一种蝶烯修饰的双三齿铱配合物、制备方法及有机电致发光器件
1.本发明涉及一类电致发光材料,具体来说涉及一类蝶烯修饰的双三齿铱配合物磷光材料及其制备方法及有机电致发光器件。
背景技术:
2.有机电致发光(oled)显示相较于传统的液晶显示(lcd)具有启明电压低、功耗低、重量轻便、厚度薄、转换时间短、不需背光源、对比度高、视角广、使用温度范围广等显著优势,有望成为下一代主流显示技术。由于重原子效应,配合物磷光材料的电致发光理论内量子效率接近100%,有应用于有机电致发光的巨大潜力。在这些配合物中,环金属铱(iii)配合物拥有效率高、激发态寿命短、颜色可调性强等优点,是目前研究最广泛的一类重金属配合物。要满足应用于oled的要求,配合物必须具有的高发光效率和强的光、热稳定性等优良性能,而配体结构决定了这些配合物性能,因此对配体的设计提出了很高的要求。
3.环金属铱(iii)配合物为八配位的球形分子,配体一般为三个二配位的配体组合而成,在构型上存在异构体,给分离提纯带来困难,而且容易在加热、光照及电场存在下脱落部分配体而分解。此外,由于三个配体的振动也容易导致激发态能量损失,降低发光效率。增加配体的配位数可以提高配体的刚性,增加配合物的稳定性和发光效率,避免异构体的生成。申请者在《journal of materials chemistry c》2015年第14卷第3期第3460-3471页,公开了一篇名为“heteroleptic ir(iii)phosphors with bis-tridentate chelating architecture for high efficiency oleds”(用于高效oled的具有双三齿螯合结构的杂配ir(iii)磷光粉)的文章,验证了双三齿铱配合物的高效性和稳定性,说明双三齿铱配合物具有很高的商业化应用价值;其他研究者也进行过相关研究,比如季昀等人在《advanced functional materials》2017年第27卷第35期第1702856页,公开了一篇名为“unprecedented homoleptic bis-tridentate iridium(iii)phosphors:facile,scaled-up production,and superior chemical stability”(前所未有的同质双三齿铱(iii)磷光体:简便、可扩大生产和优越的化学稳定性)的文章,由于三齿配体容易堆积,报道的配合物电致发光器件的最佳掺杂重量百分比浓度在4%-6%之间,明显低于一般三(二齿)铱配合物8%左右的掺杂浓度,说明存在较为严重的浓度淬灭现象。浓度淬灭严重的材料给电致发光器件带来的另外一个问题是,随着驱动电压的升高,效率滚降严重。同时,如果缺乏位阻基团的保护,这些材料的发光颜色会随浓度的变化而发生较大的变化,这就给器件的光色控制带来了较大困难,导致工艺的重现性差。因此降低材料的浓度淬灭率对铱配合物来说是非常重要的。降低材料浓度淬灭率的方法通常有高分子化、超支化以及位阻基团修饰三种途径。其中高分子化由于其结构难以控制,提纯困难,通常器件效率较低;超支化则由于合成困难,成本高,难以产业化;位阻基团修饰是最为经济有效的方法。
4.例如,申请者的中国专利申请号为201710436528.7,申请公开日为2017年9月15日的专利申请文件公开了一种含烷基位阻基团的哒嗪类铱配合物及其制备方法和应用。该专
利配合物中含有的二环烷基位阻基团,能有效降低铱配合物的浓度淬灭(cn 201710436528.7,dalton transactions,2018,47,12243-12252.)。说明通过在铱配合物结构中合理引入有效的位阻基团能大幅度提升铱配合物的性能。
