一类吲哚衍生物及其制备方法和应用
1.本发明属于制药技术领域,具体涉及一类吲哚衍生物及其制备方法和应用。
背景技术:
2.抑郁症是一种常见的精神障碍疾病,主要表现为情绪低落、兴趣减退、快感缺失等。部分严重的患者可能还会出现焦虑、自责自罪、自杀未遂等行为。据研究显示,大约有3亿人在一生中的某段时间会遭受重症抑郁症的困扰。自杀是抑郁症最严重的问题,其发生率约占抑郁症患者的15%。
[0003]5‑
羟色胺(5
‑
hydroxytryptamine,5
‑
ht或serotonin)是与抑郁密切相关的单胺类神经递质,大量研究表明抑郁症发生与脑内5
‑
ht水平低下密切相关。5
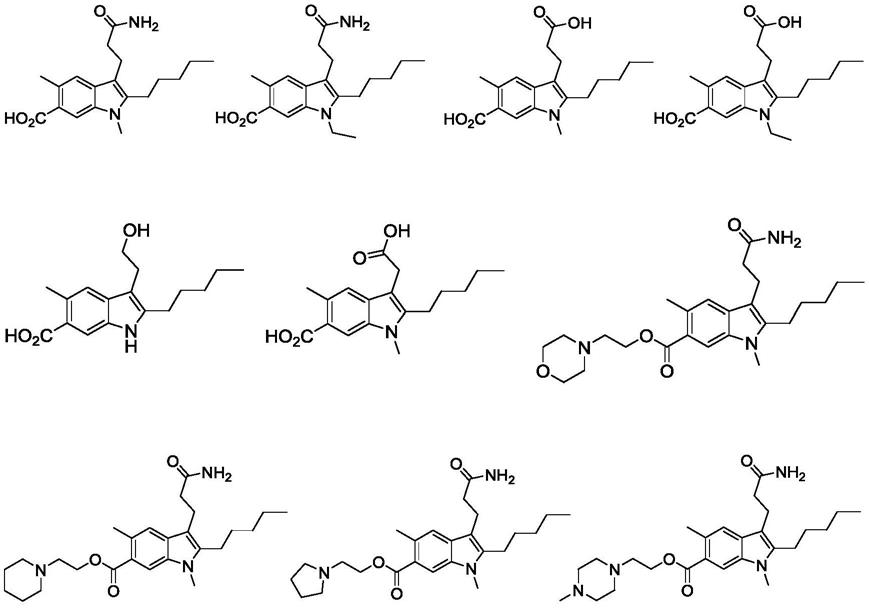
‑
羟色胺再摄取抑制剂(selective serotonin reuptake inhibitors,ssris)舍曲林、帕罗西汀、氟西汀、氟伏沙明、西酞普兰是临床上广泛使用的抗抑郁药物。这些药物通过抑制脑内5
‑
羟色胺的再摄取,增加突触间隙内单胺递质的浓度,从而发挥抗抑郁的作用。这类药物普遍存在恶心、嗜睡、发汗、眩晕、性功能障碍、高血压、焦虑、口干、头昏、便秘等副作用,导致病人的顺应性较差。起效缓慢是这类药物另外一个重要缺点,发挥疗效需要3~4周,具有明显的临床延迟效应。由于药物的副作用,有些病人在没有等到药物发挥作用,就已放弃药物治疗。因此,基于新机制的、快速起效的、副作用少的新型抗抑郁药物的研制具有重要的临床价值。
[0004]
2019年美国批准了s
‑
氯胺酮(esketamine)用于难治性抑郁症的治疗。氯胺酮是一种主要作用于nmda受体的、快速起效的抗抑郁药物(j med chem,2020,63,13514
‑
13525)。但氯胺酮存在剂量依赖的头晕、恶心、呕吐和唾液分泌过多等副作用,另外,它还可能导致心理分离(psychological dissociation)。较高剂量或氯胺酮的长期使用可引发一些持续和明显的神经精神疾病症状,例如与精神分裂症相关的症状、认知障碍等。另一个严重缺点是氯胺酮存在滥用的分险。这些不足限制了其在临床实践中的广泛应用(molecules,2020,25,5777)。
[0005]
中缝背核是前脑5
‑
羟色胺的主要来源,而血清素转运体或称5
‑
ht转运体(serotonin transporter,sert或5
‑
hydroxytryptamine,5
‑
htt)是5
‑
ht能传递信号的关键调制器,是ssris抗抑郁药物的作用靶点。sert是一种依赖na
+
/cl
‑
的高亲和力跨膜转运蛋白,具有12个跨膜区,n端和c端均位于胞质中,研究发现sert的基因多态性与抑郁症的发生密切相关,另有研究表明中缝背核sert的缺失能够促使5
‑
ht能神经元活性降低而导致抑郁。神经元型一氧化氮合酶(neuronal nitric oxide synthase,nnos)是中枢神经系统中催化合成no的合酶,它在drn有大量表达。研究表明nnos的pdz结构域可以与sert的c末端结合,从而影响sert在细胞膜上的分布。drn区的nnos
‑
sert耦联可以调节5
‑
ht能神经元放电。因此,研发一种能够解开或抑制nnos
‑
sert耦联的药物,通过增加5
‑
ht能神经元放电频率,大量释放5
‑
ht,可发挥快速抗抑郁作用。
[0006]
焦虑症以广泛、持续性焦虑或反复发作的惊恐不安为主要特征的神经症性障碍,常伴有植物神经症状和运动性紧张。苯二氮卓类化合物为临床治疗焦虑症的最主要药物,
但是该类药物同时具有镇静/催眠的副作用。目前,5
‑
羟色胺再摄取抑制剂也用于焦虑症的治疗。并且,同样的,5
‑
羟色胺再摄取抑制剂治疗焦虑症时也起效缓慢。本发明披露的能够解开或抑制nnos
‑
sert耦联的药物,也可以发挥快速抗焦虑作用。
技术实现要素:
[0007]
为克服现有抗抑郁药物存在起效慢且具有多种副作用的技术问题,本发明提供了一类吲哚衍生物及其制备方法和应用,所述吲哚衍生物可有效抑制nnos
‑
sert的偶联,从而发挥快速抗抑郁作用。
[0008]
为实现上述目的,本发明提供了如下技术方案:
[0009]
本发明提供了一类吲哚衍生物,所述吲哚衍生物为通式(ⅰ)所代表的化合物:
[0010][0011]
所述通式(ⅰ)中,r1=h、1
‑
5个碳原子的直链或支链烷基、苄基、苯乙基或烷基取代的苄基或烷基取代的苯乙基,r2=h或1
‑
6个碳原子的直链或支链烷基,r3=h、1
‑
6个碳原子的直链或支链烷基或
‑
(ch2)
n
conh2或
‑
(ch2)
m
co2r5,其中r5=h或1
‑
6个碳原子的直链或支链烷基;r4=h或1
‑
6个碳原子的直链或支链烷基、6个碳原子的直链或支链烷基、所述m取自1
‑
5的任意整数,n取自1
‑
6的任意整数。
[0012]
优选的,所述吲哚衍生物的化学结构为以下任意一种:
[0013][0014][0015]
本发明还提供了上述吲哚衍生物及其药学上可接受的盐在制备治疗抑郁症、焦虑症的药物中的应用。
[0016]
优选的,所述药学上可接受的盐包括钠盐、钾盐、盐酸盐、氢溴酸盐、硝酸盐、高氯酸盐、磷酸盐、硫酸盐、甲酸盐、乙酸盐、阿康酸盐、抗坏血酸盐、苯磺酸盐、苯甲酸盐、肉桂酸盐、柠檬酸盐、庚酸盐、富马酸盐、谷氨酸盐、乙醇酸盐、乳酸盐、马来酸盐、丙二酸盐、扁桃酸盐、甲磺酸盐、萘
‑
2磺酸盐、邻苯二甲酸盐、水杨酸盐、山梨酸盐、硬脂酸盐、琥珀酸盐、酒石酸盐或对甲苯磺酸盐。
[0017]
与现有技术相比,本发明具有以下有益效果:
[0018]
目前基于单胺假说开发的、临床上常用的抗抑郁药物普遍存在起效慢、副作用多等不足,本发明提供的吲哚衍生物是一种新型抗抑郁、抗焦虑药物,可有效抑制nnos
‑
sert的耦联,具有快速起效的特点,同时也可避免目前抗抑郁药物存在的诸多副作用。
附图说明
[0019]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例。
[0020]
图1为sw
‑
9解开nnos
‑
sert偶联的鉴定结果图,其中(a)为sw
‑
9在hek293t解开nnos
‑
sert偶联的鉴定结果;(b)为腹腔注射sw
‑
9解开nnos
‑
sert偶联的鉴定结果;
[0021]
图2为小鼠分别腹腔注射溶剂及不同浓度sw
‑
9对抑郁行为的影响结果图,其中(a)为腹腔注射溶剂、1mg/kg、2.5mg/kg、5mg/kg的sw
‑
9 2h引起tst不动时间统计结果;(b)为腹腔注射溶剂、1mg/kg、2.5mg/kg、5mg/kg的sw
‑
9 2h引起fst不动时间统计结果;(c)为腹腔注射溶剂、5mg/kg的sw
‑
9 24h对tst不动时间的影响结果;(d)为腹腔注射溶剂、5mg/kg的sw9 24h对fst不动时间的影响结果;
[0022]
图3为腹腔注射sw
‑
9快速逆转小鼠抑郁行为的研究结果图,其中(a)为腹腔注射10mg/kg的sw
‑
9 2h对cms导致的tst不动时间的减少的逆转结果;(b)为腹腔注射10mg/kg的sw
‑
9 2h对cms导致的fst不动时间的减少的逆转结果;(c)为腹腔注射10mg/kg的sw
‑
9 2h对cms导致的spt糖水偏爱率的减少的逆转结果;
[0023]
图4为灌胃sw
‑
9快速逆转小鼠抑郁行为的研究结果图,其中(a)为灌胃10mg/kg的sw
‑
9 2h对cms导致的tst不动时间的减少的逆转结果;(b)为灌胃10mg/kg的sw
‑
9 2h对cms导致的fst不动时间的减少的逆转结果;(c)为灌胃10mg/kg的sw
‑
9 2h对cms导致的spt糖水偏爱率的减少的逆转结果;
[0024]
图5为尾静脉给药小鼠sw
‑
9脑组织药物浓度
‑
时间曲线图。
具体实施方式
[0025]
现详细说明本发明的多种示例性实施方式,该详细说明可使本专业技术人员全面理解本发明,但不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。
[0026]
以下效果验证例中所采用的实验小鼠为spf级icr雄性小鼠,体重22
±
2g,购自南京医科大学动物中心,合格证号:spfnjmu001189。
[0027]
实施例1
[0028]
吲哚衍生物1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
