一种可熔性聚四氟乙烯树脂及其制备方法与流程
1.本发明属于氟化工技术领域,具体涉及可熔性聚四氟乙烯生产技术。
背景技术:
2.可熔性聚四氟乙烯(pfa)由四氟乙烯(tfe)和全氟烷基乙烯基醚(pave)共聚得到。其具有与聚四氟乙烯(ptfe)相同的优异的化学稳定性、物理机械性能、电绝缘性能、润滑性、不沾性、耐老化性、不燃性和热稳定性,而且由于主链中含有全氟烷氧基直链,增加了链的柔顺性,改善了聚合物的熔体粘度,所以可以通过一般热塑性塑料的成型加工方法进行加工。
3.基于上述优异的性能,可熔性聚四氟乙烯在制作电线电缆绝缘护套、高频超高频绝缘零件、化工管道阀门和泵的耐腐蚀衬里;机械工业用特殊零配件、轻纺工业用各种防腐材料、聚四氟乙烯防腐衬里等焊条;以及半导体行业、医药行业、电子电气设备行业、国防军工、航空航天等领域均有着广泛应用。
4.在可熔性聚四氟乙烯树脂合成上,许多专利文献有过详细的介绍。比如美国专利3635926中公开了一种可熔性聚四氟乙烯制备方法,具体为:使用过硫酸铵作为引发剂,全氟辛酸铵作为表面活性剂、氟碳作为溶剂,在70-95℃、1.7-2.4mpa下进行聚合得到可熔性聚四氟乙烯乳液。专利cn104558365中在制备可熔性聚四氟乙烯时,全氟丙基乙烯醚和四氟乙烯投入质量比约为17%,但是在产品中的全氟丙基乙烯醚的含量仅约为3.7%,聚入率约为22%;专利jp4599640b2中的一个实施例中在制备可熔性聚四氟乙烯时,ppve和tfe投入质量比约为17.39%时,产品中的ppve含量仅为3.7%,聚入率约为21%。ppve聚入率都比较低。
5.pave作为制备可熔性聚四氟乙烯中不可或缺的共聚单体,成本高,回收损失率高。在现有技术中,pave聚入率过低,回收工艺压力大,不利于工业化生产。
技术实现要素:
6.本发明所要解决的技术问题就是提供一种可熔性聚四氟乙烯树脂及其制备方法,提高pave的聚入率。
7.为解决上述技术问题,本发明采用如下技术方案:
8.一种可熔性聚四氟乙烯树脂制备方法,包括如下步骤:
9.s1:在聚合釜中加入无离子水、有机溶剂、表面活性剂和链转移剂;
10.s2:温度升至设定温度50-80℃以后,加入由四氟乙烯和全氟烷基乙烯基醚组成的聚合单体至设定压力0.7-1.5mpa后,加入引发剂开始反应,全氟烷基乙烯基醚与四氟乙烯投入质量比为1:25-1:8;
11.s3:补加聚合单体和全氟烷基乙烯基醚聚合促进剂,维持聚合釜压力在设定压力0.7-1.5mpa直到反应结束,得到可熔性聚四氟乙烯乳液,其中,全氟烷基乙烯基醚聚合促进剂为六氟丙烯,全氟烷基乙烯基醚聚合促进剂加入量为聚合单体的0.01wt%-2wt%;
12.s4:将步骤s3得到的可熔性聚四氟乙烯乳液凝聚、洗涤、造粒得到可熔性聚四氟乙烯树脂。
13.优选的,全氟烷基乙烯基醚与四氟乙烯投入质量比为1:15-1:8。
14.优选的,全氟烷基乙烯基醚聚合促进剂加入量为聚合单体的0.01wt%-1.5wt%。
15.优选的,聚合过程中四氟乙烯组分为70-95wt%,六氟丙烯组分为0.1-25wt%,全氟烷基乙烯基醚组分为2-20wt%。
16.优选的,所述全氟烷基乙烯基醚为全氟甲基乙烯基醚、全氟乙基乙烯基醚、全氟丙基乙烯基醚、全氟丁基乙烯基醚和全氟戊基乙烯基醚中的一种或多种混合物。
17.优选的,六氟丙烯在反应开始前一次加入,通过控制聚合单体加入时间调控聚合釜内压力,且保证聚合单体和六氟丙烯聚合釜内处于要求浓度下;或者六氟丙烯在反应过程中分次加入,控制聚合单体和六氟丙烯三者的加入速率,使聚合釜内三者在要求浓度下。
18.优选的,还包括对步骤s4得到的可熔性聚四氟乙烯树脂进行氟化处理,使不稳定端基<10个。
19.通过上述的一种可熔性聚四氟乙烯树脂制备方法制备得到的可熔性聚四氟乙烯树脂中pave含量为3.0-10.0wt%,六氟丙烯含量为0.03-1.0%;并且所述可熔性聚四氟乙烯树脂的熔融指数为0.1-80g/10min,熔点为280-310℃。
20.进一步的,通过连续自成核退火热分级方法将可熔性聚四氟乙烯树脂熔点峰分为>317.5℃、315