5.三蝶烯(triptycene)是由三个苯环通过桥头碳原子之间相互铰链而成的五环化合物,具有独特的结构特点(一般的阻隔基团是单键连接,而三蝶烯是双键连接)和丰富的反应性能,在分子机器、材料化学、超分子化学以及有机催化等很多领域引起了人们越来越多的关注。特别是这种独特的三位刚性结构中,三个苯环是通过饱和碳原子相连,彼此没有形成共轭,位阻面更大,三个芳环能形成巨大的立障空间。申请者前期成功将该结构融于三(二齿)铱配合物的设计合成中,并显示了较多优异性能。申请者在《dalton transactions》2019年第43卷第48期第16289-16297页,公开了一篇名为“novel phosphorescent triptycene-based ir(iii)complexes for organic light-emitting diodes”(用于有机发光二极管的新型磷光三蝶烯基铱(iii)配合物)的文章,在中国专利申请号201710347452.0公开了以下结构的有机电致磷光铱配合物:
[0006][0007]
其中y为硫、氧或取代氮,l^x为含氮辅助配体。
[0008]
这些论文和专利表明,三蝶烯结构能提高这些铱配合物的热稳定性和发光效率,降低其浓度淬灭,但是这些工作都是在三蝶烯1位上进行修饰构建的配体,它们在与铱配位后桥碳与配位杂环间存在较大的分子内排斥力,破坏了环金属苯环与配位杂环的共面性,严重降低目标配合物合成产率,并对产物稳定性造成一定影响。并且在1位构建杂环,需要通过甲基氧化、还原、再氧化、缩聚成环等多步反应,降低了合成产率,增加了合成成本,严重阻碍了其商业化应用的前景。
[0009]
因此,开发一种低浓度淬灭率的双三齿铱配合物有机电致磷光材料、制备方法及有机电致发光器件,是目前亟需解决的问题。
技术实现要素:
[0010]
1.要解决的问题
[0011]
针对现有的双三齿铱配合物浓度敏感的问题,本发明提供一种蝶烯修饰的双三齿铱配合物有机电致磷光材料,通过刚性位阻基团的修饰,在不引起明显非辐射跃迁的前提下(柔性基团能引起激发态能量以非辐射跃迁的形式损失,造成发光效率降低),降低铱配合物分子之间的相互作用,使配合物的电致发光波长和效率对浓度和电压变化不敏感,同时调控配合物的载流子传输性能,提高铱配合物的电致发光效率。同时,本发明还提供了上述有机电致磷光材料的制备方法,其目的在于将刚性位阻基团引入双三齿铱配合物的相应位点上。后续,上述有机电致磷光材料作为有机电致发光器件的发光层材料具有应用价值。
[0012]
2.技术方案
[0013]
为了解决上述问题,本发明所采用的技术方案如下:
[0014]
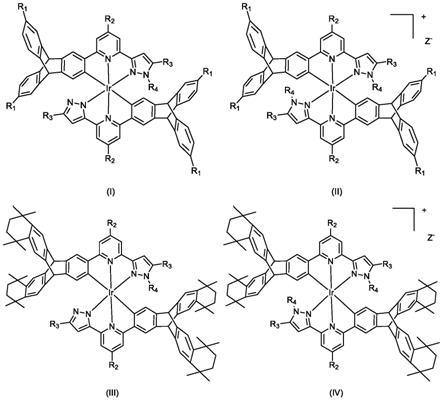
一种蝶烯修饰的双三齿铱配合物,该类配合物的结构通式如图1所示的式(i)、式(ii)、式(iii)或式(iv):
[0015][0016]
式中,r1为氢原子或含1~8个碳原子的烷基中的一种;r2为氢原子、含1~8个碳原子的烷基或氟代烷基中的一种;r3为含1~8个碳原子氟代烷基;r4为氢原子、含1~8个碳原子的烷基或取代苄基(取代基为氢原子、1~8个碳原子的烷基、咔唑基)中的一种。阴离子z-为卤素原子、六氟磷酸根、高氯酸根、四(取代苯基)硼酸根(取代基为氢原子、1-5个氟原子、1-2个三氟甲基)中的一种。
[0017]
一种蝶烯修饰的双三齿铱配合物的制备方法,其特征在于:在三蝶烯2硼酸衍生物中添加2-乙酰基-6-溴吡啶衍生物,同时在碱、催化剂和溶剂的存在下反应一段时间得到三蝶烯2位功能化的中间体;该中间体再通过偶联、环化反应得到三齿的配体l;配体l与三氯化铱反应,然后再烷基化、离子交换得到蝶烯修饰的双三齿铱配合物,其中,配体l的结构通式为式(v)或(vi):
[0018][0019]
式中,r1为氢原子或含1~8个碳原子的烷基中的一种;r2为氢原子、含1~8个碳原子的烷基或氟代烷基中的一种;r3为含1~8个碳原子氟代烷基;r4为氢原子、含1~8个碳原子的烷基或取代苄基中的一种。
[0020]
一种上述蝶烯修饰的双三齿铱配合物的制备方法,步骤为:
[0021]
(1)将三蝶烯2硼酸衍生物、2-乙酰基-6-溴吡啶衍生物、碱a、催化剂a和溶剂a加入到反应体系中,氮气保护下,50~150℃,反应2~24小时,得到6-(三蝶烯-2-)吡啶-2-乙酰衍生物中间体;
[0022]
