一种联苯二醌衍生物的制备方法
1.本发明属于化学领域,具体涉及一种联苯二醌衍生物的制备方法。
背景技术:
2.联苯二醌衍生物是一类用途广泛的重要化工中间体。它可以作为再分配反应的引发剂用于制备双羟基聚苯醚低聚物;它也是一种重要的活性中间体,可以用于合成耐热性好、力学性能优良的阻燃材料;其中,3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌还可作为偶联剂使用;除此之外,3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌经还原处理还可制备用于液晶聚合物、共聚聚苯醚、共聚碳酸酯、橡胶老化剂的3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二酚。
3.现有的联苯二醌衍生物制备方法中,一般以铜盐或铜配合物为催化剂,在氧气或双氧水的作用下,以2,6-二甲基苯酚为原料制备四甲基联苯二醌。如中国发明申请cn109046355a公开了一种利用铜基类水滑石催化合成3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的方法,该发明申请在碱性乳化液条件下,将原料2,6-二甲基苯酚、双氧水与铜基水滑石催化剂混合反应,制备得到3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌。但是这种制备方法在反应中会形成部分低分子量聚苯醚,该聚苯醚容易和四甲基联苯二醌掺杂在一起,给四甲基联苯二醌的纯化分离带来较大的困难,不利于制备高纯度四甲基联苯二醌。为了制备高纯度的联苯二醌衍生物,因此有必要开发一种新型的联苯二醌衍生物制备方法。
技术实现要素:
4.为了克服上述现有技术存在的问题,本发明的目的之一在于提供一种催化剂在制备联苯二醌衍生物中的应用;本发明的目的之二在于提供一种产率高、易分离的联苯二醌衍生物的制备方法;本发明的目的之三在于提供这种联苯二醌衍生物制备方法的应用。
5.为了实现上述目的,本发明所采取的技术方案是:
6.本发明第一方面提供一种催化剂在制备联苯二醌衍生物中的应用,所述催化剂包括四齿席夫碱-铁配合物。
7.优选的,所述四齿席夫碱-铁配合物的结构如式(ⅰ)所示;
[0008][0009]
式(ⅰ)中,r1、r2、r3、r4分别独立选自氢、取代或未取代的烷基、烷氧基或卤素;x选自卤素、no
3-或bf
4-,n选自0或1;
[0010]
进一步优选的,所述四齿席夫碱-铁配合物的结构如式(ⅰ)所示,式(ⅰ)中,r1、r2、r3、r4分别选自氢、c1-c4的烷基、卤素;n为0;
[0011]
再进一步优选的,所述四齿席夫碱-铁配合物为双水杨醛缩乙二胺铁,结构如式(ⅳ)所示;
[0012][0013]
优选的,所述催化剂还包括相转移促进剂。
[0014]
优选的,所述相转移促进剂包括脂肪胺聚氧乙烯醚;进一步优选的,所述相转移促进剂的结构如式(
ⅴ
)所示,
[0015][0016]
式(
ⅴ
)中,r选自c8-c25的烷基,m为5-25的整数;
[0017]
再进一步优选的,所述相转移促进剂的结构如式(
ⅴ
)所示,式(
ⅴ
)中,r选自c12-c18的烷基,m为8-20的整数。
[0018]
本发明第二方面提供一种联苯二醌衍生物的制备方法,包括以下步骤:
[0019]
将式(ⅱ)所示化合物、氧化剂、催化剂混合,反应,得到所述的联苯二醌衍生物;
[0020]
所述催化剂包括四齿席夫碱-铁配合物和相转移促进剂;
[0021][0022]
式(ⅱ)中,r5、r6、r7、r8分别独立选自氢、取代或未取代的烷基、烷氧基或卤素;
[0023]
所述联苯二醌衍生物的结构如式(ⅲ)所示;
[0024][0025]
式(ⅲ)中,r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
分别独立选自氢、取代或未取代的烷基、烷氧基或卤素。
[0026]
