PMO(SLLTP-POSS)亲水微球及其制备方法与应用与流程
h.x.monodispersemicrosphere-basedimmobilizedmetalaffinitychromatographyapproachforpreparingantarctickrillphospholipidsfollowedbyhilic-msanalysis.foodchemistry,2021,344:128585-128593.
11.3.costap.p.k.g.,mendest.d.,salumt.f.c.,pachecot.f.,rodriguesc.m.developmentandvalidationofhilic-uhplc-elsdmethodsfordeterminationofsugaralcoholsstereoisomersanditsapplicationforbioconversionprocessesofcrudeglycerin.journalofchromatographya,2019,1589:56-64.
12.4.liangt.,fuq.,shena.j.,wangh.,jiny.,xinh.x.,key.x.,guoz.m.,liangx.m.preparationandchromatographicevaluationofanewlydesignedsteviolglycosidemodified-silicastationaryphaseinhydrophilicinteractionliquidchromatographyandreversedphaseliquidchromatography.journalofchromatographya,2015,1388:110
–
118.
13.5.spicerv.,krokhino.v.peptideretentiontimepredictioninhydrophilicinteractionliquidchromatographyandcomparisonofseparationselectivitybetweenbaresilicaandbondedstationaryphases.journalofchromatographya,2018,1534:75-84.
14.6.huangx.,zhangm.n.,wangm.j.,liw.,wangc.,houx.j.,luans.,wangq.gold/periodicmesoporousorganosilicaswithcontrollablemesostructurebyusingcompressedco2.langmuir,2018,34:3642-3653.
15.7.kaczmareka.m.,maegaway.,abalymova.,skirtacha.g.,inagakis.,voortp.v.d.lanthanide-graftedbipyridineperiodicmesoporousorganosilicas(bpy-pmos)physiologicalrangeandwidetemperaturerangeluminescencethermometry.acsappliedmaterials&interfaces,2020,12:13540-13550.
16.8.lic.,dib.,haow.,yanf.,sum.aminopropyl-functionalizedethane-bridgedperiodicmesoporousorganosilicaspheres:preparationandapplicationinliquidchromatography.journalofchromatographya,2016,1218(3):408-415.
17.9.dembekm.,bocians.purewaterasamobilephaseinliquidchromatographytechniques.trendsinanalyticalchemistry,2020,123:115793-115806.
技术实现要素:
18.本发明的目的是提供一种pmo(slltp-poss)亲水微球及其制备方法。
19.本发明所提供的pmo(slltp-poss)亲水微球是按照包括下述步骤的方法制备得到的:
20.1)slltp桥联的硅烷(slltpbs)的合成
21.a、将百合多糖(lltp)溶解在离子液体中,然后向其中滴加无水吡啶稀释的氯磺酸,在30-70℃恒温搅拌0.5-3h后,用碱溶液调至中性,得到硫酸酯化改性的百合多糖(slltp);