(2)在6-(三蝶烯-2-)吡啶-2-乙酰衍生物中添加碱b和溶剂b,然后加入氟代烷基甲酸酯,氮气保护下,25~100℃,反应2~24小时,反应完成后调节混合物酸碱性到中性,萃取、蒸除溶剂后得到有机物,所得有机物与溶剂c和水合肼混合,氮气保护下50~200℃,反应2~24小时,得到三齿配体l;
[0023]
(3)将三齿配体l与三氯化铱、碱c、溶剂d混合,氮气保护下50~200℃反应2~24小时,得到配合物中间体。然后配合物中间体、碘代烃或溴代烃、碱d和溶剂e混合,氮气保护下0~100℃反应0.5~150小时,得到中性或离子型配合物。离子型配合物再在醇类溶剂f中与阴离子z的盐室温下,离子交换反应0.1~2小时得到离子型配合物最终产物。其中,控制烷基化试剂的用量,一个当量形成中性配合物,两个当量形成离子型配合物。
[0024]
更进一步地,所述步骤(1)中,反应体系中各反应物的摩尔份数为:
[0025][0026]
其中,所述的碱a包括碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、醋酸钠、醋酸钾、氢氧化钠、氢氧化钾中的一种或两种及以上的混合物;所述的溶剂a包括四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、乙醇、甲醇、水、苯、甲苯、二甲苯中的一种或两种及以上的混合物;所述的催化剂a包括醋酸钯、pd(pph3)4、pd(pph3)2cl2或pd(dppf)cl2。
[0027]
更进一步地,步骤(1)中,2-乙酰基-6-溴吡啶衍生物与三蝶烯2硼酸衍生物的摩尔比优选1:(1~2)。
[0028]
更进一步地,所述步骤(2)中,反应体系中各反应物的摩尔份数为:
[0029][0030]
其中,所述的碱b为氢化钠、叔丁醇钾、甲醇钠、乙醇钠中的一种或两种及以上的混合物;所述的溶剂b为四氢呋喃、1,4-二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜中的一种或两种及以上的混合物。溶剂c为甲醇、乙醇、丙醇、叔丁醇中的一种或两种及以上的混合物。
[0031]
更进一步地,所述步骤(3)中,反应体系中各反应物的摩尔份数为:
[0032][0033][0034]
其中,所述的碱c为乙酸钠、乙酸钾、甲酸钠、甲酸铵、丙酸钠、丙酸钾中的一种或混合物;所述的溶剂d为甲酸、乙酸、丙酸中的一种或混合物;所述的碱d为碳酸钾、碳酸钠、磷酸钾、磷酸钠中的一种或混合物;所述的溶剂e为四氢呋喃、二氯甲烷、三氯甲烷、四氯化碳、1,4-二氧六环,乙醚、丙醚、乙二醇二甲醚、乙二醇二乙醚中的一种或两种及以上的混合物。所述的醇类溶剂f为甲醇、乙醇、丙醇中的一种或两种及以上的混合物。所述的阴离子z的盐为钠、钾、铵盐中的一种或两种及以上的混合物。
[0035]
一种有机电致发光器件,包括阳极、空穴传输层、有机发光层、电子传输层以及阴极,所述有机发光层包含上述的蝶烯修饰的双三齿铱配合物。
[0036]
更进一步地,所述蝶烯修饰的双三齿铱配合物占所述有机发光层总质量的1%-20%。
[0037]
更进一步地,所述有机发光层的主体材料为mcp、cbp中的一种或两者的混合,其中mcp、cbp可以以任意比例混合。
[0038]
3.有益效果
[0039]
相比于现有技术,本发明的有益效果为:
[0040]
本发明的双三齿铱配合物具有比前期专利更高的稳定性和发光效率,(配体为三齿的,不容易脱落,刚性更强,非辐射跃迁率更低,因而效率更高),解决有机电致发光稳定
性差的行业瓶颈。相对于原来的双三齿铱配合物则由于大立体位阻三蝶烯的引入能降低浓度淬灭,增加溶解性,具有来说:
[0041]
(1)三蝶烯为非共轭结构,本发明的三蝶烯修饰对铱配合物的发光影响较小,例如,配合物ir1和ir4的发光波长分别为547纳米和551纳米,与参比配合物irc的发光波长545纳米接近,有利于保持原有母核的发光颜色。
[0042]