优选的,所述式(ⅱ)中,r5、r6、r7、r8分别独立选自氢、c1-c4的烷基、卤素;进一步优选的,所述式(ⅱ)中,r5、r6、r7、r8分别独立选自氢、c1-c2的烷基;再进一步优选的,所述式(ⅱ)中,r5、r6分别选自甲基,r7、r8分别选自氢。
[0027]
优选的,所述联苯二醌衍生物的结构如式(ⅲ)所示,式(ⅲ)中,r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
分别独立选自氢、c1-c4的烷基、卤素;进一步优选的,所述联苯二醌衍生物的结构如式(ⅲ)所示,式(ⅲ)中,r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
分别独立选自氢、c1-c2的烷基;再进一步优选的,所述联苯二醌衍生物的结构如式(ⅲ)所示,式(ⅲ)中,r9、r
10
、r
15
、r
16
分别选自甲基,r
11
、r
12
、r
13
、r
14
分别选自氢。
[0028]
优选的,所述四齿席夫碱-铁配合物的结构如式(ⅰ)所示;
[0029][0030]
式(ⅰ)中,r1、r2、r3、r4分别独立选自氢、取代或未取代的烷基、烷氧基或卤素;x选自卤素、no
3-或bf
4-,n选自0或1;
[0031]
进一步优选的,所述四齿席夫碱-铁配合物的结构如式(ⅰ)所示,式(ⅰ)中,r1、r2、r3、r4分别选自氢、c1-c4的烷基、卤素;n为0;
[0032]
再进一步优选的,所述四齿席夫碱-铁配合物为双水杨醛缩乙二胺铁,结构如式(ⅳ)所示;
[0033][0034]
优选的,所述相转移促进剂为脂肪胺聚氧乙烯醚;进一步优选的,所述相转移促进剂的结构如式(
ⅴ
)所示,
[0035][0036]
式(
ⅴ
)中,r选自c8-c25的烷基,m为5-25的整数;
[0037]
再进一步优选的,所述相转移促进剂的结构如式(
ⅴ
)所示,式(
ⅴ
)中,r选自c12-c18的烷基,m为8-20的整数。
[0038]
优选的,所述催化剂、助催化剂、氧化剂和苯酚衍生物的质量比为(0.001-0.1):(0.001-0.1):(0.3-1.5):1;进一步优选的,所述催化剂、助催化剂、氧化剂和苯酚衍生物的质量比为(0.01-0.05):(0.005-0.05):(0.5-1.0):1。
[0039]
优选的,所述氧化剂包括过氧化氢、氧气、臭氧、硝酸、次氯酸中的至少一种;进一步优选的,所述氧化剂包括过氧化氢、氧气、臭氧中的至少一种;再进一步优选的,所述氧化剂为过氧化氢。
[0040]
优选的,所述过氧化氢为质量浓度5%~50%的过氧化氢水溶液;进一步优选的,所述过氧化氢为质量浓度30%~50%的过氧化氢水溶液。
[0041]
优选的,所述混合步骤中,氧化剂采用滴加的方式与苯酚衍生物混合。
[0042]
优选的,所述滴加的时间为1h-4h;进一步优选的,所述滴加的时间为2h-3h。
[0043]
优选的,所述滴加的温度为60℃-100℃;进一步优选的,所述滴加的温度为70℃-90℃。
[0044]
优选的,所述反应的温度为50℃-100℃;进一步优选的,所述反应的温度为70℃-90℃。
[0045]
优选的,所述反应的时间为1h-6h;进一步优选的,所述反应的时间为3h-5h。
[0046]
优选的,所述反应的溶剂为苯类溶剂;进一步优选的,所述反应的溶剂包括甲苯、二甲苯、二氯苯中的至少一种;再进一步优选的,所述反应的溶剂为甲苯。
[0047]
优选的,所述溶剂与苯酚衍生物的质量比为(1-10):1;进一步优选的,所述溶剂与
苯酚衍生物的质量比为(3-6):1。
[0048]
优选的,所述反应结束后,还包括洗涤的步骤。
[0049]
优选的,所述洗涤剂为去离子水;进一步优选的,所述洗涤剂为50℃-100℃的去离子水;再进一步优选的,所述洗涤剂为70℃-90℃的去离子水。
[0050]
优选的,所述反应结束后,还包括干燥的步骤。
[0051]
优选的,所述干燥为真空干燥;进一步优选的,所述干燥为50℃-100℃的真空干燥;再进一步优选的,所述干燥为60℃-80℃的真空干燥。
