por的xps图谱。如图 4所示,s元素的精细图谱s 2p可以拟合出两种成分,一种是未修试的mo
‑
s键,位于161.72ev和162.93ev。另一种是修饰后的c
‑
s键,位于162.36ev和163.65 ev。同时c元素的精细图谱c1s上同样存在c
‑
s键的拟合峰,有利证实了por与 mos2纳米片间的共价键连方式。
[0051]
杂化材料mos2‑
por的tem图谱如图5所示,可以发现对应(002)、(100)and (103)的晶格条纹,间距分别是0.63nm、0.27nm和0.223nm,杂化材料的层间距没有发生变化,表明por的修饰更多的是在外层的mos2表面上。
[0052]
图6显示了杂化材料mos2‑
por和原料mos2纳米片的氯仿分散液图片,可以发现mos2‑
por的浓度更高,分散性更好。图7中,图7a显示了杂化材料mos2‑
por 和原料mos2纳米片及por的吸收。mos2纳米片显示了位于668nm和614nm的特征吸收峰,这是第一布里渊区k/k’点的激子跃迁吸收,在340nm
‑
510nm间的宽峰对应深层价带到导带的跃迁吸收。por的吸收谱图上显示了卟啉的特征吸收,即417nm处的soret带和500nm
‑
600nm的q带。杂化材料的吸收谱图与mos2纳米片的类似,仅在421nm处有对应por的soret带的凸起,相对por而言红移了 4nm,这是由于基态下por与mos2纳米片的相互作用导致的。另外,杂化材料在可见区至近红外区的吸收的相对强度略微提升,说明修饰反应及后处理过程种发生了一定程度的聚集。通过荧光测试能够进一步研究杂化材料在激发态的相互作用。由于卟啉的荧光峰位于mos2纳米片的吸收峰内,为了客观研究键连方式对杂化材料激发态的影响,参比杂化材料mos2‑
por
‑
blends被制备作为对比。在 mos2‑
por
‑
blends材料中,por与mos2纳米片通过范德华相互作用连接。其合成方法与mos2‑
por类似,合适方法具体如下:
[0053]
将1mg mos2纳米片分散于2ml n,n二甲基甲酰胺中,加入对应含量的0.89 mg/ml的por溶液,搅拌3h后,制得参比杂化材料mos2‑
por
‑
blends的分散液用于进一步的对比测试。
[0054]
荧光测试谱图如图7b所示,在417nm的激光光照射下,por的荧光发射峰为位于649nm的强吸收峰和717nm的弱肩峰。参比杂化材料mos2‑
por
‑
blends和杂化材料mos2‑
por的荧光峰的峰位置没有变化,但其强度发生很大的变化, mos2‑
por
‑
blends的谱图显示为717nm的强吸收峰和649nm的弱肩峰,这是由于 mos2的在649nm处的强吸收导致的。而mos2‑
por的荧光则发生明显猝灭,717nm 处的荧光强度猝灭了约95%。与物理吸附的参比杂化材料相比,更加佐证了共价键连的杂化产物在激发态下具有更强的电荷/能量转移
[0055]
图8给出了杂化材料mos2‑
por和参比杂化材料mos2‑
por
‑
blends以及原料 mos2纳米片的开孔z扫描测试结果。在波长为800nm,脉冲宽度为34fs,脉冲能量为90nj的激光照射下,mos2纳米片显示为弱的饱和吸收,在焦点处,其透过率增长至1.016。这是由于基态电子减少和泡利不相容导致的。而杂化材料mos2‑
por和参比杂化材料mos2‑
por
‑
blends都表现为反饱和吸收,在焦点处,其透过率分别降低至0.92和0.97。这可以归因于por和mos2纳米片间有效的电荷/能量转移导致的来自于mos2纳米片的增强的双光子吸收。而共价键连的杂化材料 mos2‑
por由于具有更有效的电荷/能量转移因而表现出了更强的反饱和吸收,也进一步证实了通过马来酰亚胺的将卟啉共价键连于mos2纳米片上能够有效的调控 mos2纳米片的非线性光学性质,为进一步开发基于mos2纳米片的新材料提供参考。
[0056]
图9给出对比例1和实施例1中的得到产物mos2‑
por的紫外吸收图谱,可以发现对比例1中对应por的421nm处吸收峰强度降低,证实了分批加入原料可以在相同反应条件下提升修饰程度。
[0057]
对比例1:
[0058]
与实施例1相比,绝大部分相同,除了二硫化钼纳米片为一次性投入,得到的mos2‑
por中修饰程度下降,在紫外吸收谱图对应卟啉峰强显著降低。
[0059]
对比例2:
[0060]
与实施例1相比,绝大部分相同,除了直接使用异丙醇:水直接对块体mos2进行超声剥离,最终获得所得mos2纳米片更少,剥离效率由10%降低至6%左右。
[0061]
以上实施例所采用的各原料试剂及其添加量、以及反应的工艺参数条件等,根据需要还可以在以下范围内任意调整(即任意调整为其端值,或任一中间点值):
[0062]
液相剥离过程中,所述异丙醇与水的体积比为(2
‑
4):(6
‑
8);
[0063]
液相剥离过程中,超声时间为4~8h;
[0064]
四苯基卟啉合成过程中,苯甲醛与吡咯的摩尔比为1:(1.5~2.5);
[0065]
硝化过程中,所用试剂为质量分数为65%
‑
68%的浓硝酸和亚硝酸钠,两者的添加量之比为6ml:(180~220)mg;
[0066]
硝基卟啉的还原过程中采用质量分数为35%
‑
37%浓盐酸作为反应溶剂,并在惰性气体保护气氛下进行(以为反应提供还原性环境),还原过程中体系温度为 60~80℃;
[0067]
por的缩合反应过程中,5
‑
(4
‑
氨基苯基)
‑
10,15,20
‑
三苯基卟啉、马来酸酐、乙酸钠的质量比为(500~700):108:(450~550);
[0068]
卟啉的马来酰亚胺衍生物por、少层二硫化钼粉末的质量比为(50~70):10 等。
[0069]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
[0070]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的
保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1