一种高纯度甘草查尔酮A的制备及检测方法
一种高纯度甘草查尔酮a的制备及检测方法
技术领域
1.本发明属于化合物单体制备技术领域,具体涉及一种高纯度甘草查尔酮a的制备及检测方法。
背景技术:
2.甘草查尔酮a是从豆科植物甘草的根中提取的黄酮类化合物,被视为胀果甘草的种属特异性成分。现代药理学表明,甘草查尔酮a 具有抑制激素非依赖性pc
‑
3前列腺癌细胞增殖,诱导凋亡,抗寄生性疾病疟疾,抗菌消炎,抗氧化等作用,在食品和医药工业中有广泛的用途。
3.然而,由于甘草查尔酮a在甘草中含量极低(0.4%左右),制备工艺繁复,成本昂贵。截至目前,我国尚无甘草查尔酮a标准样品,市场上销售的甘草查尔酮a对照品纯度也参差不齐。因此,为满足药材及成药分析检测及质量控制工作的需求,亟待开发研制甘草查尔酮a标准样品及其质量控制技术研究,为以此为原料的功能食品、保健食品提供坚实的物质基础。
4.公开号为101117311a的中国发明专利以甘草为原料,经冷渗法提取,有机溶剂萃取后再利用大孔树脂纯化,重结晶得到纯度大于 90%的甘草查尔酮a。公开号为112898146a的中国发明专利中利用 dna甲基转移酶抑制剂培育甘草毛状根,激活有利于甘草查尔酮a 积累的沉默基因,通过甲醇提取甘草查尔酮a。公开号为106431877a 的中国发明专利将甘草渣用饱和石灰水回流提取,大孔树脂吸附,所得洗脱液naoh溶解后加酸沉淀,重结晶得到浓度大于95%的样品。但已公开的方法中制备得到的甘草查尔酮a的纯度均未达到中药化学对照品的要求,即纯度大于98.5%。且已公开的方法也没有纯度与质量控制方法,无法满足甘草查尔酮a化学对照品的需要。
技术实现要素:
5.本发明的目的是提供一种高纯度甘草查尔酮a的制备及检测方法,本发明的方法以甘草渣为原料,制备得到纯度高于98.5%的甘草查尔酮a,从而满足甘草查尔酮a国家标准品的申报要求。
6.本发明首先一种高纯度甘草查尔酮a的制备方法,所提供的方法包括有如下的步骤:
7.1)在甘草渣中加入体积浓度为60%~90%的乙醇溶液,加热回流提取,再将提取的浸膏采用ab
‑
8大孔树脂纯化,分别用水、30%乙醇、75%乙醇洗脱,收集75%乙醇的洗脱部分,洗脱液加入氢氧化钠溶液溶解后,再加入盐酸溶液获得沉淀,沉淀干燥得到甘草查尔酮a 粗提物;
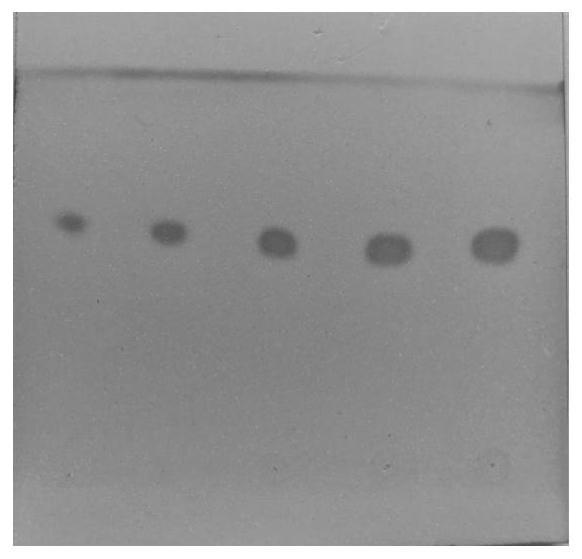
8.2)将甘草查尔酮a粗提物通过高速逆流色谱进行分离纯化,其中溶剂系统为正己烷、氯仿、甲醇和水的混合溶液,上相为固定相,下相为流动相,检测波长为254nm,收集含甘草查尔酮a组分,减压真空干燥,收集含甘草查尔酮a馏分,将含甘草查尔酮a馏分进行减压
浓缩,得到纯度>96%的甘草查尔酮a样品,最后采用甲醇进行重结晶,得到纯度>98.5%甘草查尔酮a。
9.甘草渣与乙醇溶液的料液比为1:10~1:20;
10.所述的氢氧化钠溶液的浓度为2%;盐酸溶液的浓度为5%。
11.所述的溶剂系统中正己烷:氯仿:甲醇:水的体积比为2~5:5~9: 2~10:2。
12.本发明还提供一种甘草查尔酮a纯度的检测方法,是采用分析型高效液相色谱进行检测,其中色谱柱为c18,4.6
×
250mm,5μm,流速0.8
‑