9)的合成:
[0029]
合成路线如下:
[0030][0031]2‑
甲基
‑5‑
硝基苯甲酸通过硫酸催化甲酯化(2)、硝基催化氢化成氨基(3)、氯化碘4
‑
位碘化得5
‑
氨基
‑4‑
碘
‑2‑
甲基苯甲酸甲酯(4),之后通过pd催化sonogashira偶联反应(5)和随后的pd催化的成环反应构建5
‑
甲基
‑2‑
戊基吲哚
‑6‑
甲酸甲酯(6),之后和卤代烃反应在1
‑
位发生烷基化反应(7),所得产物和丙烯酸在3
‑
位发生反应得3
‑
位羧基乙基化的产物(8),该产物和ibcf成酸酐再和氨水反应成酰胺(9),碱水解成羧酸(10),再和计算量的naoh反应生成1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
9),具体步骤如下:
[0032]
(a)2
‑
甲基
‑5‑
硝基苯甲酸甲酯(2)的合成:向1000ml烧瓶中加入2
‑
甲基
‑5‑
硝基苯甲酸(1,40g,221mmol),甲醇400ml,边搅边加入98%硫酸24ml,75℃回流8h。反应结束后,将反应瓶置于冰水浴,析出大量固体,抽滤,干燥,得黄色固体2
‑
甲基
‑5‑
硝基苯甲酸甲酯(2)32g,产率74%。1h nmr(400mhz,cdcl3)δ:8.76(d,j=4hz,1h),8.22(dd,j=4,8hz,1h),7.42(d,j=8hz,1h),3.94(s,3h),2.71(s,3h).ms(esi,m/z):196.1[m+h]
+
。
[0033]
(b)5
‑
氨基
‑2‑
甲基苯甲酸甲酯(3)的合成:向1000ml烧瓶中加入步骤(a)得到的2
‑
甲基
‑5‑
硝基苯甲酸甲酯(2,30g,182mmol),甲醇500ml,10%钯碳1.13g,氢气置换3次排除反应体系内的空气,在氢气条件下室温搅拌,不断补充氢气至气球体积不再明显变化。反应结束后,抽滤除去不溶物,收集滤液,将滤液旋干得深棕色粘稠油状液5
‑
氨基
‑2‑
甲基苯甲酸甲酯(3)22.8g,产率76%。
[0034]
(c)5
‑
氨基
‑4‑
碘
‑2‑
甲基苯甲酸甲酯(4)的合成:向500ml烧瓶中加入步骤(b)得到的5
‑
氨基
‑2‑
甲基苯甲酸甲酯(3,20g,121mmol),碳酸钙(21g,210mmol),甲醇200ml,水20ml,制成溶液1;一氯化碘(22g,123mmol)溶于132ml二氯甲烷中,制成溶液2;在冰浴条件下,将溶液2缓慢滴入溶液1中,滴加完毕后,升至室温,搅拌8h。反应结束后,向反应体系中加入25%na2so3溶液淬熄1h,过滤,滤渣用甲醇洗涤3次,收集滤液,滤液浓缩后加入乙酸乙酯溶解,饱和食盐水洗涤2次,收集有机层,无水硫酸钠干燥。过滤,滤液浓缩得深棕色油状物,硅胶柱层析(pe:ea=10:1)得淡黄色固体5
‑
氨基
‑4‑
碘
‑2‑
甲基苯甲酸甲酯(4)18g,产率45%。1h nmr(400mhz,cdcl3)δ:7.54(s,1h),7.28(s,1h),4.03(s,2h),3.85(s,3h),2.41(s,3h).ms(esi,m/z):292[m+h]
+
。
[0035]
(d)5
‑
氨基4
‑
(1
‑
庚炔基)
‑2‑
甲基
‑
苯甲酸甲酯(5)的合成:向干燥的250ml烧瓶中加入100ml重蒸dmf,步骤(c)得到的5
‑
氨基
‑4‑
碘
‑2‑
甲基
‑
苯甲酸甲酯(4,16g,55mmol),pdcl2(pph3)
2 1.92g,cui 0.528g和三乙胺240ml,在冰水浴和ar保护的条件下,不断搅拌,用注射器向反应体系中缓慢加入1
‑
庚炔(7.84ml,60mmol),滴加完毕后,升至室温,搅拌8h。反应结束后,抽滤,收集滤液,加入乙酸乙酯稀释,饱和食盐水洗涤2次,收集有机层,无水硫酸钠干燥。过滤,滤液浓缩得深棕色固体,硅胶柱层析(pe:ea=10:1)得深棕色固体5
‑
氨基
‑4‑
(1
‑
庚炔基)
‑2‑
甲基苯甲酸甲酯(5)12.34g,产率87%。1h nmr(400mhz,cdcl3)δ:7.27(s,1h),7.11(s,1h),4.11(s,2h),3.85(s,3h),2.46(t,j=7.1hz,2h),2.42(s,3h),1.63(m,2h),1.48
–
1.32(m,4h),0.92(t,j=7.2hz,3h).ms(esi,m/z):260.2[m+h]
+
。
[0036]
(e)5
‑
甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(6)的合成:向干燥的250ml烧瓶中加入100ml重蒸dmf,步骤(d)得到的5
‑
氨基
‑4‑
(1
‑
庚炔基)
‑2‑
甲基苯甲酸甲酯(5,12g,46.3mmol),pdcl2(phcn)
2 3.6g,80℃下,ar保护,搅拌4h。反应结束后,抽滤,收集滤液,加入适量乙酸乙酯稀释,饱和食盐水洗涤2次,收集有机层,无水硫酸钠干燥,过滤,滤液浓缩得黄色固体,硅胶柱层析(pe:ea=10:1)得黄色固体5
‑
甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(6)10.3g,产率86%。1h nmr(400mhz,cdcl3)δ:8.08(s,1h),7.98(s,1h),7.33(s,1h),6.17(s,1h),3.89(s,3h),2.74(t,j=7.6hz,2h),2.67(s,3h),1.77
–
1.65(m,2h),1.35(m,4h),0.90(t,j=7.0hz,3h).ms(esi,m/z):258.1[m
‑
h]
‑
。
[0037]
(f)1,5
‑
二甲基
‑2‑
戊基
‑
吲哚
‑6‑
甲酸甲酯(7)的合成:向干燥的100ml烧瓶中加入50ml重蒸dmf,步骤(e)得到的5
‑
甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(6,9g,34.7mmol),碘甲烷(6.4g,45.1mmol),边搅拌边缓慢加入60%氢化钠(1.53g,38.2mmol),产生大量气泡,80℃下搅拌过夜。反应结束后,缓慢加入冰水淬灭多余的氢化钠,加入适量乙酸乙酯稀释,用水洗涤三次,收集有机层,无水硫酸钠干燥,过滤,滤液浓缩得黄色固体,硅胶柱层析(pe:ea=10:1)得淡黄色固体,即1,5
‑
二甲基
‑2‑
戊基
‑
吲哚
‑6‑
甲酸甲酯(7)8.1g,产率85%。1h nmr(400mhz,cdcl3)δ:7.94(s,1h),7.31(s,1h),6.15(s,1h),3.89(s,3h),3.61(s,3h),2.73
–
2.60(m,5h),1.68(m,2h),1.39(m,4h),0.92(t,j=7.0hz,3h).ms(esi,m/z):296.2[m+na]
+
。
[0038]
(g)1,5
‑
二甲基
‑2‑
戊基
‑6‑
(甲氧羰基)
‑
吲哚
‑3‑
丙酸(8)的合成:向干燥的耐压瓶中加入步骤(f)得到的1,5
‑
二甲基
‑2‑
戊基
‑
吲哚
‑6‑
甲酸甲酯(7,8g,29.3mmol),丙烯酸(4.65g,62.5mmol),醋酸13.4ml,醋酸酐5.6ml,90℃下搅拌3h,冷却至室温,继续搅拌8h。反应结束后,加适量乙酸乙酯溶解,水洗3次,收集有机层,无水硫酸钠干燥,过滤,滤液浓缩得
黄色油状液,硅胶柱层析(pe:ea=4:1)得黄色粘稠油状物,冷冻干燥除去醋酸,得黄色固体1,5
‑
二甲基
‑2‑
戊基
‑6‑
(甲氧羰基)
‑
吲哚
‑3‑
丙酸(8)7.32g,产率72%。1h nmr(400mhz,cdcl3)δ:7.95(s,1h),7.31(s,1h),3.91(s,3h),3.69(s,3h),3.04(t,j=8.0hz 2h),2.76(t,j=8.0hz 2h),2.69
–
2.64(m,5h),1.66
–
1.49(m,2h),1.42
–
1.31(m,4h),0.91(t,j=7.0hz,3h).ms(esi,m/z):344.2[m
‑
h]
‑
。
[0039]
(h)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸甲酯(9)的合成:向干燥的100ml烧瓶中加入35ml干燥四氢呋喃,步骤(g)得到的1,5
‑
二甲基
‑2‑
戊基
‑6‑