±
2.5℃、310
±
2.5℃、305
±
2.5℃、300
±
2.5℃、295
±
2.5℃、290
±
2.5℃、<287.5℃八个峰;且>317.5℃处熔点峰峰面积占总峰面积的10-35%,315
±
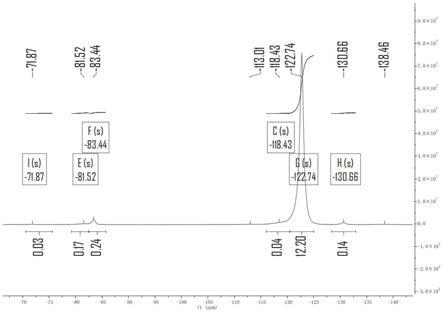
2.5℃处熔点峰峰面积占总峰面积的0.05-3%,310
±
2.5℃处熔点峰占总峰面积的5-20%,305
±
2.5℃、300
±
2.5℃、295
±
2.5℃、290
±
2.5℃处熔点峰峰面积之和占总峰面积的35-70%,<287.5℃熔点峰峰面积占总峰面积的0.01-8%。
21.本发明使用六氟丙烯作为pave聚合促进剂,通过控制六氟丙烯加入时间和聚合釜内六氟丙烯组分来调整pave相对聚合速度,这样就可以控制聚合链中两者的组分和排布,使pave聚入率达到65-90%;同时发现采用这种工艺的同时,能够得到拥有特殊的熔点峰分布可熔性聚四氟乙烯,且性能更加优异和稳定。
22.本发明的具体技术方案及其有益效果将会在下面的具体实施方式中结合附图进行详细的说明。
附图说明
23.下面结合附图和具体实施方式对本发明作进一步描述:
24.图1a为对比例1得到的可熔性聚四氟乙烯树脂的峰值示意图;
25.图1b为对比例1得到的可熔性聚四氟乙烯树脂的熔点峰分布示意图;
26.图2a为实施例1得到的可熔性聚四氟乙烯树脂的峰值示意图;
27.图2b为实施例1得到的可熔性聚四氟乙烯树脂的熔点峰分布示意图;
28.图3a为实施例2得到的可熔性聚四氟乙烯树脂的峰值示意图;
29.图3b为实施例2得到的可熔性聚四氟乙烯树脂的熔点峰分布示意图;
30.图4a为实施例3得到的可熔性聚四氟乙烯树脂的峰值示意图;
31.图4b为实施例3得到的可熔性聚四氟乙烯树脂的熔点峰分布示意图;
32.图5a为实施例4得到的可熔性聚四氟乙烯树脂的峰值示意图;
33.图5b为实施例4得到的可熔性聚四氟乙烯树脂的熔点峰分布示意图;
34.图6为实施例3得到的可熔性聚四氟乙烯树脂的流变分析图;
35.图7为实施例5得到的可熔性聚四氟乙烯树脂的流变分析图;
36.图8为实施例4得到的可熔性聚四氟乙烯树脂的核磁分析图;
37.图9为实施例5得到的可熔性聚四氟乙烯树脂的核磁分析图。
具体实施方式
38.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。以下对至少一个示例性实施例的描述实际上仅仅是说明性的,决不作为对本发明及其应用或使用的任何限制。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
39.本发明提供的一种可熔性聚四氟乙烯制备方法,使用六氟丙烯作为pave聚合促进剂,通过控制六氟丙烯加入时间和聚合釜内六氟丙烯组分来调整pave聚合速度,提高pave在聚合中的聚入率,包括以下步骤:
40.步骤一:在无氧聚合釜中按一定比例加入无离子水、有机溶剂、表面活性剂和链转移剂。
41.例如10000份无离子水、20-1000份有机溶剂、2-200份表面活性剂和0.1-200份链转移剂。
42.可以理解的是,上述的有机溶剂、表面活性剂和链转移剂均可以使用本领域常见的多种物质。由于每种的加入量都不一样,相对量差异较大,不再一一量化,具体可以参考实施例中的用量举例。
43.步骤二:温度升至设定温度50-80℃以后,加入四氟乙烯tfe和全氟烷基乙烯基醚pave组成的聚合单体至设定压力0.7-1.5mpa,且此时聚合釜内tfe组分为70-95wt%,pave组分为2-20wt%,加入引发剂开始反应。
44.步骤三:补加聚合单体和全氟烷基乙烯基醚聚合促进剂,维持聚合釜压力在设定压力0.7-1.5mpa直到反应结束,得到可熔性聚四氟乙烯乳液。
45.其中,pave聚合促进剂为六氟丙烯,采用六氟丙烯作为pave聚合促进剂可以提高pave聚入率。通过调整六氟丙烯加入时间和在聚合釜内组分控制pave在聚合链上的分布,得到可熔性聚四氟乙烯树脂的目标熔点峰分布。比如,对比例1中未加入六氟丙烯作为pave聚合促进剂,pave聚入率约为35%,熔点峰中320