22.b、在氮气保护下,将slltp的n,n-二甲基甲酰胺(dmf)溶液与氯甲基三甲氧基硅烷
(cmtms)的thf溶液,在40-80℃恒温搅拌反应3-10h,得到slltp桥联的硅烷(slltpbs);
23.2)在naoh存在下,以c18tacl为模板剂,使poss[c2h4si(oet)3]8、slltpbs、c18tacl 在水和thf的混合溶剂中进行反应;反应结束后过滤,产物依次用乙醇、去离子水、甲醇冲洗,真空干燥;然后,采用溶剂萃取法去除模板剂,得到所述pmo(slltp-poss)亲水微球。
[0024]
上述方法步骤1)的a中,所述离子液体为咪唑类离子液体,具体可为下述任意一种: 1-丁基-3-甲基咪唑氯盐、1-丁基咪唑氯盐、1,3-二甲基咪唑氯盐。
[0025]
上述方法步骤1)的a中,所述lltp与离子液体的配比可为500mg:10ml-500mg: 50ml;具体如500mg:30ml。
[0026]
上述方法步骤1)的a中,所述氯磺酸的摩尔用量为所述lltp上羟基摩尔数的40-80%,具体如60%;
[0027]
上述方法步骤1)的a中,所述氯磺酸与无水吡啶的体积比为1:10-1:20。
[0028]
上述方法步骤1)的a中,具体可在50℃恒温搅拌1h。
[0029]
上述方法步骤1)的a中,所述碱可为用氢氧化钾或氨水。
[0030]
上述方法步骤1)的a中,用氢氧化钠溶液调至中性后,还包括下述步骤:将调节至中性的体系装入截流量为10kda透析袋,在超纯水中透析24-72h(具体如48h),浓缩后,乙醇沉淀,-20℃冷冻干燥,获得slltp。
[0031]
上述方法步骤1)的b中,所述slltp与氯甲基三甲氧基硅烷的摩尔比为1:2-1:4。
[0032]
上述方法步骤1)的b中,所述氯甲基三甲氧基硅烷与thf的体积比为1:3-1:6,具体如 1:4。
[0033]
上述方法步骤1)的b中,所述n,n-二甲基甲酰胺为无水n,n-二甲基甲酰胺;所述thf 为无水thf。
[0034]
上述方法步骤1)的b中,所述反应结束后还包括下述步骤:将反应液装入截流量为10 kda的透析袋中,在超纯水中透析24-72h(具体如48h),浓缩后乙醇沉淀,-20℃冷冻干燥,获得slltpbs。
[0035]
上述方法步骤2)中,所述poss[c2h4si(oet)3]8和slltpbs的摩尔比为0.5:1-1.5:1,优选摩尔比为0.75:1-1.5:1。
[0036]
上述方法步骤2)中,所述c18tacl与slltpbs的质量比为1:17。
[0037]
上述方法步骤2)中,所述naoh与slltpbs的质量比为1:37.5。
[0038]
上述方法步骤2)中,所述水和thf的混合溶剂中水与thf的体积比可为30:30-60: 30,优选体积比为40:30-50:30。
[0039]
上述方法步骤2)中,所述反应的反应温度可为50℃-90℃,优选为60℃-70℃。
[0040]
上述方法步骤2)中,所述反应的反应时间可为10-20h,具体如15h。
[0041]
上述方法步骤2)中,所述反应在微型高压反应釜内进行。
[0042]
上述方法步骤2)中,所述真空干燥的温度可为65℃。
[0043]
上述方法步骤2)中,所述采用溶剂萃取法去除模板剂的具体方法如下:将干燥后的固体以每克加入150ml浓盐酸(质量分数36%-38%)/去离子水/乙醇(v/v=5/45/50)溶液中,加热回流8h萃取模板剂,重复两次,过滤、洗涤、干燥。
[0044]
上述方法还进一步包括对得到的pmo(slltp-poss)亲水微球进行扩孔的步骤,具体方法如下:将所述pmo(slltp-poss)亲水微球、dmda和dda在超纯水中进行反应,反应完成
后过滤,依次用超纯水、甲醇洗涤,60℃真空干燥,获得扩孔后的pmo(slltp-poss) 亲水微球。
[0045]
所述扩孔方法还包括:将扩孔后的pmo(slltp-poss)亲水微球采用溶剂萃取法去除 dmda和dda的步骤。
[0046]
其中,所述pmo(slltp-poss)亲水微球、dmda和dda的质量比可为4.0:(3.0-6.0): (0.5-1.5),具体如4.0:5.0:1.2。
[0047]
所述反应的反应条件为110℃下静置24-72h。
[0048]
本发明还提供了上述pmo(slltp-poss)亲水微球的应用。
[0049]
本发明所提供的pmo(slltp-poss)亲水微球的应用是其在制备亲水色谱固定相中的应用。
[0050]
本发明还保护一种亲水色谱柱。