1.0ml/min,记录色谱图至主成分的出峰保留时间的2.5倍以上,用面积归一化法计算含量。
13.其中流动相(体积比)乙腈:0.1%磷酸水=47:53,检测波长为 254~372nm。
14.另一种条件下,流动相(体积比)乙腈:0.1%磷酸水=25:75~90:10。检测波长为254~372nm。
15.本发明提供的方法制备的甘草查尔酮a纯度可达到98.5%以上,纯度符合化学对照品要求,同时提供了甘草查尔酮a化学对照品完整的基础化学依据、化学信息和分析测试方法,有利于甘草查尔酮a 的进一步开发。此外,采用薄层色谱和高效液相色谱进行纯度检查、含量测定及质量控制,从而建立甘草查尔酮a化学对照品的技术标准,确保产品质量,为甘草及其相关制品的质量控制及高技术、高附加值的产品的开发提供科学基础和保证。本发明采用大孔树脂结合高速逆流色谱制备高纯度甘草查尔酮a,样品纯度高,分离速度快,生产周期短,适合工业化生产,有较好的应用前景。
附图说明
16.图1
‑
1、1
‑
2、1
‑
3分别是甘草查尔酮a三种展开系统薄层色谱图:
17.其中,图1
‑
1中展开剂为石油醚:乙酸乙酯:甲醇(5:4:1,v/v/v),图1
‑
2中展开剂为石油醚:乙酸乙酯:甲醇(4:5:1,v/v/v),图1
‑
3中展开剂为二氯甲烷:甲醇(8:1,v/v)。
18.图2是本发明甘草查尔酮a分离纯化后的hplc图谱,图中保留时间为11min的色谱图,图中峰为甘草查尔酮a。
具体实施方式
19.本发明所提供的方法,其基本步骤如下:
20.1)取胀果甘草(glycyrrhiza inflata bat.)提取甘草酸后所得甘草渣,加入体积浓度为60%~90%乙醇,料液比1:10~1:20,加热回流提取3
‑
5次,每次1~2h,提取浸膏采用ab
‑
8大孔树脂纯化,分别用水、30%乙醇、75%乙醇洗脱,富集75%乙醇洗脱部分,加入2%氢氧化钠溶液溶解后,加入5%盐酸酸化至无再沉淀生成,所得沉淀干燥得到甘草查尔酮a粗提物。进一步通过高速逆流色谱技术对其进行分离纯化,溶剂系统为正己烷:氯仿:甲醇:水,上相为固定相,下相为流动相,检测波长为254nm,收集含甘草查尔酮a组分,减压真空干燥,收集含甘草查尔酮a馏分,减压浓缩,得到纯度>96%的甘草查尔酮a样品,最后采用甲醇进行重结晶,得到纯度>98.5%甘草查尔酮a。
21.所述的溶剂系统正己烷
‑
氯仿
‑
甲醇
‑
水体积比为2~5:5~9:2~10:2。
22.2)采用分析型高效液相色谱测定步骤(1)收集的甘草查尔酮a 产品纯度,最高可达99.85%以上。色谱柱:c18,4.6
×
250mm,5μm,流速0.8
‑
1.0ml/min,面积归一法定量;系统
条件为以下两个条件的其中之一。
23.条件一:流动相(体积比)乙腈:0.1%磷酸水=47:53。检测波长为254~372nm。
24.条件二:流动相(体积比)乙腈:0.1%磷酸水=25:75~90:10。检测波长为254~372nm。
25.3)本发明对于产品甘草查尔酮a的质量控制方法如下:
26.含量与纯度检验:分别用2个流动相溶剂系统和2个检测波长,记录色谱图至主成分的出峰保留时间的2.5倍以上,用面积归一化法计算含量,结果系统测定样品含量均在98.5%以上,杂质检查,分别在不同系统记录的色谱图中,处溶剂峰外,杂质峰面积总和结果均小于1.5%。