(2)三蝶烯为刚性结构,稳定性高,不会造成激发态能量的非辐射跃迁损失,本发明的三蝶烯修饰能提高铱配合物的热稳定性及发光效率。因此配合物ir1和ir4有机电致发光掺杂器件最大电流效率都超过了75cd a-1
,有较大的实用化前景。
[0043]
(4)本发明的蝶烯修饰的双三齿铱配合物分子间相互作用较弱,大大降低了由浓度引起的发光淬灭,纯固体粉末的发光效率在10%以上,最高能达到30.5%,浓度淬灭明显低于位阻不充分的双三齿铱配合物(irc,5.6%)。同时还通过引入1,3-二咔唑基苯等载离子传输单元,在降低浓度淬灭率的同时提高配合物的载流子传输性能。
[0044]
(5)本发明的蝶烯修饰的双三齿铱配合物能用于高效非掺杂有机电致发光器件的制备。例如以配合物ir1和ir4的非掺杂器件的最大外量子效率都超过5%,而相同条件下对比配合物irc的仅为1.6%,充分说明了三蝶烯修饰能改善双三齿铱配合物的器件性能。
[0045]
(6)与发明人前期保护的三蝶烯基铱配合物(中国专利申请号为201810961164.9)相比,前期产品是通过在三蝶烯的1位上进行功能化,生成的是二齿配体,路线较长,配体的合成产率低,并且三蝶烯基团与邻近的官能团存在较大分子内排斥作用,影响最终配合物的产率和稳定性,而本发明是在三蝶烯的2位功能化,2位功能化消除了三蝶烯骨架与连接芳环的分子内位阻,不会使配位结构发生扭曲而降低配位能力,提高了产物的稳定性,生成的三齿配体,合成更为便捷,且不存在分子内的排斥作用。
[0046]
总之,以上综合因素使本发明的铱配合物具有优异的光电性能。
附图说明
[0047]
图1为本发明蝶烯修饰的双三齿铱配合物的结构通式;
[0048]
图2为本发明中铱配合物ir1在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
[0049]
图3为本发明中铱配合物ir2在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
[0050]
图4为本发明中铱配合物ir3在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
[0051]
图5为本发明中铱配合物ir4在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
[0052]
图6为本发明中铱配合物ir5在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱;
[0053]
图7为对比配合物irc的化学结构;
[0054]
图8为本发明铱配合物ir1的电致发光器件的亮度-外量子效率图;
[0055]
图9为对比配合物ir4的电致发光器件的亮度-外量子效率图;
[0056]
图10为对比配合物irc的电致发光器件的亮度-外量子效率图。
具体实施方式
[0057]
下面结合具体实施例对本发明进一步进行描述。
[0058]
实施例1
[0059]
称取三蝶烯-2-硼酸片呐醇酯3.0g(7.89mmol),1.6g(7.89mmol)2-溴-6-乙酰基吡啶,2.6g(23.67mmol)无水碳酸钠,催化剂pd(dppf)cl20.45g加入封管中,加入thf30ml和水10ml,氮气氛围下110℃反应24h。冷却室温,旋蒸除去thf后用二氯甲烷萃取,石油醚:乙酸乙酯=8:1作为洗脱剂,硅胶柱层析分离,得到z1的白色固体2.15g,产率73%。
[0060]1h nmr(400mhz,cdcl3)δ8.13(s,1h),7.92(s,1h),7.82(s,2h),7.70(d,j=7.7hz,1h),7.52(s,1h),7.43(s,4h),7.02(d,j=8.5hz,4h),5.57(s,1h),5.51(s,1h),2.82(s,3h)。
[0061][0062]