[0051]
本发明所述亲水色谱柱,其固定相为本发明所述的pmo(slltp-poss)亲水微球。
[0052]
本发明还保护上述亲水色谱柱在分离和/或检测极性物质中的应用。
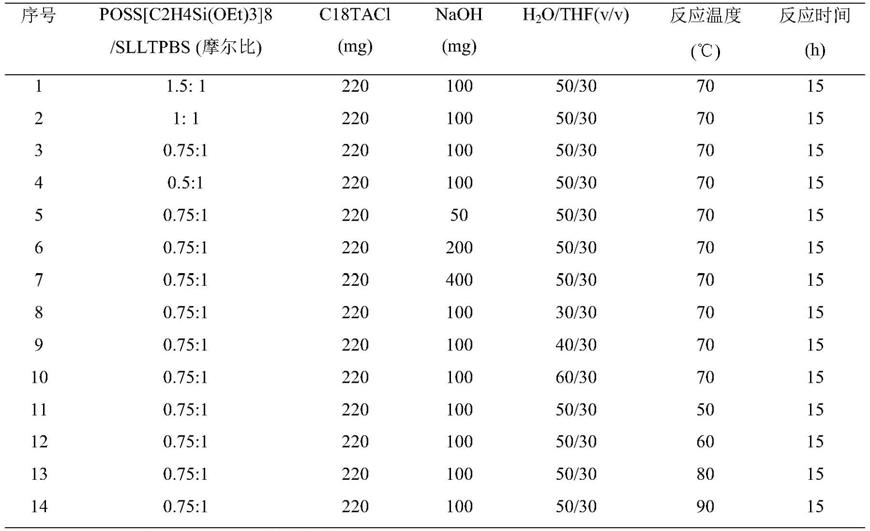
[0053]
具体的,所述极性物质选自有机酸类、糖、糖醇、氨基酸、甜味剂(包括人工和天然甜味剂)中至少一种;
[0054]
所述有机酸类具体选自草酸、酒石酸、奎宁酸、苹果酸、莽草酸、抗坏血酸、乙酸和马来酸中至少一种;
[0055]
所述甜味剂具体选自糖精钠、三氯蔗糖、甜蜜素、阿斯巴甜、安赛蜜、阿力甜、纽甜、甘草酸、甘草次酸、甜菊糖苷和甜菊双糖苷。
[0056]
上述分离或检测方法中,待测样品具体可为食品;更具体的可为含有或可疑含有上述有机酸类、单糖、糖醇、氨基酸或甜味剂的食品。
[0057]
本发明制备了pmo(slltp-poss)填料作为亲水固定相,利用红外光谱、元素分析和扫描电镜等对其进行了表征,并研究了它的色谱行为。该新型固定相具有hilic和palc的特征。与hilic类似,palc也可以对极性化合物产生保留,而且耐酸碱能力得到较大提高。该固定相可以分离有机酸类,糖、糖醇和氨基酸混合物,以及甜味剂类等极性化合物,分离度大、选择性好。与常见的c18柱相比,该固定相对非极性和弱极性化合物有更弱的保留,而对强极性化合物有更强的保留。从发展绿色色谱的角度,以及acn对环境的危害来讲, palc作为一种绿色的色谱模式,有望成为hilic的替代模式和rplc的补充模式。
附图说明
[0058]
图1为pmo(slltp-poss)亲水微球的合成反应流程图。
[0059]
图2为pmo(slltp-poss)亲水微球合成各个阶段的红外光谱图;(a)lltp;(b)slltp; (c)slltpbs;(d)未除模板的pmo(slltp-poss);(e)除模板的pmo(slltp-poss)。
[0060]
图3为不同条件下,获得的pmo(slltp-poss)微球的扫描电镜图。a和b对应表1中的实验3;c,d和e分别对应表1中的实验8,9和10。
[0061]
图4为pmo(slltp-poss)微球的热重分析曲线。
[0062]
图5为pmo(slltp-poss)微球的低角xrd图谱。
[0063]
图6为不同poss[c2h4si(oet)3]8/slltpbs摩尔比制备的pmo(slltp-poss)色谱柱与商品化c18色谱柱柱压/流速性能对比。
[0064]
图7为在ph=11情况下,pmo(slltp-poss)色谱柱与商业化c18色谱柱保留因子(a)和塔板数(b)的变化。(流动相:三乙胺水溶液/acn=95:5,流速:1.0ml/min.);在ph=1.0情况下,pmo(slltp-poss)色谱柱与商业化c18色谱柱保留因子(c)和塔板数(d)的变化。(流动相: 1%tfa/acn=92:8,流速:1.0ml/min.)
[0065]
图8为在plac模式下,八种有机酸的分离色谱图。(a)为pmo(slltp-poss)柱分离色谱图,(b)为c18柱色谱分离图,(c)为连续冲柱30天后,在pmo(slltp-poss)柱分离色谱图,(d)为连续冲柱30天后,c18柱分离色谱图。色谱峰:(1)草酸;(2)酒石酸;(3)奎宁酸;(4)苹果酸;(5)莽草酸;(6)抗坏血酸;(7)乙酸;(8)马来酸。
具体实施方式
[0066]
下面结合具体实施例对本发明作进一步阐述,但本发明并不限于以下实施例。所述方法如无特别说明均为常规方法。所述原材料如无特别说明均能从公开商业途径获得。
[0067]
下述实施例中使用的实验材料如下所示:
[0068]
百合多糖购自陕西蓝禾生物科技有限公司。1-丁基-3-甲基氯、氯磺酸、氯甲基三甲氧基硅烷、十八烷基三甲基氯化铵(octadecyltrimethylammonium chloride,c18tacl)、八(三乙氧基甲硅烷基乙基)寡聚倍半硅烷(poss[c2h4si(oet)3]8)、n,n-二甲基癸胺 (n,n-dimethyldecylamine,dmda)和十二胺(dodecylamine,dda)购自德国alfa aesar公司。吡啶、四氢呋喃、n,n-二甲基甲酰胺、甲醇和乙醇均购自于南京化学试剂有限公司(南京,中国)。
[0069]
用于亲水和富水色谱模式评价的化合物腺嘌呤、咖啡因、克伦特罗、水杨酸、山梨酸钾、富马酸、硫脲、柠檬黄、落日黄、亮蓝、新红、维生素b2(vb2)和维生素b6(vb6)均购自于阿拉丁公司(北京,中国)。8种有机酸化合物,包括草酸,酒石酸,奎宁酸,苹果酸,莽草酸,抗坏血酸,乙酸,马来酸;核糖,甘露糖醇,蔗糖,麦芽糖醇,雷诺糖,松三糖,苯丙氨酸,蛋氨酸,谷氨酸和组氨酸购自于sigma公司。