[0027][0028]
峰纯度检测:取对照品适量,按所述高效液相色谱条件,在高效液相色谱仪上,用二极管阵列dad检测器进行峰纯度检查,hplc 色谱图(>98.5%),色谱峰的紫外吸收光谱图、三维图谱以及5点光谱图完全重合,表明为单一纯物质峰。
[0029]
本发明高纯度化合物的质量控制方法还包括:薄层色谱法检测,取甘草查尔酮a样品用甲醇配置1mg/ml溶液,在同一硅胶g上,按不同的点样量梯度点样,点样量分别为20μg、40μg、60μg、80μg、 100μg;分别以石油醚
‑
乙酸乙酯
‑
甲醇重量比例为5:4:1为展开剂,石油醚:乙酸乙酯:甲醇重量比例4:5:1为展开剂,或者二氯甲烷:甲醇重量比例8:1为展开剂,展距为3.5cm;碘显色,在5个不同浓度的梯度点样及三个展开系统薄层色谱中,均为黄色的单一的荧光斑点,则代表纯度符合要求,否则不符合要求。
[0030]
结果:本发明分离、纯化的甘草查尔酮a化学对照品,经红外光谱、紫外光谱、核磁共振、质谱确认化学结构,经三个展开系统5 个不同浓度的tlc检测,2个流动相系统和3个不同波长的hplc 检测,同时对色谱峰用dad做纯度检查,表明符合国家标准样品要求,含量大于98.5%。
[0031]
下面结合实施例对本发明进行详细的描述。
[0032]
实施例1高纯度甘草查尔酮a制备
[0033]
取胀果甘草渣1kg,用70%乙醇加热回流提取2次(50℃),料液比为1:10,每次1h。提取液减压抽滤,55℃旋转蒸发浓缩。所得甘草提取物采用ab
‑
8大孔树脂纯化(2.6
×
50cm,树脂柱床高约30 cm),分别用水、30%乙醇、75%乙醇洗脱,洗脱流速1.5bv/h,收集75%乙醇洗脱部分。减压浓缩后,加入2%氢氧化钠溶液提取甘草黄酮成分,至溶液澄清后,加入5%盐酸并不停搅拌至ph=1,产生大量沉淀,过滤,收集沉淀冷水洗至中性,50℃烘干得甘草查尔酮a 粗提物。采用高速逆流色谱技术对所得甘草查尔酮a粗提物进一步纯化,实验采用tbe
‑
300a型高速逆流色谱仪,配有tbp5002泵、 tbd
‑
2000紫外检测器和dc
‑
0506低温恒温槽,溶剂系统为正己烷: 氯仿:甲醇:水(5:6:3:2,v/v/v/v),配置后静置过夜,次日分离后超声脱气15min。将上相(固定相)在最大流速下泵入并充满分离螺线管,开启循环水浴并将温度设定为25℃。然后将下相(流动相)以1.8 ml/min泵入,同时开启检测器并按800rpm/min的转速启动主机。称取上述样品溶于下相溶液,待流动相从管柱出口流出且基线稳定后
将样品溶液由进样圈注入管柱。紫外检测器检测并由色谱工作站和记录仪记录,检测波长为254nm,收集含甘草查尔酮a组分,减压真空干燥,得到纯度为96%的甘草查尔酮a样品。取甘草查尔酮a样品置于烧杯中,加入少量甲醇,加热溶解制成饱和溶液,趁热过滤,室温静置待结晶析出,用少量的甲醇溶液洗涤,室温干燥,得到纯度 99.2%的甘草查尔酮a黄色粉末。
[0034]
用hplc分析方法对所有制备液进行检测:色谱柱:c18柱;流动相:乙腈:0.1%磷酸水体积比为47:53;检测波长为372nm,流速为1ml/min。
[0035]
实施例2:高纯度甘草查尔酮a制备