(甲氧羰基)
‑
吲哚
‑3‑
丙酸(8,7g,20.3mmol),三乙胺3.1ml,冰浴条件下,加入氯甲酸异丁酯(2.8ml,21.3mmol),搅拌30分钟,反应液变浑浊,加入26%氨水(7.56ml,50.7mmol),反应液变澄清,室温下搅拌4h,出现大量沉淀。反应结束后,旋干溶剂,加适量乙酸乙酯溶解,水洗2次,收集有机层,无水硫酸钠干燥,浓缩至小体积,冰浴下快速加入大量石油醚,析出白色固体,抽滤,干燥,得白色固体1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸甲酯(9)5.27g,产率75%。1h nmr(400mhz,cdcl3)δ:7.94(s,1h),7.31(s,1h),5.30(s,2h),3.91(s,3h),3.69(s,3h),3.05(t,j=8hz,2h),2.77(t,j=8hz,2h),2.69(s,3h),2.53(t,j=8hz,2h),1.59
–
1.50(m,2h),1.42
–
1.30(m,4h),0.90(t,j=7.1hz,3h).ms(esi,m/z):367.2[m+na]
+
。
[0040]
(i)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸(10)的合成:向干燥的100ml烧瓶中加入50ml四氢呋喃:水:甲醇=6:2:1的混合溶剂,步骤(h)得到的1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸甲酯(9,2g,6.06mmol),氢氧化锂(0.44g,18.2mmol),室温条件下,搅拌48h。反应结束后,反应液浓缩至小体积,加适量水溶解,加入2n盐酸溶液调节ph至1,析出白色固体,抽滤,适量水洗涤,干燥,得黄白色固体,硅胶柱层析(ch2cl2:meoh=20:1)得白色固体1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸(10)1.52g,产率79%。1h nmr(400mhz,dmso
‑
d6)δ:7.90(s,1h),7.32(s,1h),7.28(s,1h),6.73(s,1h),3.66(s,3h),2.85(t,j=8hz,2h),2.76(t,j=8hz,2h),2.59(s,3h),2.29(t,j=8hz,2h),1.58
–
1.46(m,2h),1.36
–
1.31(m,4h),0.88(t,j=7.0hz,3h).ms(esi,m/z):329.2[m
‑
h]
‑
。
[0041]
(j)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
9)的合成:向干燥的50ml烧瓶中加入20ml重蒸四氢呋喃,步骤(i)得到的1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸(10,1g,3.03mmol),1n氢氧化钠溶液(3.03ml,3.03mmol),室温下搅拌2h。反应结束后,旋干溶剂,冷冻干燥,得白色固体1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
9)1.06g,产率99%。
[0042]
实施例2
[0043]
吲哚衍生物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
14)的合成:
[0044]
合成路线如下:
[0045][0046]
中间体6在1位发生烷基化反应(12),再和丙烯酸反应,得到3
‑
位丙酸化的产物(13),随后酰胺化(14),6
‑
位酯的水解(15)和成盐反应得到1
‑
乙基
‑5‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
14),具体步骤如下:
[0047]
(a)5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑
吲哚
‑6‑
甲酸甲酯(12)的合成:从实施例1得到的化合物6(2.5g,9.7mmol)出发,合成方法同实施例1步骤(f),区别在于,将碘甲烷换成溴乙烷,反应后硅胶柱层析(pe:ea=10:1),得到化合物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑
吲哚
‑6‑
甲酸甲酯(12)1g,产率36%。1h nmr(400mhz,cdcl3)δ:7.97(s,1h),7.34(s,1h),6.19(s,1h),4.15(q,j=7.2hz,2h),3.91(s,3h),2.72(t,j=8.0hz,2h),2.67(s,3h),1.80
–
1.72(m,2h),1.48
–
1.38(m,4h),1.35(t,j=7.2hz,3h),0.93(t,j=7.1hz,3h).ms(esi,m/z):310.2[m+na]
+
。
[0048]
(b)5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑6‑
(甲氧羰基)
‑
吲哚
‑3‑
丙酸(13)的合成:从化合物12(1g,3.5mmol)出发,合成方法同实施例1步骤(g),区别在于,反应后硅胶柱层析(pe:ea=4:1),得到化合物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑6‑
(甲氧羰基)
‑
吲哚
‑3‑
丙酸(13)0.89g,产率71%。ms(esi,m/z):358.2[m
‑
h]
‑
。
[0049]
(c)5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸甲酯(14)的合成:
[0050]
从化合物13(0.89g,2.5mmol)出发,合成方法同实施例1步骤(h),得到化合物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸甲酯(14)0.48g,产率54%。1h nmr(400mhz,dmso
‑
d6)δ:7.90(s,1h),7.36(s,1h),7.28(s,1h),6.75(s,1h),4.16(q,j=6.9hz,2h),3.82(s,3h),2.85(t,j=8.0hz,2h),2.75(t,j=8.0hz,2h),2.59(s,3h),2.35
–
2.23(m,2h),1.61
–
1.47(m,2h),1.37
–
1.34(m,4h),1.24(t,j=7.1hz,3h),0.88(t,j=7.0hz,3h).ms(esi,m/z):381.3[m+na]
+
。
[0051]
(d)5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
丙酰胺基
‑
吲哚
‑6‑
甲酸(15)的合成:从化合物14(0.48g,1.3mmol)出发,合成方法同实施例1步骤(i),反应后硅胶柱层析(ch2cl2:meoh=20:1),得到化合物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸(15)0.34g,产率73%。1h nmr(400mhz,dmso
‑
d6)δ:7.89(s,1h),7.31(s,1h),7.28(s,1h),6.74(s,1h),4.14(q,j=7.2hz,2h),2.84(t,j=8.0hz,2h),2.74(t,j=8.0hz,2h),2.59(s,3h),2.29
(t,j=8.0hz,2h),1.60
–
1.47(m,2h),1.36
–
1.34(m,4h),1.24(t,j=7.1hz,3h),0.88(t,j=7.0hz,3h).ms(esi,m/z):343.2[m
‑
h]
‑
。
[0052]
(e)5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
14)的合成:从化合物15(0.34g,0.99mmol)出发,合成方法同实施例1步骤(j),得到化合物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
氨甲酰基乙基)
‑
吲哚
‑6‑
甲酸钠(sw
‑