[0070]
实验过程中使用的超纯水均来自bwt超纯水系统(best water technology,德国)。高效液相色谱仪器的流动相为乙腈(色谱级,merck,德国)和超纯水的混合物,流动相和测试化合物在使用前均使用0.22μm的滤膜过滤。
[0071]
实施例1、pmo(slltp-poss)亲水微球的制备
[0072]
此过程共分三步,具体内容如下:
[0073]
1、slltp桥联的硅烷(slltpbs)的合成
[0074]
首先,采用绿色合成技术,离子液体—氯磺酸吡啶法制备slltp。500mg百合多糖 (lltp)溶解在30ml1-丁基-3-甲基咪唑氯盐([c4mim]cl)中,然后滴加5ml无水吡啶稀释的氯磺酸(氯磺酸含量相当于lltp上羟基摩尔数的60%),50℃恒温搅拌1h后,用氢氧化钠溶液调至中性。10kda透析袋在超纯水中透析48h,浓缩后,乙醇沉淀,-20℃冷冻干燥,获得slltp。
[0075]
然后,上述slltp溶解在30ml无水n,n-二甲基甲酰胺(n,n-dimethylformamide,dmf) 中。2ml氯甲基三甲氧基硅烷(chloromethyltrimethoxysilane,cmtms)溶于8ml无水thf 后,滴加入上述dmf溶液中。在氮气保护下,60℃恒温搅拌5h。反应结束后,将反应液装入截流量为10kda的透析袋中,超纯水中透析48h,浓缩后乙醇沉淀,-20℃冷冻干燥,获得
slltpbs。
[0076]
2、pmo(slltp-poss)亲水微球的制备
[0077]
称取一定量的c18tacl于烧瓶中,依次加入thf和超纯水,充分搅拌。然后,加入naoh 并溶解。将poss[c2h4si(oet)3]8和slltpbs分别溶于thf和水溶液中,并加入上述反应体系内。室温搅拌5h后,在微型高压反应釜内加热反应。反应结束后过滤,产物依次用乙醇、去离子水、甲醇冲洗,65℃真空干燥。
[0078]
通过考察体系中各种反应参数的变化,确定最佳的反应条件,反应参数设置见表1。然后,采用溶剂萃取法去除模板剂。将干燥后的固体以每克加入150ml浓盐酸(质量分数 36%-38%)/去离子水/乙醇(v/v=5/45/50)溶液中,加热回流8h萃取模板剂,重复两次。过滤、洗涤、干燥后,最终获得pmo(slltp-poss)亲水微球,合成流程图见图1。
[0079]
表1反应参数梯度表
[0080][0081]
其中,c18tacl与slltpbs的质量比为1:17。
[0082]
3、pmo(slltp-poss)亲水微球的扩孔
[0083]
称取4.0g pmo(slltp-poss)亲水微球与5.0g dmda和1.2g dda于烧瓶中,向其中加入120ml超纯水,在室温下搅拌1h。装入微型高压反应釜中,放入电热恒温箱中110℃下静置48h。反应完成后过滤,依次用超纯水、甲醇洗涤,60℃真空干燥,获得扩孔后的 pmo(slltp-poss)亲水微球。然后采用溶剂萃取法去除模板剂dmda和dda。去除dmda 和dda的过程与上述2中去除模板剂的方法相同。
[0084]
实施例2、pmo(slltp-poss)亲水微球的表征
[0085]
pmo(slltp-poss)亲水微球的表面化学结构变化通过nicolet is 10型傅里叶红外光谱仪 (thermo fisher,usa)进行分析。c、h和s元素含量变化的结果由vario el型元素分析仪 (elementar co.,germany)获得。材料的热稳定性和有序性分别在sta 409pc型热重分析仪 (netzsch,germany)和ultimate iv型x射线衍射仪(xrd)上分析(rigaku,japan)。 jsm-6360lv扫描电子显微镜(japan)观察亲水微球的表面形貌和测定颗粒尺寸。
bi-200smdls型动态光散射仪(brookhaven,usa)考察不同条件制备的pmo(slltp-poss)亲水微球的粒径分布情况。采用asap-2460型氮吸附比表面积分析仪(micromeritics instrumentscorporation,usa)考察pmo(slltp-poss)亲水微球的比表面积、孔径和孔体积的变化情况。
[0086]
结果如下所示。
[0087]
1、首先,lltp、slltp、slltpbs和pmo(slltp-poss)亲水微球的红外光谱和元素分析结果见图2和表2。lltp在3400cm-1