[0052]
本发明第三方面提供根据本发明第二方面所述联苯二醌衍生物的制备方法在精细化学品领域中的应用。
[0053]
本发明的有益效果是:
[0054]
本发明提供的联苯二醌衍生物的制备方法采用四齿席夫碱-铁配合物提高反应原料的c-c偶联选择性,采用相转移促进剂提高氧化剂与反应原料的反应效率,采用该制备方法制备得到的联苯二醌衍生物产率和纯度高,不含低分子量的聚苯醚,解决了低分子量的聚苯醚与联苯二醌衍生物难以分离的问题,该制备方法可广泛应用于精细化学品领域。
[0055]
具体来说,本发明具有如下优点:
[0056]
1.本发明提供的联苯二醌衍生物的制备方法中,催化剂四齿席夫碱-铁配合物对苯酚衍生物的c-c偶联反应具有很高的选择性,助催化剂脂肪胺聚氧乙烯醚作为相转移促进剂,能促进水相中的过氧化氢进入油相溶剂中,提高了苯酚衍生物c-c偶联反应效率,从而提高了联苯二醌衍生物的产率。
[0057]
2.本发明提供的联苯二醌衍生物的制备方法不会形成低分子量的聚苯醚,制得产物纯度高,解决了低分子量聚苯醚与联苯二醌衍生物难以分离的问题,该方法可广泛应用于精细化学品领域。
附图说明
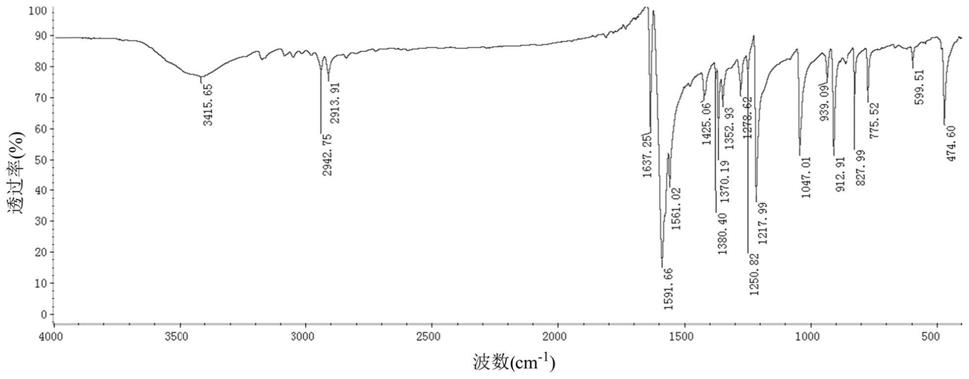
[0058]
图1为实施例1制备的3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的红外光谱图。
具体实施方式
[0059]
以下结合实例对本发明的具体实施作进一步说明,但本发明的实施和保护不限于此。需指出的是,以下若有未特别详细说明之过程,均是本领域技术人员可参照现有技术实现或理解的。所用试剂或仪器末注明生产厂商者,视为可以通过市售购买得到的常规产品。
[0060]
实施例中所用的双水杨醛缩乙二胺铁购买广东金柏化学有限公司;所用的相转移促进剂烷基胺聚氧乙烯醚结构式如式(1)所示,式(1)中,r为c12-c18的烷基,m为聚合度,取8~20;
[0061][0062]
实施例1
[0063]
本例3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的制备方法如下:
[0064]
在带有温度计、回流冷凝器和机械搅拌桨的四颈圆底烧瓶中依次加入80g甲苯、25g 2,6-二甲酚、1.0g双水杨醛缩乙二胺铁、0.1g十二烷基胺聚氧乙烯醚(如式(1)所示,m
=10),加热搅拌溶解,在80℃下,2小时内均匀滴加30%过氧化氢水溶液40g,滴加完双氧水溶液后,保持温度继续搅拌3小时,停止搅拌,冷却至10℃,过滤,用80℃去离子水洗涤滤渣二次,60℃真空干燥得21.6g红棕色固体。
[0065]
图1为实施例1制备的3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的红外光谱图,图1表明该红棕色固体为3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌。用hplc测得纯度为99.1%,产率87.8%。
[0066]
实施例2
[0067]
本例3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的制备方法如下:
[0068]
在带有温度计、回流冷凝器和机械搅拌桨的四颈圆底烧瓶中依次加入80g二甲苯、15g 2,6-二甲酚、0.5g双水杨醛缩乙二胺铁、0.06g十八烷基胺聚氧乙烯醚(如式(1)所示,m=15),加热搅拌溶解,在90℃下,3小时内均匀滴加30%过氧化氢水溶液30g,滴加完双氧水溶液后,保持温度继续搅拌2小时,停止搅拌,冷却至0℃,过滤,用90℃去离子水洗涤滤渣二次,70℃真空干燥得13.5g红棕色固体。用hplc测得红棕色固体纯度为99.0%,产率91.8%。
[0069]
实施例3
[0070]
本例3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的制备方法如下:
[0071]