14)0.35g,产率97%。
[0053]
实施例3
[0054]
1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
羧基乙基)
‑
吲哚
‑6‑
甲酸二钠(sw
‑
10)和5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
羧基乙基)
‑
吲哚
‑6‑
甲酸二钠(sw
‑
15)的合成:
[0055]
合成路线如下:
[0056][0057]
中间体8和13通过水解(17,19)、成盐反应分别得到1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
羧基乙基)吲哚
‑6‑
甲酸二钠盐(sw
‑
10)和1
‑
乙基
‑5‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羧基乙基)吲哚
‑6‑
甲酸二钠盐(sw
‑
15),具体步骤如下:
[0058]
(a)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸(17)的合成:向干燥的50ml烧瓶中加入18ml甲醇,1,5
‑
二甲基
‑2‑
戊基
‑6‑
(甲氧羰基)
‑
吲哚
‑3‑
丙酸(8,2g,5.79mmol),1n氢氧化钠溶液(18ml,18mmol),室温条件下,搅拌12h。反应结束后,反应液浓缩至小体积,加适量水溶解,2n盐酸溶液调节ph至1,析出白色固体,抽滤,适量水洗涤,干燥,得白色固体1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸(17)1.66g,产率86%。1h nmr(400mhz,dmso
‑
d6)δ:12.20(s,2h),7.91(s,1h),7.32(s,1h),3.67(s,3h),2.89(t,j=7.6hz,2h),2.76(t,j=7.6hz,2h),2.59(s,3h),2.46(t,j=7.6hz,2h),1.58
–
1.46(m,2h),1.38
–
1.26(m,4h),0.88(t,j=8.0hz,3h).ms(esi,m/z):329.2[m
‑
h]
‑
。
[0059]5‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸(19)的合成:合成方法同化合物17的合成方法,从化合物13(0.3g,0.83mmol)出发,得到化合物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸(19)0.26g,产率89%。1hnmr(400mhz,dmso
‑
d6)δ:12.21(s,2h),7.91(s,1h),7.32(s,1h),4.16(q,j=6.9hz,2h),2.88(t,j=7.7hz,2h),2.75(t,j=8.0hz,2h),2.59(s,3h),2.46(t,j=8.0hz,2h),1.59
–
1.47(m,2h),1.40
–
1.29(m,4h),1.24(t,j=7.1hz,3h),0.88(t,j=6.9hz,3h).ms(esi,m/z):344.2[m
‑
h]
‑
。
[0060]
(b)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸二钠(sw
‑
10)的合成:向干燥的50ml烧瓶中加入20ml甲醇,1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸(17,1g,3.02mmol),1n氢氧化钠溶液(6.04ml,6.04mmol),室温条件下,搅拌2h。反应结束后,旋干溶剂,冷冻干燥,得白色固体,即1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸二钠(sw
‑
10)1.13g,产率99%。
[0061]5‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸二钠(sw
‑
15)的合成:合成方法同sw
‑
10的合成方法,从化合物19(0.26g,0.75mmol)出发,得到化合物5
‑
甲基
‑1‑
乙基
‑2‑
戊基
‑3‑
(2
‑
羧乙基)
‑
吲哚
‑6‑
甲酸二钠(sw
‑
15)0.29g,产率99%。
[0062]
实施例4
[0063]5‑
甲基
‑2‑
戊基
‑6‑
(羧基甲基)
‑
1h
‑
吲哚
‑6‑
甲酸二钠(sw
‑
20)和5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸钠(sw
‑
21)的合成:
[0064]
合成路线如下:
[0065][0066]
中间体6和草酰氯反应得到3
‑
位草酸甲酯修饰的化合物21,和苯环酰肼缩合(22)再用nabh4还原同时得到5
‑
甲基
‑2‑
戊基
‑3‑
(甲氧碳基甲基)吲哚
‑6‑
甲酸酯(23)和5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟基乙基)吲哚
‑6‑
甲酸酯(24),23和24经水解、再成盐分别得到5
‑
甲基
‑2‑
戊基
‑3‑
(羧基甲基)吲哚
‑6‑
甲酸二钠盐(sw
‑
20)和5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟基乙基)吲哚
‑6‑
甲酸二钠盐(sw
‑
21),具体步骤如下:
[0067]
(a)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代乙酰基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(21)的合成:向干燥的100ml烧瓶中加入5
‑
甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(6,5g,19.3mmol),重蒸乙醚50ml,在冰浴条件下,逐滴滴加草酰氯(4.9ml,57.9mmol),温度回升至室温,搅拌3h,在冰浴条件下,加入甲醇(3.85ml,96.5mmol),搅拌15min。反应结束后,旋干溶剂,加适量乙酸乙酯溶解,水洗三次,收集有机层,无水硫酸钠干燥。过滤,滤液浓缩,硅胶柱层析(pe:ea=3:1)得淡黄色固体5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代乙酰基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(21)4.75g,产率71%。1h nmr(400mhz,cdcl3)δ:9.93(s,1h),8.01(s,1h),7.75(s,1h),3.97(s,3h),3.88(s,3h),3.00(t,j=8.0hz,2h),2.66(s,3h),1.76
–
1.65(m,2h),1.37
–
1.26(m,4h),0.86(t,j=7.1hz,3h).ms(esi,m/z):368.2[m+na]
+
。
[0068]
(b)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代
‑1‑
(2
‑
(苯磺酰基)亚肼基)乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(22)的合成:向干燥的250ml烧瓶中加入5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代乙酰基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(21,4g,11.59mmol),对甲苯磺酰肼(5.40g,28.97mmol),甲醇150ml,ar保护条件下,加热至80℃,回流16h。反应结束后,旋干有机溶剂,加入适量乙酸乙酯溶解,水洗3次,收集有机层,无水硫酸钠干燥。过滤,滤液浓缩,硅胶柱层析(pe:ea=5:1)得淡黄色固体5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代
‑1‑
(2
‑