处的宽峰是o-h的伸缩振动,2910cm-1
处的吸收峰是c-h的伸缩振动,1380cm-1
处的吸收峰为c-h的弯曲振动,1020cm-1
处的吸收峰是c-o 伸缩振动,这些均为多糖的特征峰(图2a)。与lltp相比,slltp出现了两个新的特征峰,一个是1198cm-1
处为不对称s=o伸缩振动峰,另一个在820cm-1
处为对称的c-o-s伸缩振动峰(图2b)。表2的元素分析表明,slltp中发现了元素s(9.02%),这是由于引入了磺酸基团所导致的。以上结果都表明,硫酸酯化多糖slltp的合成是成功的。
[0088]
图2c为slltpbs的红外光谱。从图中可以看出,在1080cm-1
处出现了新的吸收峰,为 si-o-si伸缩振动峰,表明硅氧基的存在;并且与lltp的c-o伸缩振动峰(1020cm-1
)基本重合,变成了更宽更大的吸收峰。3400cm-1
处吸收峰明显减弱,这是因为lltp上的大部分羟基己被磺酸基团和硅烷取代。元素分析结果表明,与slltp相比,引入硅烷使得slltpbs 的c、h和s的含量均略有下降。与slltpbs相比,引入poss后,未除模板剂的 pmo(slltp-poss)亲水微球在1080cm-1
处si-o-si伸缩振动峰明显增强;同时,2920和2818 cm-1的吸收峰也相应增强,这主要是引入模板剂c18tacl导致的(图2d)。图2e为 pmo(slltp-poss)纯化后的红外图谱。由图可知,模板剂c18tacl(2920和2818cm-1
) 的红外吸收峰强度大大削弱了,说明通过溶剂萃取法可除去大部分c18tacl。除此之外,红外图谱变化不大,可见除去模板后,pmo(slltp-poss)亲水微球基本化学结构不变。表2表明,与未除模板剂的pmo(slltp-poss)亲水微球相比,除模板的pmo(slltp-poss)的c 和h含量明显下降,s含量略有上升,也证明了大部分c18tacl被除去。
[0089]
表2固定相合成各个阶段的元素分析结果
[0090][0091]
2、表3和图3分别为不同条件下得到的pmo(slltp-poss)微球的粒径分布和形貌情况。采用控制变量法,在其他合成条件不变的情况下,poss[c2h4si(oet)3]8/slltpbs的摩尔比从 0.5:1变化到1.5:1时,pmo(slltp-poss)微球均可保持球形形态,而且平均粒径均在4.4-5.1 μm之间。表明摩尔比在此区间内时,微球平均粒径变化不大。图3a和b显示的是 poss[c2h4si(oet)3]8/slltpbs的摩尔比为0.75:1时的sem结果,发现获得的微球表面光滑,平均粒径约为5μm且粒度分布均匀。
[0092]
3、为探索naoh含量的变化对所合成材料形貌的影响,其他条件不变,naoh含量从 50mg到400mg,结果如表3所示。可以看出,当naoh含量为50mg时,微球的平均粒径仅为2.1μm左右。随着naoh含量的增加,微球的粒径逐渐增大,当naoh增大到400mg 时,最终所得微球的平均粒径达到10.2μm左右。
[0093]
4、硅源的水解速率受水含量影响也很大,水量稍微增加,硅源的水解速率就会显著增加。二氧化硅颗粒形成的过程是水解、成核及颗粒生长三者的复杂竞争过程,其中水解是整个反应的关键,因此水含量的变化也是影响最终颗粒形貌的主要因素之一。当h2o/thf=30/30时,所得材料具有不规则的形貌(图3c);随着h2o/thf的体积比的逐渐增加(40/30和50/30),材料的形貌逐渐趋于规则的球形(图3a、b和d),平均粒径分别为4.2μm和4.9μm(表3),且微球粒径分布比较均一;进一步增加体积比至60/30时,微球的平均粒径变小且粒径尺寸分布变宽(图3e)。水含量对材料形貌的影响主要与硅源的水解缩聚速率有关。当水含量较低时,naoh的相对含量变高,此时,硅源缩聚速率增大,且缩聚速率远大于水解速率,得到的材料粗大且不规则;随着水含量的逐渐增加,硅源的水解缩聚速率逐渐趋于平衡,使得材料的形貌逐渐趋于规则均一;水含量进一步增加时,硅源水解速率变大,此时形核速率要快于生长速率,使得新老晶核一起生长,导致最终所得颗粒的尺寸不均。
[0094]
5、反应温度对介孔杂化二氧化硅材料形貌的形成也有较大的影响。研究了反应温度分别为50℃、60℃、70℃、80℃和90℃时微球的粒径变化,见表3。从中可以看出,反应温度为50℃时,得到平均粒径在5.9μm左右的pmo(slltp-poss)微球。随着温度的逐渐升高,微球的粒径呈逐渐减小的趋势。当温度达到90℃时,微球的平均粒径减小到1.3μm左右。