±
2.5℃处峰面积超过总峰面积的50%,性能严重下降;而在实施例1中,通过调整六氟丙烯加入时间和用量,控制聚合釜内六氟丙烯浓度,提升pave聚入率的同时控制产品熔点峰的分布,使性能最优化。
46.其中,tfe和pave可以连续加入,也可通过分次加入;比如实施例2中聚合釜内每降低0.1mpa再补加聚合单体,配合六氟丙烯的适当时机加入,也能得到理想中的熔点峰分布。
47.其中,聚合单体为四氟乙烯和全氟烷基乙烯基醚,且全氟烷基乙烯基醚与四氟乙烯在反应过程中的总计投入质量比为1:25-1:8。可以理解的是,两者一般都是分开计量加入的,只要保证最终加入量在这个范围内即可。
48.其中全氟烷基乙烯基醚(pave)为全氟甲基乙烯基醚、全氟乙基乙烯基醚、全氟丙基乙烯基醚、全氟丁基乙烯基醚和全氟戊基乙烯基醚中的一种或多种混合物。
49.其中,六氟丙烯可以在反应开始前一次加入,也可在反应过程中分次加入;比如实施例2和实施例3,实施例3中,六氟丙烯聚合开始时一次性加入,但是可以通过控制tfe和pave加入时间调控聚合釜内压力在0.7-1.5mpa,且保证tfe、pave和六氟丙烯聚合釜内处于要求浓度下,也能得到理想中的熔点峰分布和高pave聚入率。
50.其中,六氟丙烯也可连续加入,但是要控制tfe、pave和六氟丙烯三者的加入速率,使聚合釜内三者在要求浓度下,比如实施例4。其中,六氟丙烯可以和聚合单体混合后加入,也可单独加入。
51.其中,要求浓度是指聚合釜内四氟乙烯组分为70-95wt%,六氟丙烯组分为0.1-20wt%,全氟烷基乙烯基醚组分为2-20wt%。全氟烷基乙烯基醚聚合促进剂加入量为聚合单体的0.01wt%-2wt%。
52.步骤四:将得到的可熔性聚四氟乙烯凝聚、洗涤、造粒得到可熔性聚四氟乙烯树脂。
53.在本发明中,所制得的pfa树脂的熔融指数为0.1-80g/10min;
54.在本发明中,所制得的pfa树脂的熔点为280-310℃;
55.在本发明中,所制得的pfa树脂的力学性能为30-38mpa;
56.在本发明中,所制得的pfa树脂的断裂伸长率为300%-410%;
57.在本发明中,所制得的pfa树脂的pave含量为3.0-10.0wt%;
58.在本发明中,所制得的pfa树脂的六氟丙烯含量为0.03-1.0%。
59.上述步骤四得到可熔性聚四氟乙烯树脂,还进一步进行氟化处理,使其不稳定端基<10个。
60.测量方法
61.1.熔体流动速率测定
62.根据astmd 1238所述的方法,采用熔体流动速率仪(rl-z1b1,上海思尔达科学仪器有限公司)测定。测试温度372℃,测试载荷5kg。
63.2.力学性能测定
64.根据astmd 638所述的方法,采用万能拉力机(etm503a,深圳万测试验设备有限公司)测定模制样品的拉伸强度和断裂伸长率。实验环境温度23
±
2℃,拉伸速度为50mm/min
±
5mm/min,夹具间距为24mm。
65.3.熔点
66.根据astmd 3418所述的方法,采用差示扫描量热仪(dsc823e,mettler)测定pfa的熔点:称取20mg
±
0.5mg的样品,在氮气氛围下,以10℃/min的升温速率升温至400℃,取dsc图谱熔融峰的峰顶温度为聚合物的熔点。
67.4.全氟烷基乙烯基醚含量测定
68.通过已知加工工艺制得0.05-0.3mm厚的薄片,采用傅里叶变换红外光谱仪(spectrum two,perkine lmer)扫描,全氟烷基乙烯基醚含量根据特征峰的吸光度(a)通过公式计算得到,其中全氟甲基乙烯基醚含量通过波数893cm-1处吸光度确定,全氟乙基乙烯基醚含量通过波数1089cm-1处吸光度确定,全氟丙基乙烯基醚含量通过波数990cm-1处吸
光度确定,公式如下:
69.pmve含量wt%=7
×
(a1/a0);
70.peve含量wt%=0.75+1.28
×
(a2/a0);
71.ppve含量wt%=0.97