[0036]
取胀果甘草渣1kg,用90%乙醇加热回流提取3次(50℃),料液比为1:20,每次2h。提取液减压抽滤,55℃旋转蒸发浓缩。所得甘草提取物采用ab
‑
8大孔树脂纯化(2.6
×
50cm,树脂柱床高约30 cm),分别用水、30%乙醇、75%乙醇洗脱,洗脱流速1.5bv/h,收集75%乙醇洗脱部分。减压浓缩后,加入2%氢氧化钠溶液提取甘草黄酮成分,至溶液澄清后,加入5%盐酸并不停搅拌至ph=1,产生大量沉淀,过滤,收集沉淀冷水洗至中性,50℃烘干得甘草查尔酮a 粗提物。采用逆流色谱技术对所得甘草查尔酮a粗提物进一步纯化,实验采用tbe
‑
300a型高速逆流色谱仪,配有tbp5002泵、tbd
‑
2000 紫外检测器和dc
‑
0506低温恒温槽,溶剂系统为正己烷:氯仿:甲醇: 水(2:9:10:2,v/v/v/v),配置后静置过夜,次日分离后超声脱气15min。将上相(固定相)在最大流速下泵入并充满分离螺线管,开启循环水浴并将温度设定为25℃。然后将下相(流动相)以1.8ml/min泵入,同时开启检测器并按800rpm/min的转速启动主机。称取上述样品溶于下相溶液,待流动相从管柱出口流出且基线稳定后将样品溶液由进样圈注入管柱。紫外检测器检测并由色谱工作站和记录仪记录,检测波长为254nm,收集含甘草查尔酮a组分,减压真空干燥,得到纯度为96%的甘草查尔酮a样品。取甘草查尔酮a样品置于烧杯中,加入少量甲醇,加热溶解制成饱和溶液,趁热过滤,室温静置待结晶析出,用少量的甲醇溶液洗涤,室温干燥,得到纯度98.7%的甘草查尔酮a黄色粉末。
[0037]
用hplc分析方法对所有制备液进行检测:色谱柱:c18柱;流动相:乙腈:0.1%磷酸水体积比为47:53;检测波长为372nm,流速为1ml/min。
[0038]
实施例3:甘草查尔酮a检测
[0039]
取实施例1
‑
2制得的含量在98.5%以上的甘草查尔酮a,通过以下质量控制方法进行检测: 1薄层色谱检测方法:
[0040]
采用3种展开体系进行检测,薄层板硅胶254g板,设计五个点样量分别为20μg(点1)、40μg(点2)、60μg(点3)、80μg(点4)、 100μg(点5)。利用以下3种不同的展开剂进行的薄层色谱检测,展距为3.5cm。
[0041]
展开剂系统1:石油醚:乙酸乙酯:甲醇(5:4:1,v/v/v)(i,图1)
[0042]
展开剂系统2:石油醚:乙酸乙酯:甲醇(4:5:1,v/v/v)(ii,图2)
[0043]
展开剂系统3:二氯甲烷:甲醇(8:1,v/v)(iii)
[0044]
展开后取出,晾干,碘显色,置白光下,结果显示,甘草查尔酮 a样品在20~100μg的点样量范围内,仅见明显的一个显色斑点,未见杂质的斑点,说明该样品的纯度较高。
[0045]
2.高效液相色谱法
[0046]
色谱条件用十八烷基键合硅胶为填充剂
[0047]
分别采用以下流动相系统,
[0048]
流动相系统1:乙腈:0.1%磷酸水
‑
溶液体积比为47:53。
[0049]
流动相系统2:乙腈(a):0.1%磷酸水(b)溶液体积比为0~8min 25~32%a;8~30min 32~45%a;30~42min 45~60%a;42~50min 60~90%a。