[0095]
表3不同条件下,获得的pmo(slltp-poss)微球的粒径分布情况
[0096][0097][0098]
6、未扩孔的pmo(slltp-poss)和扩孔后的pmo(slltp-poss)微球的比表面积和孔径等物理参数见表4。结果表明,未扩孔的pmo(slltp-poss)的比表面积、孔径和孔体积分别为517m2/g、3.7nm和0.74cm3/g。而扩孔后的pmo(slltp-poss)孔径为9.6nm,远大于未扩孔的
pmo(slltp-poss)的孔径,并且比表面积也略有增加。因此,扩孔后的 pmo(slltp-poss)具有高表面积,大孔径和球形形态,适合作为hplc填料。
[0099]
表4不同反应阶段微球的比表面积和孔结构参数
[0100][0101]
7、从应用的角度来看,pmo(slltp-poss)微球的热稳定性是一个重要方面,图4显示了其在空气中的热重分析曲线。从图4中可以看出,从50℃到320℃,pmo(slltp-poss) 微球的重量几乎没有损失,这保证了材料满意的热稳定性。开始热解的温度在330℃左右,归属于杂化微球骨架中的有机成分的分解和燃烧,最终pmo(slltp-poss)微球的无机残留 (主要是由硅烷交联的sio2)为46%,保证了微球较强的机械强度。pmo(slltp-poss)微球较高的热稳定性主要是因为poss具有规整的笼状结构,其中的无机硅氧框架结构对微球的热稳定性和机械强度具有明显的增强效果。
[0102]
8、通过xrd评估有机无机杂化微球的结构有序性。图5显示了pmo(slltp-poss)微球的xrd图谱。在低角度图谱中,在2θ=0.74
°
观察到的主峰可以归结为(100)面的衍射峰,另外,在2θ=1.63
°
和2.02
°
观察到的两个小峰分别归结为(110)和(200)面的衍射峰,这是典型的六角形介孔材料的结构,表明pmo(slltp-poss)微球具有高度有序的特征。
[0103]
实施例3、pmo(slltp-poss)亲水微球作为亲水固定相的色谱评价
[0104]
色谱分析在安捷伦1260高效液相色谱仪(美国)上完成。
[0105]
1、pmo(slltp-poss)亲水色谱柱的制备
[0106]
采用匀浆法制备pmo(slltp-poss)亲水色谱柱。以异丙醇/三氯甲烷=1:3(v/v)为匀浆液,将4.0g pmo(slltp-poss)亲水微球加入到上述匀浆液中,超声10min使其均匀分散,倒入匀浆罐中。甲醇为顶替液,在370bar的压力下装填于不锈钢柱管(150mm
×
4.6mm)中,得到新型亲水色谱柱。
[0107]
2、pmo(slltp-poss)亲水色谱柱机械强度的考察
[0108]
在palc模式下,通过考察pmo(slltp-poss)色谱柱的流速和柱压降的关系,判断微球的机械强度是否符合色谱柱填料要求。在流动相为100%甲醇、柱温为室温的条件下,改变流速,即0.25ml/min,0.5ml/min,0.75ml/min,1.0ml/min,1.25ml/min,1.5ml/min,1.75 ml/min,2.0ml/min,2.5ml/min,3.0ml/min,3.5ml/min和4.0ml/min来测量柱压。
[0109]
色谱柱的机械强度对于hplc和uplc分离来说非常重要。可以观察柱压与流速是否成正比关系,若符合则说明其具有较好的机械强度。将表1中1-4号反应条件下制备的 pmo(slltp-poss)填料分别标记为pmo(slltp-poss)-1、pmo(slltp-poss)-2、 pmo(slltp-poss)-3和pmo(slltp-poss)-4,并填装成色谱柱。在不同流速下测量柱压,并选择商品化同规格c18色谱柱(5μm,150mm
×
4.6mm)作对比。结果见图6。由图6可知, poss[c2h4si(oet)3]8/slltpbs的摩尔比为1.5:1、1:1和0.75:1时,对应pmo(slltp-poss)-1、 pmo(slltp-poss)-2和pmo(slltp-poss)-3,流速和柱压呈现出良好的线性关系。在流速达到4ml/min的情况下,柱压和流速之间的关系依然没有偏离线性,说明三种杂化填料具有良好的机械稳
定性,并且色谱柱得到了较好的填充。但是,摩尔比为0.5:1时,对应 pmo(slltp-poss)-4,当流速从0.2-2.5ml/min时,呈现出良好的线性关系。但是,流速超过2.5ml/min时,柱压迅速增加,严重偏离线性。可能是由于poss含量较少,材料刚性较差,色谱柱压力过大时造成部分微球颗粒坍塌。