×
(a3/a0);
72.其中:a0为波数2353cm-1处吸光度,a1为波数893cm-1处吸光度,a2为波数1089cm-1处吸光度,a3为波数990cm-1处吸光度。
73.当存在其它改性单体的情况下,可能会影响全氟正丙基乙烯基醚特征吸光度的测定,此时使用核磁进行测定。
74.5.六氟丙烯含量测定
75.使用核磁共振氟谱测定。
76.6.耐弯折次数测定
77.已知塑料加工工艺工艺制备0.2mm厚的薄片,裁剪成120mm
×
15mm大小的长条。根据astm d2176所述的方法,采用mit耐折度测定仪(pn-nz135,杭州品享科技有限公司)测定。载荷1kg,弯曲速度175次/min。
78.7.连续自成核退火热分级(ssa)
79.采用差示扫描量热仪(dsc823e,mettler)测定,称取20mg
±
0.5mg的样品,在氮气氛围下,从200℃以10℃/min的升温速率升温至400℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至320℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至315℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至310℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至305℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至300℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至295℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至290℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至285℃,保温30min,以10℃/min的降温速率降温至200℃,保温30min,以10℃/min的升温速率升温至280℃,保温30min,以10℃/min的降温速率降温至50℃,保温30min,以10℃/min的升温速率升温至400℃,取最后一次升温图谱得到200-350℃各段温度对应的峰面积。
80.对比例1:
81.步骤一:在20l的带有搅拌装置的卧式反应釜中加入10l无离子水、100g氟碳溶剂、20g分散剂x,抽空反应釜至反应釜内氧含量<30ppm;
82.步骤二:向反应釜内加入0.4g高纯氢,升温至60℃后,投入100g全氟正丙基乙烯基醚和适量四氟乙烯至反应釜压力1.0mpa;
83.步骤三:加入5g过硫酸钾开始反应,连续补加四氟乙烯和全氟正丙基乙烯基醚时压力稳定在1.0-1.2mpa;
84.步骤四:共补加4000g四氟乙烯和200g全氟正丙基乙烯基醚时停止反应,排空反应釜内未反应气体,得到可熔性聚四氟乙烯乳液;
85.步骤五:对步骤五得到的可熔性聚四氟乙烯乳液进行凝聚、洗涤、造粒得到不可熔
性聚四氟乙烯树脂3892g;
86.步骤六:对步骤六得到的可熔性可熔性聚四氟乙烯树脂进行氟化处理,使其不稳定端基<10个。
87.对比例1得到的可熔性聚四氟乙烯树脂分析测试结果如表1所示。
88.表1:
[0089][0090][0091]
峰值和熔点峰分布如图1a和图1b所示。
[0092]
实施例1:
[0093]
步骤一:在20l的带有搅拌装置的卧式反应釜中加入10l无离子水、100g氟碳溶剂、20g分散剂x,抽空反应釜至反应釜内氧含量<30ppm;
[0094]
步骤二:向反应釜内加入0.4g高纯氢,升温至60℃后,投入100g全氟正丙基乙烯基醚和适量四氟乙烯至反应釜压力1.0mpa;
[0095]
步骤三:加入5g过硫酸钾开始反应,连续补加四氟乙烯和全氟正丙基乙烯基醚时压力稳定在1.0-1.2mpa;当四氟乙烯补加量500g时,一次补加4g六氟丙烯,当四氟乙烯补加量为2000g时,一次补加10g六氟丙烯,当四氟乙烯补加量为3300g时,一次补加6g六氟丙烯;