[0050]
检测波长分别为254nm、372nm。
[0051]
测定法取于105℃干燥至恒重的本品适量,精密称定,配置浓度为0.5mg/ml溶液,注入液相色谱仪,记录色谱图至主成分的出峰保留时间的2.5倍以上,用面积归一化法计算含量,结果样品测定甘草查尔酮a含量均在98.5%以上,杂质检查,分别在不同系统记录的色谱图中,除溶剂峰外,杂质峰面积总和结果均小于1.5%。
[0052]
峰纯度检测:取对照品适量,分别按以上两个流动相系统,在高效液相色谱仪上,用二极管阵列dad检测器进行峰纯度检查,甘草查尔酮a hplc色谱峰>98.5%,其色谱峰紫外吸收光谱图,三维图谱完全重合,表明为单一纯物质峰。
[0053]
结构确认
[0054]
uv(meoh)λmax(logε)=254(3.97),312(4.21),377(4.29)
[0055]
ir v kbr max cm
‑1:3452cm
‑1:
‑
oh;1604cm
‑1:
‑
o=c
‑
;1641, 1588,1510cm
‑1:多共轭双键的伸缩振动;1291cm
‑1:不对称c
‑
o
‑
c 的伸缩振动;836,755cm
‑1:芳香c
‑
h面外弯曲振动。
[0056]1h
‑
nmr(400mhz,in cd3od)δppm:7.48(1h,s,h
‑
3);6.45(1 h,s,h
‑
6);7.95(2h,d,j=8.5hz,h
‑2′
);6.88(2h,d,j=8.5hz,h
‑3′
); 7.57(1h,d,j=8.5hz,h
‑
α);7.99(1h,d,j=16hz,h
‑
β);6.21(1h,dd, j=18,10hz,h
‑2″
);5.00(1h,dd,j=18.1hz,h
‑3″
);4.96(1h,dd, j=10.1hz,h
‑3″
);1.47(3h,s,h
‑4″
);1.47(3h,s,h
‑5″
);3.86(3h,s, och3)
[0057]
13
c
‑
nmr(400mhz,in cd3od)δppm:115.0(c
‑
1);159.2(c
‑
2); 99.4(c
‑
3);160.1(c
‑
4);127.0(c
‑
5);128.8(c
‑
6);117.9(c
‑
α);140.9(c
‑
β); 130.1(c
‑1′
);130.7(c
‑2′
);114.4(c
‑3′
);162.1(c
‑4′
);114.4(c
‑5′
);130.7 (c
‑6′
);39.7(c
‑1″
);147.7(c
‑2″
);109.3(c
‑3″
);26.1(c
‑4″
);54.7(c
‑5″
); 190.5(c
‑6″
)
[0058]
ms(esi
‑
ms)m/z:337.1460[m
‑
h]
‑
[0059]
实验结果表明:本发明分离、纯化的甘草查尔酮a化学对照品,经红外光谱、紫外光谱、核磁共振、质谱及理化检测确认化学结构,经本发明公开的3个展开系统tlc检测,2个流动相系统和2个不同波长的hplc检测,同时对色谱峰用dad做纯度检查,表明符合国家标准样品的要求,含量大于98.5%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1