称取2.0g(5.36mmol)z-1和1.55g(32.16mmol)的50%矿物油/nah粉末放进三口圆底烧瓶中,用真空泵抽真空充n2后,在冰水浴条件下注射适量干燥的四氢呋喃作为溶剂;待反应瓶中稳定后,再缓慢注射2.5ml的三氟乙酸乙酯,反应30分钟后撤掉冰水浴,加热至80℃反应12h。反应结束后,冰水浴条件下淬灭。向反应瓶里注射乙醇,中和过量未反应的nah直至没有气泡产生。后加入少量蒸馏水,确保nah反应完全后,滴加盐酸调节ph至6~7。最后萃取收集有机相,旋干得到中间产物,再加入30ml无水乙醇和3ml 85%的水合肼,在n2保护下100℃反应12h。反应结束后,除去溶剂并萃取,收集有机相旋干过硅胶柱,洗脱剂比例为石油醚:乙酸乙酯=9:1,产物l1为白色固体,约1.5g,产率为60.0%。
[0063]1h nmr(400mhz,cdcl3)δ11.56(s,1h),8.06(s,1h),7.76(t,j=7.8hz,1h),7.64
–
7.57(m,2h),7.51
–
7.40(m,6h),7.03(d,j=8.5hz,4h),6.94(s,1h),5.55(s,1h),5.50(s,1h)。
19
f nmr(377mhz,cdcl3)δ-62.23(s)。hrms((+)-esi):m/z=466.1523(calcd.466.1453for[c
29h18
f3n3][m+h]
+
)。
[0064][0065]
将配体l1(4.5g,9.6mmol)与ircl3·
h2o(1.5g,4.5mmol)和醋酸钠(3.6g,43.5mmol)溶于置于圆底烧瓶,加入30ml乙酸,搅拌回流24h。反应结束冷却至室温,减压蒸馏除乙酸,用二氯甲烷重结晶得黄色中间体irz1。将中间体溶于30mlthf溶剂中,加入k2co3(1.2g,9.6mmol),n2保护,室温下搅拌15min,注射入ch3i(0.6g,4.8mmol),室温下反应4h,旋干溶剂,用二氯甲烷:乙酸乙酯=20:1洗脱剂硅胶柱层析分离,用二氯甲烷与正己烷混合液重结晶,得ir1黄色产物0.1g,产率为11.1%。
[0066]1h nmr(400mhz,cd2cl2)δ8.16(d,j=6.3hz,1h),8.06(d,j=5.6hz,2h),7.85
–
7.71(m,3h),7.58(d,j=6.4hz,1h),7.42(d,j=7.6hz,1h),7.36
–
7.26(m,3h),7.16(d,j=
8.3hz,2h),7.09(d,j=6.7hz,1h),7.03(d,j=7.7hz,1h),6.97
–
6.76(m,8h),6.53
–
6.43(m,2h),6.30-6.19(m,2h),6.02(s,1h),5.31(d,j=1.1hz,4h),3.09(s,3h)。
19
f nmr(377mhz,cd2cl2)δ-60.13(s,3f),-61.07(s,3f)。hrms((+)-esi):m/z=1135.2547(calcd.1135.2457for[c
59h36
f6irn6][m+h
+
])。
[0067][0068]
在二氯甲烷中,0.00001mol/l浓度下ir1的最大发射波长为547纳米,发光量子效率ф=75.4%,空气中纯固体的发光量子效率为ф=10.6%。氮气保护下5%热失重温度为405℃。在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图2所示。
[0069]
实施例2
[0070]
将配体l1(3.0g,6.4mmol)与ircl3·
h2o(1.0g,3.0mmol)和醋酸钠(2.4g,18.9mmol)溶于置于圆底烧瓶,加入30ml乙酸,搅拌回流24h。反应结束冷却至室温,减压蒸馏除乙酸,用二氯甲烷重结晶得黄色中间体irz1。将中间体溶于thf溶剂中,加入k2co3(0.8g,6.4mmol),n2保护,室温下搅拌15min,加入1,3-二咔唑基5溴甲基苯(0.4g,1.9mmol),60℃下反应5d,反应完毕冷却至室温,旋干溶剂,用二氯甲烷:乙酸乙酯=25:1洗脱剂硅胶柱层析分离,用二氯甲烷与正己烷混合液重结晶,得ir2黄色产物0.1g,产率为11.5%。