[0096]
步骤四:共补加4000g四氟乙烯、150g全氟正丙基乙烯基醚和20g六氟丙烯时停止反应,排空反应釜内未反应气体,得到可熔性聚四氟乙烯乳液;
[0097]
步骤五:对步骤五得到的可熔性聚四氟乙烯乳液进行凝聚、洗涤、造粒得到不稳定端基<50个的可熔性聚四氟乙烯树脂3866g;
[0098]
步骤六:对步骤六得到的可熔性可熔性聚四氟乙烯树脂进行氟化处理,使其不稳定端基<10个。
[0099]
实施例1得到的可熔性聚四氟乙烯树脂分析测试结果如表2所示。
[0100]
表2:
[0101]
项目结果数据熔指(g/10min)6.2熔点(℃)304.1
拉伸强度(mpa)35.6断裂伸长率(%)388pave含量(%)4.82hfp含量(%)0.31耐弯折次数(次)61万临界剪切速率(s-1
)50pave聚入率(%)74.54
[0102]
峰值和熔点峰分布如图2a和图2b所示。
[0103]
实施例2:
[0104]
步骤一:在20l的带有搅拌装置的卧式反应釜中加入10l无离子水、100g氟碳溶剂、20g本技术人专利cn106366230中所述的混合表面活性剂(下文称分散剂x),抽空反应釜至反应釜内氧含量<30ppm;
[0105]
步骤二:向反应釜内加入0.4g高纯氢;升温至60℃后,投入40g全氟甲基乙烯基醚、60g全氟正丙基乙烯基醚和适量四氟乙烯至反应釜压力1.0mpa;
[0106]
步骤三:加入5g过硫酸钾开始反应,聚合釜内每降低0.1mpa,补加四氟乙烯和全氟正丙基乙烯基醚使压力回升到1.0mpa;当四氟乙烯补加量为1000g时,一次补加6g六氟丙烯,当四氟乙烯补加量为2000g时,一次补加3g六氟丙烯,当四氟乙烯补加量为3000g时,一次补加8g六氟丙烯;
[0107]
步骤四:共补加4000g四氟乙烯、170g全氟正丙基乙烯基醚和17g六氟丙烯时停止反应,排空反应釜内未反应气体,得到可熔性聚四氟乙烯乳液;
[0108]
步骤五:对步骤五得到的可熔性聚四氟乙烯乳液进行凝聚、洗涤、造粒得到不稳定端基<50个的可熔性聚四氟乙烯树脂3893g;
[0109]
步骤六:对步骤六得到的可熔性可熔性聚四氟乙烯树脂进行氟化处理,使其不稳定端基<10个。
[0110]
实施例2得到的可熔性聚四氟乙烯树脂分析测试结果如表3所示。
[0111]
表3:
[0112]
项目结果数据熔指(g/10min)10.4熔点(℃)301.1拉伸强度(mpa)34.3断裂伸长率(%)379pave含量(%)5.05hfp含量(%)0.32耐弯折次数(次)13万临界剪切速率(s-1
)120pave聚入率(%)72.81
[0113]
峰值和熔点峰分布如图3a和图3b所示。
[0114]
实施例3:
[0115]
步骤一:在20l的带有搅拌装置的卧式反应釜中加入10l无离子水、100g氟碳溶剂、
20g分散剂x,抽空反应釜至反应釜内氧含量<30ppm;
[0116]
步骤二:向反应釜内加入0.5g高纯氢,升温至60℃后,投入45g全氟乙基乙烯基醚、75g全氟正丙基乙烯基醚、20g六氟丙烯和适量四氟乙烯至反应釜压力1.0mpa;
[0117]
步骤三:加入5g过硫酸钾开始反应,聚合釜内每降低0.1-0.3mpa压力,补加适量的聚合单体,使聚合釜内压力回升到1.0-1.2mpa,保证聚合釜内tfe、ppve和六氟丙烯在规定浓度范围内;
[0118]
步骤四:补加4000g四氟乙烯和190g全氟正丙基乙烯基醚时停止反应,排空反应釜内未反应气体,得到可熔性聚四氟乙烯乳液;
[0119]
步骤五:对步骤五得到的可熔性聚四氟乙烯乳液进行凝聚、洗涤、造粒得到不稳定端基<50个的可熔性聚四氟乙烯树脂3926g;
[0120]
步骤六:对步骤六得到的可熔性可熔性聚四氟乙烯树脂进行氟化处理,使其不稳定端基<10个。
[0121]
实施例3得到的可熔性聚四氟乙烯树脂分析测试结果如表4所示。
[0122]
表4:
[0123]
