含聚有机硅氧烷的聚合物粒子群、组合物、树脂组合物及成形体的制作方法
1.本发明涉及含有聚有机硅氧烷的聚合物粒子群、组合物、树脂组合物及成形体。本技术基于2020年6月12日向日本技术的特愿2020-102397号要求优先权,并将其内容援用于此。
背景技术:
2.乙烯基单体与橡胶质聚合物聚合而成的含橡胶的聚合物能够在维持规定的橡胶粒径、橡胶结构的状态下分散在多种多样的树脂中,因此适合用于要求冲击强度的树脂。通常,从冲击强度改良方面考虑,优选使用橡胶的弹性模量低、泊松比高的橡胶质聚合物。丁二烯橡胶和硅酮系橡胶的泊松比为极高的0.5,弹性模量也低,因此适合作为橡胶质聚合物使用。其中,硅酮系橡胶与丁二烯橡胶相比,不易引起由热或紫外线导致的固化或着色,耐久性优异,因此适合用于建材或汽车部件等要求长期维持机械特性的用途。作为硅酮系橡胶,可以使用以聚二甲基硅氧烷为代表的聚有机硅氧烷。
3.然而,聚有机硅氧烷比丁二烯橡胶昂贵。另外,将含有聚有机硅氧烷聚合物混合于与聚有机硅氧烷聚合物相比折射率高的树脂(聚碳酸酯、聚甲基丙烯酸甲酯、苯乙烯-丙烯腈共聚物等)中,制成成形体时,成形体的透明性降低,存在难以表现出着色外观、特别是难以表现出深浓色色调的问题。
4.专利文献1中记载了含有氯乙烯树脂、聚碳酸酯树脂、聚酯树脂等各种热塑性树脂和含有聚有机硅氧烷的接枝共聚物的树脂组合物。在专利文献1中,通过使含有聚有机硅氧烷接枝共聚物的数均粒径为5~80nm,且使大于100nm的粒子的体积为总粒子体积的10%以下,从而改良了所得成形体的颜料着色性。
5.专利文献2中记载了含有苯乙烯-丙烯腈共聚物和含聚有机硅氧烷的接枝共聚物的树脂组合物。另外,记载了使含聚有机硅氧烷的接枝共聚物的重量平均粒度为110nm以下。在专利文献3中记载了含有聚甲基丙烯酸甲酯树脂和含聚有机硅氧烷的接枝共聚物的树脂组合物。现有技术文献专利文献
6.[专利文献1]日本特开平6-116471号公报[专利文献2]日本特开2009-155421号公报[专利文献3]日本特开2000-327880号公报
技术实现要素:
发明要解决的技术问题
[0007]
但是,在任意现有技术中,均存在得到的成形体的冲击强度不充分的课题。本发明的目的在于提供能够得到冲击强度优异的成形体的含聚有机硅氧烷聚合物粒子群、组合物及树脂组合物。本发明另外一目的在于提供冲击强度优异的成形体。解决技术问题的手段
[0008]
本发明具有以下方式。〔1〕一种含聚有机硅氧烷的聚合物粒子群,是含有聚合物(a)和第二乙烯基聚合物(b)的含聚有机硅氧烷的聚合物粒子群,所述聚合物(a)包含聚有机硅氧烷(a1)和第一乙烯基聚合物(a2),将使所述含聚有机硅氧烷的聚合物粒子群分散在树脂中而得到的树脂片的截面用透射型电子显微镜观察时的所述含聚有机硅氧烷的聚合物粒子群的各粒子的直径设为l,将所述聚有机硅氧烷(a1)区域的最大长度设为m时,满足下述式(1)的粒子的比例小于60%。m/l>0.1
···
(1)〔2〕根据〔1〕所述的含聚有机硅氧烷的聚合物粒子群,相对于所述含聚有机硅氧烷的聚合物粒子群100质量%,所述聚有机硅氧烷(a1)的比率为1质量%以上50质量%以下。〔3〕根据〔1〕或〔2〕所述的含聚有机硅氧烷的聚合物粒子群,相对于所述含聚有机硅氧烷的聚合物粒子群100质量%,所述聚有机硅氧烷(a1)的比率为1质量%以上10质量%以下。〔4〕根据〔1〕~〔3〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,数均粒径为10nm以上150nm以下。〔5〕根据〔1〕~〔4〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,所述含聚有机硅氧烷的聚合物粒子群的各粒子中的一部分不溶于四氢呋喃,相对于所述含聚有机硅氧烷的聚合物粒子群100质量%,不溶于四氢呋喃的所述含聚有机硅氧烷的聚合物粒子群的比率为80质量%以上小于100质量%。〔6〕根据〔1〕~〔5〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,所述含聚有机硅氧烷的聚合物粒子群的各粒子中的一部分可溶于四氢呋喃,可溶于四氢呋喃的所述含聚有机硅氧烷的聚合物粒子群的重均分子量为2万以上50万以下。〔7〕根据〔1〕~〔6〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,通过透射型电子显微镜观察所述树脂片截面时,所述聚合物(a)具有将所述聚有机硅氧烷(a1)作为海成分、将所述第一乙烯基聚合物(a2)作为岛成分的海岛结构,在所述海岛结构中,所述聚有机硅氧烷(a1)区域中含有多个所述第一乙烯基聚合物(a2)区域。〔8〕根据〔1〕~〔7〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,通过透射型电子显微镜观察所述树脂片的截面时,所述含聚有机硅氧烷的聚合物粒子群的各粒子具有将所述聚有机硅氧烷(a1)作为海成分、将所述第一乙烯基聚合物(a2)作为第一岛成分、将所述第二乙烯基聚合物(b)作为第二岛成分的海岛结构。〔9〕根据〔1〕~〔8〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,相对于所述含聚有机硅氧烷的聚合物粒子群100质量%,所述聚合物(a)的比率为60质量%以上95质