(苯磺酰基)亚肼基)乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(22)4.23g,产率73%。1h nmr(400mhz,dmso
‑
d6)δ:11.64(s,1h),11.09(s,1h),7.93(s,1h),7.72(d,j=8.3hz,2h),7.41(d,j=8.0hz,2h),6.84(s,1h),3.83(s,3h),3.70(s,3h),2.52(s,3h),2.45(t,j=9.6hz,2h),2.39(s,3h),1.54
–
1.43(m,2h),1.19
–
1.12(m,4h),0.80(t,j=6.9hz,3h).ms(esi,m/z):536.2[m+na]
+
。
[0069]
(c)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(23)和5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(24)的合成:
[0070]
向干燥的250ml烧瓶中加入5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代
‑1‑
(2
‑
(苯磺酰基)亚肼基)乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(22,3g,6.01mmol),重蒸四氢呋喃50ml,nabh4(4.73g,125mmol),ar保护的条件下,加热至80℃,回流10h。反应结束后,冷却至室温,冰浴条件下缓慢滴加2n盐酸溶液,直至无气泡产生,旋干有机溶剂,加适量乙酸乙酯溶解,水洗三次,收集有机层,无水硫酸钠干燥,过滤,滤液浓缩,硅胶柱层析(pe:ea=3:1)得白色固体5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(23)120mg,产率12.06%。1h nmr(400mhz,dmso
‑
d6)δ:11.12(s,1h),7.87(s,1h),7.26(s,1h),3.81(s,3h),3.68(s,2h),3.57(s,3h),2.70(t,j=9.6hz,2h),2.57(s,3h),1.67
–
1.57(m,2h),1.31
–
1.25(m,4h),0.87(t,j=6.0hz,3h).ms(esi,m/z):330.2[m
‑
h]
‑
。得到5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(24)180mg,淡黄色固体,产率19.76%;1h nmr(400mhz,cdcl3)δ:8.30(s,1h),7.97(s,1h),7.33(s,1h),3.89(s,3h),3.83(t,j=6.6hz,2h),2.95(t,j=6.7hz,2h),2.73(t,j=8.0hz,2h),2.68(s,3h),1.69
–
1.61(m,2h),1.34
–
1.30(m,4h),0.89
–
0.87(m,3h).ms(esi,m/z):302.2[m
‑
h]
‑
。
[0071]
(d)5
‑
甲基
‑2‑
戊基
‑3‑
(羧甲基)
‑
1h
‑
吲哚
‑6‑
甲酸(25)的合成:向干燥的50ml烧瓶中加入5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
甲氧基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(23,120mg,0.396mmol),甲醇2.6ml,1n氢氧化钠溶液(2.6ml,2.6mmol),室温条件下,搅拌24h。反应结束后,旋干有机溶剂,加适量水溶解,2n盐酸溶液调节ph至1,析出白色固体,抽滤,适量水洗涤,干燥,得白色固体5
‑
甲基
‑2‑
戊基
‑3‑
(羧甲基)
‑
1h
‑
吲哚
‑6‑
甲酸(25)107mg,产率97%。1h nmr(400mhz,dmso
‑
d6)δ:12.20(s,2h),11.03(s,1h),7.86(s,1h),7.23(s,1h),3.55(s,2h),2.69(t,j=7.6hz,2h),2.56(s,3h),1.71
–
1.56(m,2h),1.37
–
1.24(m,4h),0.86(t,j=6.8hz,3h).ms(esi,m/z):302.1[m
‑
h]
‑
。
[0072]
(e)5
‑
甲基
‑2‑
戊基
‑3‑
(羧基甲基)
‑
1h
‑
吲哚
‑6‑
甲酸二钠(sw
‑
20)的合成:向干燥的50ml烧瓶中加入5
‑
甲基
‑2‑
戊基
‑3‑
(羧甲基)
‑
1h
‑
吲哚
‑6‑
甲酸(25,107mg,0.353mmol),甲醇1ml,1n氢氧化钠溶液(0.706ml,0.706mmol),室温条件下,搅拌2h。反应结束后,旋干有机溶剂,冷冻干燥,得淡黄色固体5
‑
甲基
‑2‑
戊基
‑3‑
(羧基甲基)
‑
1h
‑
吲哚
‑6‑
甲酸二钠(sw
‑
20)122mg,产率99%。
[0073]
(f)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸(27)的合成:向干燥的50ml烧瓶中加入5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(24,180mg,0.594mmol),甲醇2ml,1n氢氧化钠溶液(1.93ml,1.93mmol),室温条件下,搅拌12h。反应结束后,旋干有机溶剂,加适量水溶解,2n盐酸溶液调节ph至1,乙酸乙酯萃取三次,收集有机层,无水硫酸钠干燥,过滤,旋干滤液得棕色粘稠油状物5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸(27)157mg,产率91%。1h nmr(400mhz,dmso
‑
d6)δ:12.15(s,1h),10.89(s,1h),7.84(s,1h),7.24(s,1h),3.50(t,j=7.5hz,2h),2.76(t,j=7.5hz,2h),2.68(t,j=7.6hz,2h),2.57
(s,3h),1.70
–
1.57(m,2h),1.38
–
1.26(m,4h),0.86(t,j=6.9hz,3h).ms(esi,m/z):288.2[m
‑
h]
‑
。
[0074]
(g)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸钠(sw
‑
21)的合成:向干燥的50ml烧瓶中加入5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸(27,157mg,0.543mmol),甲醇1ml,1n氢氧化钠溶液(0.543ml,0.543mmol),室温条件下,搅拌2h。反应结束后,旋转蒸发除去有机溶剂,冷冻干燥,得棕色固体5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
羟乙基)
‑
1h
‑
吲哚
‑6‑
甲酸钠(sw
‑
21)165mg,产率98%。
[0075]
实施例5
[0076]
1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
吗啉基)乙基酯(sw
‑
9a)、1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
(哌啶
‑1‑
基))乙基酯(sw
‑
9b)、1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
(四氢吡咯
‑1‑
基))乙基酯(sw
‑
9c)及1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
(4
‑
甲基哌嗪
‑1‑
基))乙基酯(sw
‑
9d)的合成:
[0077]
合成路线如下:
[0078][0079]
中间体9分别和4
‑
羟乙基吗啉、n
‑
羟乙基哌啶、n
‑
羟乙基吡咯烷、1
‑
羟乙基
‑4‑
甲基哌嗪发生酯交换反应得到1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
吗啉基)乙基酯(sw
‑