项目结果数据熔指(g/10min)18.1熔点(℃)297.5拉伸强度(mpa)32.6断裂伸长率(%)366pave含量(%)5.41hfp含量(%)0.38耐弯折次数(次)7万临界剪切速率(s-1
)150pave聚入率(%)68.52
[0124]
峰值和熔点峰分布如图4a和图4b所示,流变分析如图6所示。
[0125]
实施例4:
[0126]
步骤一:在20l的带有搅拌装置的卧式反应釜中加入10l无离子水、100g氟碳溶剂、20g分散剂x,抽空反应釜至反应釜内氧含量<30ppm;
[0127]
步骤二:向反应釜内加入0.9g高纯氢,升温至60℃后,投入150g全氟正丙基乙烯基醚和适量四氟乙烯至反应釜压力1.0mpa;
[0128]
步骤三:加入6g过硫酸钾开始反应,连续补加tfe、ppve和六氟丙烯维持聚合釜内压力0.9-1.2mpa;
[0129]
步骤五:补加4000g四氟乙烯、260g全氟正丙基乙烯基醚和65g六氟丙烯时停止反应,排空反应釜内未反应气体,得到可熔性聚四氟乙烯乳液;
[0130]
步骤六:对步骤五得到的可熔性聚四氟乙烯乳液进行凝聚、洗涤、造粒得到不稳定端基<50个的可熔性聚四氟乙烯树脂4102g;
[0131]
步骤七:对步骤六得到的可熔性可熔性聚四氟乙烯树脂进行氟化处理,使其不稳定端基<10个。
[0132]
实施例4得到的可熔性聚四氟乙烯树脂分析测试结果如表5所示。
[0133]
表5:
[0134]
项目结果数据熔指(g/10min)58.7熔点(℃)291拉伸强度(mpa)30.3断裂伸长率(%)379pave含量(%)8.12hfp含量(%)0.56耐弯折次数(次)4000临界剪切速率(s-1
)400pave聚入率(%)81.24
[0135]
峰值和熔点峰分布如图5a和图5b所示,核磁分析如图8所示。
[0136]
实施例5:
[0137]
步骤一:在20l的带有搅拌装置的卧式反应釜中加入10l无离子水、100g氟碳溶剂、20g分散剂x,抽空反应釜至反应釜内氧含量<30ppm;
[0138]
步骤二:向反应釜内加入0.5g高纯氢,升温至60℃后,投入140g全氟正丙基乙烯基醚和适量四氟乙烯至反应釜压力1.0mpa;
[0139]
步骤三:加入5g过硫酸钾开始反应,连续补加四氟乙烯和全氟正丙基乙烯基醚时压力稳定在1.0-1.2mpa;当四氟乙烯补加量500g时,一次补加8g六氟丙烯,当四氟乙烯补加量为2000g时,一次补加16g六氟丙烯,当四氟乙烯补加量为3300g时,一次补加6g六氟丙烯;
[0140]
步骤四:补加4000g四氟乙烯、200g全氟正丙基乙烯基醚和30g六氟丙烯时停止反应,排空反应釜内未反应气体,得到可熔性聚四氟乙烯乳液;
[0141]
步骤五:对步骤五得到的可熔性聚四氟乙烯乳液进行凝聚、洗涤、造粒得到不稳定端基<50个的可熔性聚四氟乙烯树脂3955g;
[0142]
步骤六:对步骤六得到的可熔性可熔性聚四氟乙烯树脂进行氟化处理,使其不稳定端基<10个。
[0143]
实施例5得到的可熔性聚四氟乙烯树脂分析测试结果如表6所示。
[0144]
表6:
[0145]
项目结果数据熔指(g/10min)25.2熔点(℃)296.7拉伸强度(mpa)30.3断裂伸长率(%)379pave含量(%)5.89hfp含量(%)0.52临界剪切速率(s-1
)250pave聚入率(%)68.51
[0146]
流变分析和核磁分析如图7和图9所示。
[0147]
通过上述实施例表明,采用本发明制备方法,能够得到拥有特殊的熔点峰分布可
熔性聚四氟乙烯,且性能更加优异和稳定。
[0148]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,熟悉该本领域的技术人员应该明白本发明包括但不限于上面具体实施方式中描述的内容。任何不偏离本发明的功能和结构原理的修改都将包括在权利要求书的范围中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1