量%以下。〔10〕根据〔1〕~〔9〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,构成所述第一乙烯基聚合物(a2)的乙烯基单体成分(a2)含有单官能(甲基)丙烯酸酯单体。〔11〕根据〔1〕~〔10〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,构成所述第二乙烯基聚合物(b)的乙烯基单体成分(b)含有选自由(甲基)丙烯酸酯单体及芳香族乙烯基单体构成的组中的至少一种,相对于所述乙烯基单体成分(b)100质量%,所述(甲基)丙烯酸酯单体及所述芳香族乙烯基单体的合计比率为50质量%以上。〔12〕根据〔1〕~〔11〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,构成所述第二乙烯基聚合物(b)的乙烯基单体成分(b)含有甲基丙烯酸甲酯,相对于所述乙烯基单体成分(b)100质量%,所述甲基丙烯酸甲酯的比率为50质量%以上。〔13〕根据〔1〕~〔12〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,所述聚合物(a)是在含有所述聚有机硅氧烷(a1)的胶乳的存在下由构成所述第一乙烯基聚合物(a2)的乙烯基单体成分(a2)聚合而成的聚合物。〔14〕一种组合物,含有〔1〕~〔13〕中任一项所述的含聚有机硅氧烷的聚合物粒子群,和选自由磷酸化合物及其碱金属盐构成的组中的至少一种成分。〔15〕根据〔14〕所述组合物,所述磷酸化合物的碱金属盐是选自由烷基磷酸的碱金属盐和烷基芳基磷酸的碱金属盐构成的组中的至少一种。〔16〕根据〔14〕或〔15〕所述组合物,其中,所述磷酸化合物的碱金属盐是聚氧化烯烷基醚磷酸的碱金属盐。〔17〕根据〔14〕~〔16〕中任一项所述组合物,相对于所述含聚有机硅氧烷的聚合物粒子群和选自由所述磷酸化合物及其碱金属盐构成的组中的至少一种成分的合计100质量%,所述成分中所含的磷原子的比率为100质量ppm以上。〔18〕一种树脂组合物,含有〔1〕~〔13〕中任一项所述的含聚有机硅氧烷的聚合物粒子群和热塑性树脂。〔19〕一种树脂组合物,含有〔14〕~〔17〕中任一项所述的组合物和热塑性树脂。〔20〕根据〔18〕或〔19〕所述树脂组合物,所述热塑性树脂含有选自由芳香族聚碳酸酯、聚甲基丙烯酸甲酯、苯乙烯-丙烯腈共聚物、聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚氯乙烯、聚苯硫醚及聚缩醛构成的组中至少一种。〔21〕一种成形体,含有〔1〕~〔13〕中任一项所述的含聚有机硅氧烷的聚合物粒子群。发明的效果
[0009]
根据本发明的含聚有机硅氧烷的聚合物粒子群,可以得到冲击强度优异的成形体。根据本发明的组合物,可以得到冲击强度优异的成形体。根据本发明的树脂组合物,可以得到冲击强度优异的成形体。本发明成形体冲击强度优异。
附图说明
[0010]
[图1]是[实施例]中制造的重合体粒子群(c-1)的透射型电子显微镜(tem)图像。[图2]是对[实施例]中制造的重合体粒子群(c-1)的透射型电子显微镜(tem)图像进行明度图像解析的图像。[图3]是对[实施例]中制造的重合体粒子群(c-3)的透射型电子显微镜(tem)图像进行明度图像解析的图像。[图4]是对[实施例]中制造的重合体粒子群(c-4)的透射型电子显微镜(tem)图像进行明度图像解析的图像。[图5]是对[实施例]中制造的重合体粒子群(c-6)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图6]是对[实施例]中制造的重合体粒子群(c-7)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图7]是对[实施例]中制造的重合体粒子群(c-8)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图8]是对[实施例]中制造的重合体粒子群(c-11)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图9]是对[实施例]中制造的重合体粒子群(c-12)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图10]是对[实施例]中制造的重合体粒子群(c-13)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