9a)、1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
(哌啶
‑1‑
基))乙基酯(sw
‑
9b)、1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
(四氢吡咯
‑1‑
基))乙基酯(sw
‑
9c)及1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
吲哚
‑6‑
甲酸
‑
(2
‑
(4
‑
甲基哌嗪
‑1‑
基))乙基酯(sw
‑
9d),具体步骤如下:
[0080]
sw
‑
9a的合成:向干燥的100ml烧瓶中加入4
‑
羟乙基吗啉(1.06ml,8.76mmol),重蒸四氢呋喃15ml,冰浴条件下,缓慢加入60%氢化钠(0.23g,5.82mmol),搅拌1h,取1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基甲酰基乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(1.00g,2.92mmol)溶于15ml干燥四氢呋喃中,缓慢加入上述溶液,滴加完毕后,室温条件下搅拌3h。反应结束后,缓慢加入冰水,直至无气泡产生,加入适量乙酸乙酯溶解,水洗三次,收集有机层,无水硫酸钠干燥。过滤,滤液浓缩,硅胶柱层析(dcm:meoh=20:1)得白色固体0.51g,产率为39.3%。1h nmr(400mhz,cdcl3)δ7.92(s,1h),7.31(s,1h),5.33(d,j=20.3hz,2h),4.46(t,j=5.9hz,2h),3.76
–
3.70(m,4h),3.68(s,3h),3.05(t,j=7.6hz,2h),2.78(dt,j=15.4,6.9hz,4h),2.68(s,3h),2.59(s,4h),2.52(t,j=7.6hz,2h),1.75(s,2h),1.36(d,j=3.2hz,4h),0.90(t,j=6.8hz,3h).13c nmr(101mhz,cdcl3)δ174.93,168.54,142.06,134.88,130.42,121.88,119.99,112.24,109.45,67.09,61.70,57.35,53.96,36.96,31.76,29.87,29.82,24.66,22.59,20.36,14.08.ms(esi,m/z):444.2883[m+h]
+
。
[0081]
sw
‑
9b的合成:参照sw
‑
9a的合成方法,得白色固体产物,产率35.1%。1h nmr
(400mhz,cdcl3)δ7.94(s,1h),7.30(s,1h),5.37(d,j=29.1hz,2h),4.47(t,j=6.1hz,2h),3.68(s,3h),3.04(t,j=7.6hz,2h),2.78(dt,j=15.7,7.0hz,4h),2.68(s,3h),2.53(dd,j=16.7,8.7hz,6h),1.60(dd,j=19.6,14.2hz,6h),1.45(s,2h),1.39
–
1.31(m,4h),0.89(t,j=6.8hz,3h).13c nmr(101mhz,cdcl3)δ175.03,168.52,142.01,134.89,130.47,130.38,121.89,119.95,112.32,109.41,61.99,57.48,54.84,36.97,31.76,29.86,29.81,25.88,24.65,24.16,22.58,20.38,14.08.ms(esi,m/z):442.3187[m+h]
+
。
[0082]
sw
‑
9c的合成:参照sw
‑
9a的合成方法,得白色固体产物,产率33.2%。1h nmr(400mhz,cdcl3)δ7.96(s,1h),7.30(s,1h),5.39(d,j=26.7hz,2h),4.49(s,2h),3.68(s,3h),3.00(d,j=29.8hz,4h),2.70(d,j=17.3hz,9h),2.51(s,2h),1.83(s,4h),1.54(s,2h),1.35(s,4h),0.89(s,3h).13c nmr(101mhz,cdcl3)δ175.01,168.39,142.10,134.87,130.59,130.43,121.60,119.97,112.40,109.42,63.10,54.74,54.61,36.97,31.76,29.91,29.82,24.66,23.60,22.61,20.36,14.10.ms(esi,m/z):428.3020[m+h]
+
。
[0083]
sw
‑
9d的合成:参照sw
‑
9a的合成方法,得白色固体产物,产率37.5%。1h nmr(400mhz,cdcl3)δ7.91(s,1h),7.31(s,1h),5.38(d,j=21.0hz,2h),4.45(t,j=5.8hz,2h),3.68(s,3h),3.04(t,j=7.5hz,2h),2.83(t,j=5.8hz,2h),2.80
–
2.71(m,5h),2.67(s,8h),2.51(t,j=7.6hz,2h),2.41(s,3h),1.55(s,2h),1.35(s,4h),0.89(t,j=6.6hz,3h).13c nmr(101mhz,cdcl3)δ174.93,168.47,142.10,134.86,130.42,121.73,120.00,112.26,109.45,61.86,56.65,54.86,52.46,45.43,36.97,31.76,29.91,29.83,24.66,22.63,22.60,20.35,14.11.ms(esi,m/z):457.3177[m+h]
+
。
[0084]
实施例6
[0085]5‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
羧酸钠(sw
‑
31)、1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
羧酸钠(sw
‑
35)的合成:
[0086]
合成路线如下:
[0087][0088]
中间体6和7和2
‑
((乙氧基碳硫酰)硫代)乙酸反应分别得到3
‑
位羧甲基化的中间体28和32,生成的羧酸用ibcf/氨水制得酰胺29和33,6
‑
位酯基水解得30和34,之后成盐得到5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸钠(sw
‑
31)和1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸钠(sw
‑
35),具体步骤如下:
[0089]
(a)2
‑
((乙氧基碳硫酰)硫代)乙酸的合成:向100ml的圆底烧瓶中加入溴乙酸(0.91g,6.55mmol),蒸馏水5ml,冰浴条件下,分批加入乙基黄原酸钾(1.00g,6.24mmol),撤去冰浴,室温搅拌1h,然后将反应也冷却至0℃,搅拌3h,反应结束后,用稀盐酸酸化,乙酸乙酯萃取,无水硫酸钠干燥,浓缩得到白色固体,直接进行下一步反应。
[0090]
(b)6
‑
(甲氧羰基)
‑5‑
甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑3‑
乙酸(28)的合成:向干燥的100ml圆底烧瓶中加入5
‑
甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(6,2g,7.72mmol),((乙氧基硫代甲基)硫代)乙酸(1.39g,7.72mmol),干燥的乙酸乙酯10ml,在氩气保护下,90℃回流10min后加入过氧化十二酰(lpo,3.08g,7.72mmol),反应过夜,反应结束后乙酸乙酯萃取3次,收集有机相,无水硫酸钠干燥,浓缩得红色油状物,硅胶柱层析(pe:ea=6:1)得红色固体0.57g,产率为23.3%。1h nmr(400mhz,cdcl3)δ8.03(s,1h),7.83(s,1h),6.23(s,1h),3.96(s,2h),3.88(s,3h),2.74(t,j=7.7hz,2h),2.59(s,3h),1.79
–
1.64(m,2h),1.42
–
1.29(m,4h),0.90(t,j=6.9hz,3h).