在带有温度计、回流冷凝器和机械搅拌桨的四颈圆底烧瓶中依次加入80g甲苯、35g 2,6-二甲酚、0.8g双水杨醛缩乙二胺铁、0.8g十二烷基胺聚氧乙烯醚(如式(1)所示,m=20),加热搅拌溶解,在85℃下,3小时内均匀滴加30%过氧化氢水溶液50g,滴加完双氧水溶液后,保持温度继续搅拌4小时,停止搅拌,冷却至5℃,过滤,用90℃去离子水洗涤滤渣二次,50℃真空干燥得30.3g红棕色固体。用hplc测得红棕色固体纯度为99.3%,产率88.1%。
[0072]
实施例4
[0073]
本例3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的制备方法如下:
[0074]
在带有温度计、回流冷凝器和机械搅拌桨的四颈圆底烧瓶中依次加入80g二甲苯、45g 2,6-二甲酚、1.5g双水杨醛缩乙二胺铁2.0g十八烷基胺聚氧乙烯醚(如式(1)所示,m=8),加热搅拌溶解,在75℃下,2小时内均匀滴加30%过氧化氢水溶液60g,滴加完双氧水溶液后,保持温度继续搅拌5小时,停止搅拌,冷却至15℃,过滤,用80℃去离子水洗涤滤渣二次,60℃真空干燥得39.5g红棕色固体。用hplc测得红棕色固体纯度为99.1%,产率89.2%。
[0075]
对比例1
[0076]
本例3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的制备方法如下:
[0077]
在带有温度计、回流冷凝器和机械搅拌桨的四颈圆底烧瓶中依次加入80g甲苯、25g 2,6-二甲酚、1.0g n,n-二甲基丁胺铜、0.1g十二烷基胺聚氧乙烯醚(如式(1)所示,m=10),加热搅拌溶解,在80℃下,2小时内均匀滴加30%过氧化氢水溶液40g,滴加完双氧水溶液后,保持温度继续搅拌3小时,停止搅拌,冷却至10℃,过滤,用80℃去离子水洗涤滤渣二次,60℃真空干燥得19.8g红棕色固体。用hplc测得红棕色固体纯度为96.3%,产率80.5%。
[0078]
对比例2
[0079]
本例3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的制备方法如下:
[0080]
在带有温度计、回流冷凝器和机械搅拌桨的四颈圆底烧瓶中依次加入80g甲苯、35g 2,6-二甲酚、0.8g双水杨醛缩乙二胺铁,加热搅拌溶解,在85℃下,3小时内均匀滴加30%过氧化氢水溶液50g,滴加完双氧水溶液后,保持温度继续搅拌4小时,停止搅拌,冷却
至5℃,过滤,用90℃去离子水洗涤滤渣二次,50℃真空干燥得30.3g红棕色固体。用hplc测得红棕色固体纯度为99.0%,产率82.3%。
[0081]
对比例1与实施例1相比,反应仅不含催化剂双水杨醛缩乙二胺铁,反应产物3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的纯度比实施例1低2.8%,产率比实施例1低7.3%;对比例2与实施例3相比仅不含助催化剂十二烷基胺聚氧乙烯醚,反应产物3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌的纯度比实施例3低0.3%,产率比实施例3低5.8%。上述结果表明催化剂双水杨醛缩乙二胺铁能提高反应的产率和纯度,助催化剂十二烷基胺聚氧乙烯醚能提高反应的产率。
[0082]
实施例1~4和对比例1~2的实验结果表明,本发明提供的制备方法能够高效地合成高纯度3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌。该制备方法制备的3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二醌可以经还原处理制备高纯度3,3’,5,5
’‑
四甲基-4,4
’‑
联苯二酚,也可用于制备双羟基聚苯醚。
[0083]
上述实例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1