[0071]1h nmr(400mhz,cdcl3)δ8.17(d,j=7.8hz,4h),7.88(s,1h),7.79-7.27(m,22h),7.25-6.65(m,13h),6.58(d,j=6.5hz,1h),6.43(dd,j=23.4,9.4hz,2h),6.31-6.17(m,2h),6.00(dd,j=48.9,5.9hz,2h),5.56(s,2h),5.18(s,2h),4.73(d,j=21.0hz,2h)。
19
f nmr(377mhz,cdcl3)δ-59.33(s,3f),-59.82(s,3f)。hrms((+)-esi):m/z=1541.4054(calcd.1541.3927for[c
89h54
f6irn8][m+h
+
])。
[0072][0073]
在二氯甲烷中,0.00001mol/l浓度下ir2的最大发射波长为572纳米,发光量子效率ф=84.3%,空气中纯固体的发光量子效率为ф=23.9%。氮气保护下5%热失重温度为410℃。在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图3所示。
[0074]
实施例3
[0075]
称取0.3g(0.62mmol)配体l1、0.1g(0.28mmol)ircl3·
3h2o和0.5g(6.0mmol)naoac放进反应瓶中,加入20ml乙酸,n2气氛下110℃反应24h。反应结束后除去乙酸,得到得黄色中间体。接着向反应瓶中加入0.39g(2.8mmol)无水k2co3和10mlthf,n2环境下室温反应15分钟后缓慢注入0.5ml的ch3i,继续反应4h。反应结束后,除去溶剂,再加入10ml甲醇,0.5克高氯酸钠,搅拌1h,旋干溶剂后通过硅胶柱层析将产物分离。洗脱剂为二氯甲烷:乙酸乙酯=1:4,得到ir3的黄色固体0.072g,产率为20.6%。
[0076]1h nmr(400mhz,cdcl3)δ8.24(s,2h),8.09(s,4h),7.88(s,2h),7.55(d,j=15.2hz,4h),7.29(s,4h),7.14(d,j=5.8hz,2h),6.94(s,4h),6.82(d,j=10.2hz,4h),5.78(s,2h),5.27(s,2h),4.96(s,2h),3.20(s,6h)。
19
f nmr(377mhz,cdcl3)δ-60.33(s)。hrms((+)-esi):m/z=1149.2677(calcd.1149.2691for[c
60h38
f6irn6][m-clo
4-]
+
)。
[0077][0078]
在二氯甲烷中,0.00001mol/l浓度下ir3的最大发射波长为547纳米,发光量子效率ф=81.2%,空气中纯固体的发光量子效率为ф=14.1%。氮气保护下5%热失重温度为407℃。在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图4所示。
[0079]
实施例4
[0080]
称取4.8g(8.00mmol)环烷基取代的三蝶烯-2-硼酸片呐醇酯1,1.6g(7.89mmol)2-溴-6-乙酰基吡啶,2.6g(23.67mmol)无水碳酸钠,催化剂pd(dppf)cl20.45g加入封管中,加入1,4-二氧六环30ml和水10ml,氮气氛围下110℃反应24h。冷却室温,旋蒸除去1,4-二氧六环后用二氯甲烷萃取,石油醚:乙酸乙酯=8:1作为洗脱剂,硅胶柱层析分离,得到z2的白色固体2.5g,产率为64%。
[0081]1h nmr(400mhz,cdcl3)δ7.96(d,j=1.7hz,1h),7.92(dd,j=6.4,2.3hz,1h),7.84
–
7.81(m,2h),7.71(dd,j=7.6,1.8hz,1h),7.66(s,2h),7.57(d,j=8.1hz,2h),7.38(d,j=7.6hz,1h),5.22(s,1h),5.16(s,1h),2.83(s,3h),1.73(s,4h),1.64(s,2h),1.35(dd,j=18.2,5.6hz,14h),1.08(s,12h)。