[0110]
因此,pmo(slltp-poss)-1、pmo(slltp-poss)-2和pmo(slltp-poss)-3均可作为色谱固定相使用。但是,考虑到有机含量越高,色谱柱的分离能力越强,因此,选择 pmo(slltp-poss)-3作为后续研究对象。
[0111]
3、pmo(slltp-poss)亲水色谱柱耐酸碱能力和稳定性的考察
[0112]
色谱柱耐酸和耐碱能力考察。保持流速为1.0ml/min,柱温为室温条件下,耐酸测试流动相采用acn与质量分数1%的三氟乙酸水溶液,调ph为1.0;耐碱测试流动相采用acn/50 mmol/l三乙胺水溶液,调ph为11.0。富马酸和硫脲分别作为测试探针,每隔8h进样一次,持续15次,共120h。通过富马酸和硫脲的保留分别相对于各自初始保留剩余的百分比来判断色谱柱的耐酸耐碱稳定性。
[0113]
3.1碱性稳定性
[0114]
对于硅胶基质填料,普遍被接受的高ph下的破坏机理是硅胶颗粒会在碱催化下溶解。在高ph条件下,通过连续洗涤色谱柱,研究了色谱柱的碱稳定性。图7a和b所示,以硫脲的保留因子和理论塔板数相对于初始值剩余的百分比对冲洗时间做图。在120h内,硫脲在色谱柱上的保留因子和塔板数分别为初始值的93.8%和95.2%,均保持在90%以上,虽然有所下降,但并不明显。相比c18-sio2柱,相同时间内,保留因子和塔板数下降较快,分别为初始值的69.2%和60.1%,这可能是因为硅胶颗粒有部分溶解造成了分离能力严重下降。因此,pmo(slltp-poss)固定相具有良好的碱性稳定性。
[0115]
3.2酸性稳定性
[0116]
在酸性条件下,硅胶键合固定相的降解机理是si-o-si型的键合相在酸催化下的水解,从而导致色谱柱保留能力降低。为了提高填料在酸性条件下的稳定性,利用pmo(slltp-poss) 色谱柱来克服这一困难。在酸性条件下,通过连续冲洗来测试此色谱柱的稳定性。如图7c 和d所示,以富马酸的保留因子和塔板数相对于初始值剩余的百分比对冲洗时间作图。结果表明,在连续冲洗120h后,在商业化的c18-sio2柱上,富马酸的保留因子和塔板数对比初始值分别减少了17%和21%,说明c18键合相在ph=1.0的流动相冲洗过程中有明显地流失。而在pmo(slltp-poss)色谱柱上,富马酸的保留因子和塔板数分别为初始值的94.2%和 97.7%,下降均小于5%,说明这个新制备的固定相的酸性稳定性明显有所提升。以上结果证明了这种杂化色谱柱具有良好的耐酸性。
[0117]
水中稳定性考察。在流动相水/acn=90:10(v/v)、检测波长为260nm、流速为1.0ml/min 的条件下,将vb2、vb6、腺嘌呤和咖啡因的混合物(各物质浓度均为30mg/ml)在色谱柱上进行分离,每隔一天进样1次,期间用流动相连续冲柱,持续三个月,通过考察固定相对上述化合物保留因子和峰面积的变化情况,分别计算相应的rsd,以此来验证填料在长时间富水流动相条件下的稳定性。
[0118]
结果表明,四种化合物的保留因子rsd值分别为3.7%,3.2%,2.3%和4.1%,峰面积 rsd分别为3.5%,3.9%,2.3%和3.9%,均小于5%,证明此色谱柱在palc模式下使用三个月后,分离性能仍十分稳定。
[0119]
5、色谱柱工艺重复性考察
[0120]
按实施例1的方法合成了20批pmo(slltp-poss)-3填料(表1中3号反应条件下制备的产品),并填装成色谱柱。随机抽取其中的7批,分离柠檬黄、落日黄、亮蓝和新红四种合成色素的混合物(各物质浓度均为30mg/ml)。在流动相为水/acn=80:20(v/v),流速为 1.0ml/min以及检测波长为254nm情况下,采用保留时间、峰面积、峰宽、峰不对称性和保留因子五个色谱参数的相对标准偏差(relative standard deviation,rsd),评价此色谱柱填料的合成工艺和装柱技术的重复性。由表5可知,7批填料制备的色谱柱对四种合成色素的保留时间、峰宽和峰不对称性的rsd均小于7%,峰面积rsd在5%到8%之间,保留因子rsd 在6%到9.2%之间,证明7根色谱柱的分离能力和柱效基本一致,同时也说明填料的合成工艺和装柱技术稳定性较好。
[0121]
表5 7批pmo(slltp-poss)亲水固定相分离4种合成色素的5个色谱参数rsd值的比较
[0122][0123]
rt:retention time;pa:peak area;pw:peak width;paf:peak asymmetry factor;rf:retention factor.