13
c nmr(101mhz,cdcl3)δ176.71,169.53,143.91,132.98,132.58,
128.95,123.94,123.42,112.98,98.39,51.85,35.73,31.59,28.73,28.49,22.51,16.66,14.05.ms(esi)calcd for c
18
h
23
no4[m+h]
+
:318.1705;found:m/z 318.1690.
[0091]
(c)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
羧酸甲酯(29)的合成:向干燥的100ml烧瓶中加入5ml干燥四氢呋喃,2
‑
(6
‑
(甲氧羰基)
‑5‑
甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑3‑
乙酸(28,0.55g,1.73mmol),三乙胺0.26ml,冰浴条件下,加入氯甲酸异丁酯(0.24ml,1.82mmol),搅拌30min,反应液变浑浊,加入26%氨水(0.65ml,4.33mmol),反应液变澄清,室温下搅拌4h,出现大量沉淀。反应结束后,旋干溶剂,加适量乙酸乙酯溶解,水洗2次,收集有机层,无水硫酸钠干燥,浓缩至小体积,硅胶柱层析(pe:ea=1:1)得橙色固体5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
羧酸甲酯(29)0.41g,产率75.0%。1h nmr(400mhz,cdcl3)δ8.46(s,1h),7.83(s),6.22(s,1h),5.46(d,j=66.0hz,1h),3.88(d,j=1.6hz,2h),2.77
–
2.70(m,5h),2.56(s,2h),1.73
‑
1.68(m,2h),1.38
–
1.30(m,4h),0.88(dd,j=9.8,4.3hz,3h).
13
c nmr(101mhz,cdcl3)δ173.64,169.55,144.52,133.18,132.41,128.70,124.48,124.21,113.15,98.01,52.00,38.21,31.60,28.74,28.48,22.52,16.56,14.08.ms(esi)calcd for c
18
h
24
n2o3[m+h]
+
:317.1865;found:m/z 317.1872.
[0092]
(d)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
羧酸(30)的合成:向干燥的100ml烧瓶中加入5ml甲醇,5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
羧酸甲酯(29,0.35g,1.11mmol),2.2ml 1n naoh溶液,加热至60℃,反应过夜,反应结束后,旋去溶剂,加入适量水,用稀盐酸酸化,乙酸乙酯萃取3次,无水硫酸钠干燥,浓缩的黄色油状物,硅胶柱层析(dcm:meoh=32:1)得到黄色固体0.24g,产率为71.6%。1h nmr(400mhz,dmso
‑
d6)δ12.20(s,1h),10.99(d,j=1.2hz,1h),7.65(s,1h),7.05(d,j=150.3hz,2h),6.21(s,1h),3.64(s,2h),2.67(t,j=7.6hz,2h),2.43(s,3h),1.71
–
1.59(m,2h),1.31
–
1.25(m,4h),0.84(t,j=6.8hz,3h).
13
c nmr(101mhz,dmso
‑
d6)δ172.64,170.81,143.83,133.40,132.72,127.41,126.27,123.98,112.39,98.10,37.31,31.54,28.90,28.31,22.46,16.88,14.47.ms(esi)calcd for c
17
h
22
n2o3[m+h]
+
:303.1709;found:m/z303.1725.
[0093]
(e)5
‑
甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
羧酸钠(sw
‑
31)的合成:得到化合物30后,按照实施例4步骤(g)化合物sw
‑
21的合成方法制得sw
‑
31,产率98%。
[0094]
(f)6
‑
(甲氧羰基)
‑
1,5
‑
二甲基
‑2‑
戊基
‑
1h
‑
吲哚
‑3‑
乙酸(32)的合成:以化合物7为起始物,按照步骤(b)中化合物28的合成方法,得红色固体(32),产率为20.5%。1h nmr(400mhz,cdcl3)
13
c nmr(101mhz,cdcl3)δ7.83(s,1h),6.23(s,1h),3.97(s,2h),3.90(s,3h),3.67(d,j=2.8hz,3h),2.74
–
2.68(m,2h),2.59(s,3h),1.77
–
1.68(m,2h),1.46
–
1.36(m,4h),0.93(t,j=7.1hz,3h).
13
c nmr(101mhz,cdcl3)δ177.57,169.65,145.38,134.59,131.53,128.66,123.33,111.62,97.44,51.87,35.75,31.76,29.66,28.17,27.10,22.56,16.66,14.12.ms(esi)calcd for c19h25no4[m+h]
+
:332.1862;found:m/z 332.1868.
[0095]
(g)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸甲酯(33)的合成:得到化合物32后,按照步骤(c)中化合物29的合成方法,得橙色固体(33),产率为82.4%。1h nmr(400mhz,cdcl3)
13
c nmr(101mhz,cdcl3)δ7.82(s,1h),6.24(s,1h),5.34(d,j=27.3hz,2h),3.90(d,j=8.3hz,5h),3.69(s,3h),2.76
–
2.69(m,2h),2.58(s,3h),1.79
‑
1.69(m,2h),1.44
–
1.34(m,4h),0.92(t,j=7.1hz,3h).
13
c nmr(101mhz,cdcl3)δ177.54,169.65,145.41,134.59,131.52,128.67,123.34,123.28,111.65,97.43,51.89,35.73,
31.76,29.67,28.17,27.10,22.57,16.67,14.12.ms(esi)calcd for c
19
h
26
n2o3[m+h]
+
:331.2022;found:m/z 331.2022.