[0082][0083]
称取3.2g(5.36mmol)z2和1.55g(32.16mmol)的50%矿物油/nah粉末放进三口圆底烧瓶中,用真空泵抽真空充n2后,在冰水浴条件下注射50ml干燥的四氢呋喃作为溶剂;待反应瓶中稳定后,再缓慢注射2.5ml的三氟乙酸乙酯,反应30分钟后撤掉冰水浴,加热至70℃反应12h。反应结束后,冰水浴条件下淬灭。向反应瓶里注射乙醇,中和过量未反应的nah
直至没有气泡产生。后加入少量蒸馏水,确保nah反应完全后,滴加盐酸调节ph至6~7。最后萃取收集有机相,旋干得到中间产物,再加入30ml无水乙醇和3ml 85%的水合肼,在n2保护下90℃反应15h。反应结束后,除去溶剂并萃取,收集有机相旋干过硅胶柱,洗脱剂比例为石油醚:乙酸乙酯=10:1,产物l2为白色固体,约1.75g,产率为47.6%。
[0084]1h nmr(400mhz,cdcl3)δ11.58(s,1h),7.94(d,j=1.7hz,1h),7.80(t,j=7.8hz,1h),7.70
–
7.55(m,6h),7.50(d,j=7.7hz,1h),7.39(d,j=7.7hz,1h),6.97(s,1h),5.23(s,1h),5.17(s,1h),1.74(s,4h),1.65(s,6h),1.38(d,j=7.2hz,10h),1.10(dd,j=5.1,3.5hz,12h)。
19
f nmr(377mhz,cdcl3)δ-62.29(s)。hrms((+)-esi):m/z=686.3714(calcd.686.3644for[c
45h46
f3n3][m+h]
+
)。
[0085][0086]
称取0.37g(0.62mmol)配体l2、0.1g(0.28mmol)ircl3·
3h2o和0.5g(6.0mmol)naoac放进反应瓶中,加入20ml乙酸,n2气氛下110℃反应24h。反应结束后除去乙酸,得到得黄色中间体irz2。接着向反应瓶中加入0.39g(2.8mmol)无水k2co3和10mlthf,n2环境下室温反应15分钟后缓慢注入0.04g(0.3mmol)的ch3i,继续反应4h。反应结束后,除去溶剂。洗脱剂为二氯甲烷:乙酸乙酯=1:4,硅胶柱层析得到ir4的黄色固体0.056g,产率为12.6%。
[0087]1h nmr(400mhz,cdcl3)δ8.08(d,j=6.1hz,1h),7.97(d,j=8.8hz,2h),7.77(t,j=8.3hz,1h),7.65(s,1h),7.57(s,2h),7.38(d,j=7.7hz,1h),7.24(s,1h),7.14(s,1h),7.08-6.92(m,7h),6.80(s,1h),6.59(d,j=11.4hz,2h),4.94(d,j=9.8hz,2h),4.67(s,1h),4.27(s,1h),3.09(s,3h),1.65
–
1.52(m,16h),1.29
–
1.05(m,49h)。
19
f nmr(377mhz,cdcl3)δ-59.92(s,3f),-60.97(s,3f)。hrms((+)-esi):m/z=1575.6947(calcd.1575.6839for[c
91h92
f6irn6][m+h
+
])。
[0088][0089]
在二氯甲烷中,0.00001mol/l浓度下ir4的最大发射波长为551纳米,发光量子效率ф=86.7%,空气中纯固体的发光量子效率为ф=25.8%。氮气保护下5%热失重温度为401℃。在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图5所示。
[0090]
实施例5
[0091]
称取0.37g(0.62mmol)配体l2、0.1g(0.28mmol)ircl3·