[0124]
6、重现性实验
[0125]
采用vb2和vb6为测试探针,通过日内和日间保留因子和峰面积的rsd来评价色谱柱的重现性。在流动相为水/acn=90:10(v/v),流速为1.0ml/min以及检测波长为260nm情况下,将vb2和vb6的混合物(各物质浓度均为30mg/ml)一日内重复进样10次,连续6 天,分别计算日内和日间rsd值。结果表明,vb2和vb6的日内保留因子的rsd分别为2.1%和2.7%,峰面积的rsd分别为1.9%和3.1%,日间保留因子rsd分别为2.8%和3.5%,峰面积rsd分别为3.9%和4.3%,均小于5%,说明制备的色谱柱具有很好的重现性。
[0126]
实施例4、pmo(slltp-poss)亲水色谱柱对八种有机酸的分离
[0127]
以草酸、酒石酸、奎宁酸、苹果酸、莽草酸、抗坏血酸、乙酸和马来酸这八种有机酸为研究对象,考察了pmo(slltp-poss)亲水固定相(pmo(slltp-poss)-3)在palc条件下的分离能力。
[0128]
流动相由ⅰ:磷酸氢二钠-磷酸缓冲液(0.01mol/l,ph=2.2)和ⅱ:acn组成。梯度洗脱程序:0-2min,95%
ⅰ→
90%ⅰ;2.1-7min,90%
ⅰ→
70%ⅰ,7.1-13min,70%
ⅰ→
95%ⅰ, 13.1-15min,95%ⅰ。在室温下,流速为1.0ml/min,检测波长为210nm。
[0129]
pmo(slltp-poss)-3亲水色谱柱的制备按照实施例3中1项下进行制备。使用c18色谱柱(150mm
×
4.6mm,5μm)作为对比。然后,在上述流动相条件下,将两种色谱柱连续冲柱30天,再次分离八种有机酸,以评估保留行为。
[0130]
在分离有机酸时,因其极性较大,流动相中水的比例很高时,常发生酸的电离,从而影响分离效果。通常的做法是选择低ph值的酸性条件,减少目标化合物的电离,此方法能
很好的改善酸性化合物的峰型和保留。如图8a,在流动相ph值为2.2的情况下,在新固定相上八种有机酸混合物在10min之内获得了良好的分离,有较高的分离度。相比于c18柱,在12min内,草酸、酒石酸、奎宁酸、乙酸和马来酸达到基线分离,但是苹果酸、莽草酸和抗坏血酸色谱峰部分重叠,未达到基线分离(图8b)。然后,连续冲柱30天后,如图8c, pmo(slltp-poss)色谱柱对有机酸的分离没有明显变化。但是,在c18柱上,八种有机酸的分离效率明显下降,酒石酸和奎宁酸的色谱峰部分重叠,苹果酸、莽草酸和抗坏血酸色谱峰完全堆积在一起,无法分离(图8d)。这主要是因为过低的ph值会严重影响c18色谱柱的使用寿命。因此,pmo(slltp-poss)色谱柱具有较好的耐酸性,分离有机酸具有明显的优势。
[0131]
实施例5、pmo(slltp-poss)亲水色谱柱对单糖、糖醇和氨基酸混合物的分离
[0132]
选择10种单糖、糖醇和氨基酸混合物为测试探针,包括核糖,甘露醇,蔗糖,麦芽糖醇,雷诺糖,松三糖,苯丙氨酸,蛋氨酸,谷氨酸和组氨酸,研究pmo(slltp-poss)亲水固定相(pmo(slltp-poss)-3)对它们的分离能力。pmo(slltp-poss)-3亲水色谱柱的制备按照实施例3中1项下进行制备。
[0133]
同时,选择c18色谱柱和hilic色谱柱作对照,规格均为(150mm
×
4.6mm,5μm)。流动相,ⅰ:甲酸铵(200mm)和ⅱ:acn。pmo(slltp-poss)色谱柱的最佳分离条件(梯度洗脱程序):0-2min,98%
ⅰ→
90%ⅰ;2.1-6min,90%
ⅰ→
65%ⅰ,6.1-11min,65%ⅰ,11.1-12min,65%
ⅰ→
98%ⅰ,12.1-14min,98%ⅰ;hilic柱的最佳分离条件:0-1min, 5%ⅰ;1.1-10min,25%ⅰ,10.1-12min,25%
ⅰ→
5%ⅰ,12.1-14min,5%ⅰ;c18柱的最佳分离条件:0-2min,90%ⅰ;2.1-8min,90%
ⅰ→
75%ⅰ,8.1-12min,75%ⅰ, 12.1-13min,75%
ⅰ→
90%ⅰ,13.1-15min,90%ⅰ。柱温为室温,流速为1.0ml/min,蒸发光散射检测器的漂移管温度分别设置为55℃,高纯氮气流量为2.2l/min。
[0134]
以核糖,蔗糖,雷诺糖,松三糖,麦芽糖醇,甘露醇,苯丙氨酸,蛋氨酸,谷氨酸和组氨酸10种极性化合物为研究对象,比较pmo(slltp-poss)色谱柱、hilic柱和c18柱,在各自最佳的分离条件下,三种色谱柱对测试化合物的分离能力。
[0135]
在pmo(slltp-poss)色谱柱上,这10种极性化合物在18min内能够基线分离,这可能是由于多糖结构能提供大量的亲水作用位点及离子交换位点,能极大地提高色谱柱的柱效,对极性化合物的保留和分离能力较强。在hilic色谱柱上,核糖和苯丙氨酸的色谱峰部分重叠,其余化合物基本达到基线分离。对于上述10种极性化合物,pmo(slltp-poss)色谱柱的理论塔板数明显高于hilic色谱柱的理论塔板数。而在c18色谱柱上,10种化合物的所有峰均在10min内出现,但组氨酸、麦芽糖醇、蔗糖、甘露糖醇和蛋氨酸五个化合物的色谱峰堆积在一起,不能分离;同时,松三糖和雷诺糖的色谱峰没有达到基线分离,这表明商用c18 色谱柱对上述测试探针的保留和分离能力均较差。此外,考虑到绿色色谱法和acn对环境的影响,palc可以替代hilic法分离以上10种糖、糖醇和氨基酸的混合物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1