[0096]
(h)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸(34)的合成:得到化合物33后,按照步骤(d)中化合物30的合成方法,得黄白色固体(34),产率为70.0%。1h nmr(400mhz,dmso
‑
d6)δ12.32(s,1h),7.71(s,1h),7.05(d,j=153.4hz,2h),6.27(s,1h),3.63(d,j=13.1hz,5h),2.69(t,j=7.1hz,2h),2.44(s,3h),1.64(s,2h),1.34(s,4h),0.86(s,3h).
13
c nmr(101mhz,dmso
‑
d6)δ172.53,170.91,144.80,134.56,131.57,127.57,126.53,124.40,110.77,97.89,37.15,31.64,29.83,28.15,26.67,22.48,16.86,14.48.ms(esi)calcd for c
18
h
24
n2o3[m+h]
+
:317.1865;found:m/z 317.1857。
[0097]
(i)1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸钠(sw
‑
35)的合成:向干燥的50ml烧瓶中加入5ml重蒸四氢呋喃,1,5
‑
二甲基
‑2‑
戊基
‑3‑
(2
‑
氨基
‑2‑
氧代乙基)
‑
1h
‑
吲哚
‑6‑
甲酸(34,120mg,0.38mmol),1n氢氧化钠溶液(0.38ml,0.38mmol),室温下搅拌过夜。反应结束后,旋干溶剂,冷冻干燥,得白色固体122mg,产率95.1%。
[0098]
效果验证例1
[0099]
细胞及动物模型sw
‑
9解开nnos
‑
sert偶联的鉴定
[0100]
hek293t细胞中同时转染nnos与sert质粒,体外培养的hek293t孵育10μm的小分子化合物sw
‑
9,对照组(vehicle组)在体外培养的hek293t中加入等量的溶剂,于3h后使用免疫共沉淀实验检验小分子化合物sw
‑
9对nnos
‑
sert偶联的影响。结果如图1a所示,显示sw
‑
9能够明显降低nnos
‑
sert复合物的水平。进一步的,腹腔注射给予小鼠sw
‑
9(10mg/kg),对照组注射等量的生理盐水(saline),3h后检测drn区的nnos和sert偶联情况,结果如图1b所示,发现sw
‑
9能够显著解开nnos
‑
sert偶联。
[0101]
效果验证例2
[0102]
sw
‑
9的抗抑郁样作用
[0103]
将40只小鼠随机均分为4组,分别为溶剂组(vehicle组)、sw
‑
9低剂量组、sw
‑
9中剂量组和sw
‑
9高剂量组,对sw
‑
9低剂量组、sw
‑
9中剂量组和sw
‑
9高剂量组的小鼠腹腔注射sw
‑
9,注射量分别为1mg/kg、2.5mg/kg、5mg/kg,对溶剂组注射等量溶剂。2h后进行悬尾测试(tst)、强迫游泳实验(fst)、糖水偏爱实验(spt)。结果发现腹腔给予2.5mg/kg、5mg/kg 2h即能观察到抗抑郁样的表型,其fst和tst的不动时间均明显减少,如图2a所示;而腹腔注射24h,仅有5mg/kg的剂量的sw
‑
9能够减少fst和tst的不动时间,如图2b所示。这些结果表明腹腔注射sw
‑
9具有极为快速的抗抑郁样作用,但在24h后药效即会减弱,低剂量的sw
‑
9(1mg/kg)抗抑郁样作用不明显。
[0104]
效果验证例3
[0105]
sw
‑
9腹腔注射的快速起效抗抑郁作用
[0106]
将30只小鼠随机均分为3组,分别为空白对照组(control)、cms组及cms+sw
‑
9组,对cms组及cms+sw
‑
9组的小鼠进行为期1个月的cms抑郁症造模,1个月后检测造模成功,对cms+sw
‑
9组小鼠腹腔注射sw
‑
9(10mg/kg),对空白对照组及cms组小鼠腹腔注射等量溶剂,2h后进行tst、fst、spt行为学检测。结果表明,sw
‑
9能够快速逆转cms模型小鼠在tst(如图3a所示)和fst(如图3b所示)的不动时间延长。cms小鼠对糖水摄取的选择性明显下降,但是sw
‑
9腹腔注射快速升高抑郁模型的糖水偏爱率(如图3c所示)。这些结果证明sw
‑
9腹腔注射
能够发挥快速抗抑郁作用。
[0107]
效果验证例4
[0108]
sw
‑
9灌胃的快速起效抗抑郁作用
[0109]
将30只小鼠随机均分为3组,分别为空白对照组(control)、cms组及cms+sw
‑
9组,对cms组及cms+sw
‑
9组的小鼠进行为期1个月的cms抑郁症造模,1个月后检测造模成功,对cms+sw
‑
9组小鼠灌胃sw
‑
9(10mg/kg),对空白对照组及cms组小鼠灌胃等量溶剂,2h后小鼠后进行tst、fst、spt行为学检测。结果表明,sw
‑
9能够快速逆转cms模型小鼠在tst(图4a)和fst(图4b)的不动时间延长。cms小鼠对糖水摄取的选择性明显下降,但是sw
‑
9灌胃快速升高抑郁模型的糖水偏爱率(图4c)。这些结果证明sw
‑
9灌胃能够发挥快速抗抑郁作用。
[0110]
效果验证例5
[0111]
sw
‑
9通过血脑屏障能力的试验
[0112]
对小鼠尾静脉给药sw
‑
9(25mg/kg),给药后分别于1min、3min、5min、7min、9min、11min、13min、15min、17min、20min、30min时检测脑组织匀浆液中sw
‑
9的浓度,如表1和图5所示。
[0113]
表1
[0114]
时间/min药物浓度/(ng/g)1919.63912.825584.977583.519446.3711484.4513389.7615353.6117230.3420292.3530307.02
[0115]
通过实验,研究了小鼠给药剂量为25mg/kg时,化合物sw
‑
9灌胃给药的生物利用度,以及小鼠静脉给药25mg/kg时化合物透过血脑屏障的能力的初步判断。结果表明,小鼠静脉给药25mg/kg时,主要药动学参数如下:auc(0
‑
∞):2001.815mg/l*min、t1/2z:4.903min、tmax:1min、cmax:159.39mg/l;小鼠灌胃给药25mg/kg时,主要药动学参数如下:auc(0
‑
∞):1784.779mg/l*min、t1/2z:57.095min、tmax:3min、cmax:27.99mg/l。小鼠给药剂量为25mg/kg时,化合物sw
‑
9灌胃给药的生物利用度为89.16%。小鼠静脉给药25mg/kg后,30min内脑内均能检测出一定浓度的sw
‑
9。
[0116]
以上结果显示,化合物sw
‑
9小鼠灌胃给药吸收讯速,生物利用度高,能够透过血脑屏障,具有良好的药代动力学性质,有望克服目前临床常用抗抑郁药仅对部分患者有效且起效时间缓慢的问题,成为口服有效且起效迅速的新型抗抑郁药。
[0117]
采用化合物sw
‑
10、sw
‑
14、sw
‑
15、sw
‑
20、sw
‑
21、sw
‑
9a、sw
‑
9b、sw
‑
9c、sw
‑
9d、sw
‑
31及sw
‑
35替换sw
‑
9进行上述效果验证,所得结果与sw
‑
9的验证结果相当。
[0118]
以上所述,仅为本发明较佳的具体实施方式,本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1