3h2o和0.5g(6.0mmol)naoac放进反应瓶中,加入20ml乙酸,n2气氛下110℃反应24h。反应结束后除去乙酸,得到得黄色中间体irz2。接着向反应瓶中加入0.39g(2.8mmol)无水k2co3和10mlthf,n2环境下室温
反应15分钟后缓慢注入0.5ml的ch3i,继续反应4h。反应结束后,除去溶剂,再加入10ml甲醇,0.2克六氟磷酸铵,搅拌1h,旋干溶剂后通过硅胶柱层析将产物分离。洗脱剂为二氯甲烷:乙酸乙酯=1:5,得到ir5的黄色固体0.057g,产率为11.8%。
[0092]1h nmr(400mhz,cdcl3)δ8.26(d,j=8.6hz,4h),8.12(d,j=7.9hz,4h),7.88(d,j=13.1hz,4h),7.44(d,j=3.1hz,2h),7.36(s,2h),7.31(d,j=2.0hz,2h),5.73(s,2h),5.66(s,2h),5.51(s,2h),3.29(s,6h)。
19
f nmr(377mhz,cdcl3)δ-60.58(s,6f),-69.27(s,6f)。hrms((+)-esi):m/z=1589.7124(calcd.1589.7073for[c
92h94
f6irn6][m-pf
6-]
+
).
[0093][0094]
在二氯甲烷中,0.00001mol/l浓度下ir5的最大发射波长为556纳米,发光量子效率ф=90.6%,空气中纯固体的发光量子效率为ф=30.5%。氮气保护下5%热失重温度为403℃。在二氯甲烷溶液中的紫外可见吸收光谱和光致发光光谱如图6所示。
[0095]
性能测试
[0096]
本测试是对实施例1、4中的配合物ir1、ir4的有机电致发光器件性能测试并在相同条件下与位阻不充分的铱配合物irc(图7)做了对比。本实验的器件结构ito/moo3(2nm)/moo3:mcp(20nm)/mcp(20nm)/铱配合物(30nm)/tmpypb(40nm)/liq(1nm)/al(100nm)。
[0097]
基于ir1的器件开启电压为2.7v,最大亮度为1034cd/m2,最大功率效率、电流效率和外量子效率(图8)分别为10.2lm w-1
,14.7cd a-1
,6.3%。而且以mcp为主体材料,掺杂浓度为10%时,器件的最大亮度、最大电流效率、最大功率效率和外量子效率分别高达6337cd m-2
、80.2cd a-1
、44.2lm w-1
、14.1%,表现出了较好的商业化应用潜力。
[0098]
基于ir4的器件开启电压为4.1v,最大亮度为570cd/m2,最大功率效率、电流效率和外量子效率(图9)分别为5.0lm w-1
,10.2cd a-1
,5.5%。而且以mcp为主体材料,掺杂浓度为10%时,器件的最大亮度、最大电流效率、最大功率效率和外量子效率分别高达4372cd m-2
、76.4cd a-1
、47.3lm w-1
、15.5%,表现出了较好的商业化应用潜力。
[0099]
基于irc的器件开启电压为4.6v,最大亮度为21cd/m2,最大功率效率、电流效率和外量子效率(图10)分别为1.7lm w-1
,2.0cd a-1
,1.6%;同时,irc的发光效率:在二氯甲烷中,0.00001mol/l浓度下irc的最大发射波长为545纳米,发光量子效率ф=57.0%,空气中纯固体的发光量子效率为ф=5.6%。氮气保护下5%热失重温度为372℃。
[0100]
以上结果表明在相同条件下配合物ir1和ir4的浓度淬灭显著低于irc,同样条件下具有更高的发光效率、器件效率及热稳定性。
[0101]
值得说明的是,以上实施例的反应条件均为反应的最佳反应条件,在权利要求叙述范围内的反应条件均可合成本发明的配合物,为了避免赘述,此处不再举例说明;另外,本领域技术人员根据本发明配合物的通式以及列举的相关溶剂、催化剂等试剂,可再现本发明,由于列举实施例过多,此处挑选具有代表性的几个实施例,足以证明本发明的配合物
具有优异的发光效率和低浓度淬灭率,故此处不再赘述实施例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1