图11]是对[实施例]中制造的重合体粒子群(c-14)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图12]是对[实施例]中制造的重合体粒子群(c-15)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图13]是对[实施例]中制造的重合体粒子群(c-17)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图14]是对[实施例]中制造的重合体粒子群(c-18)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图15]是对[实施例]中制造的重合体粒子群(c-19)的透射型电子显微镜(tem)像进行明度图像解析的图像。[图16]是对[实施例]中制造的重合体粒子群(c-20)的透射型电子显微镜(tem)像进行明度图像解析的图像。
具体实施方式
[0011]
以下,对本发明的实施方式进行详细说明。在本发明中,乙烯基单体是具有聚合性双键的化合物。“(甲基)丙烯酸酯”是丙烯酸酯或甲基丙烯酸酯。在本说明书中,将“透射型电子显微镜”也称为“tem”。
[0012]
〔含聚有机硅氧烷的聚合物粒子群〕
本发明的一个实施方式涉及的含聚有机硅氧烷的聚合物粒子群(以下,也称为“聚合物粒子群(c)”)包含聚合物(a)和第二乙烯基聚合物(b)(以下也称为“乙烯基聚合物(b)”)。聚合物(a)包含聚有机硅氧烷(a1)和第一乙烯基聚合物(a2)(以下也称为“乙烯基聚合物(a2)”)。关于聚有机硅氧烷(a1)、乙烯基聚合物(a2)、聚合物(a)、乙烯基聚合物(b)将在后面进行详细说明。典型地,聚合物粒子群(c)的各粒子含有聚合物(a)和乙烯基聚合物(b)。
[0013]
聚合物粒子群(c)是将使聚合物粒子群(c)分散在树脂中而得到的树脂片的截面用tem观察时的聚合物粒子群(c)的各粒子的直径(μm)设为l,将聚有机硅氧烷(a1)区域的最大长度(μm)设为m时,满足下述式(1)的粒子的比例(以下也称为“z值”)小于60%。m/l>0.1
···
(1)z值小于60%时,含有聚合物粒子群(c)的成形体的冲击强度、着色外观及耐候性优异。z值优选为50%以下,更优选为30%以下,进一步优选为20%以下,特别优选为10%以下。z值越小越好,下限没有特别限定。
[0014]
聚合物粒子群(c)优选在用tem观察上述树脂片的截面时,聚合物(a)具有将聚有机硅氧烷(a1)作为海成分、将乙烯基聚合物(a2)作为岛成分的海岛结构。如上所述,通过采用聚有机硅氧烷(a1)和乙烯基聚合物(a2)复合而成的海岛结构,含有聚合物粒子群(c)的成形体的冲击强度、着色外观及耐候性更优异。
[0015]
在用tem观察上述树脂片的截面时,聚合物粒子群(c)的各粒子优选具有将聚有机硅氧烷(a1)作为海成分、将乙烯基聚合物(a2)作为第一岛成分、将乙烯基聚合物(b)作为第二岛成分的海岛结构。通过这样具有聚有机硅氧烷(a1)、乙烯基聚合物(a2)和乙烯基聚合物(b)复合海岛结构,成形体的冲击强度和着色外观更优异。
[0016]
如上所述,聚有机硅氧烷(a1)、乙烯基聚合物(a2)和乙烯基聚合物(b)复合而成的海岛结构例如可以通过在制造聚合物粒子群(c)时,将构成乙烯基聚合物(b)的乙烯基单体成分(b)的至少一部分含浸于聚合物(a)中并聚合而形成。为了使乙烯基单体成分(b)的至少一部分含浸在聚合物(a)中,例如只要乙烯基单体成分(b)的至少一部分使用在30℃的水中的溶解度为1.0g/l以下的乙烯基单体即可。作为30℃下在水中的溶解度为1.0g/l以下的乙烯基单体,例如可以举出后述的乙烯基单体中的苯乙烯、烷基取代苯乙烯、烷基取代异丙烯基苯、1,1-二苯基乙烯等。作为区分第一岛成分和第二岛成分方法,例如可以举出调整了染色条件(具体而言为染色剂种类、染色时间)的tem观察、能量分散型x射线分光法、电子能量损失谱法等的元素分析。聚合物粒子群(c)的各粒子可以包含没有形成第二岛成分的乙烯基聚合物(b)。没有形成第二岛成分的乙烯基聚合物(b)(没有含浸于聚合物(a)中的乙烯基单体成分(b)聚合而成的乙烯基聚合物(b)),在聚合物粒子群(c)的各粒子中,作为接枝聚合物(与聚合物(a)共价键合的聚合物)或自由聚合物(与聚合物(a)物理吸附而非共价键合的聚合物)存在于聚合物(a)的外表面。
[0017]
具体而言,上述树脂片的截面tem像可以按照以下的顺序取得。(1)首先,将聚合物粒子群(c)取到聚乙烯胶囊前端,注入液状的未固化环氧树脂,
在25℃静置12小时使其固化,得到树脂片。(2)接着,对得到的树脂片用四氧化锇水溶液(23℃、12小时)进行染色。(3)接着,对用四氧化锇水溶液染色后的树脂片,用四氧化钌水溶液(23℃、5小时)进行染色。(4)接着,从用四氧化钌水溶液染色后的树脂片中,使用切片机切出切片,回收到带支撑膜的铜栅上。切片的厚度设为50nm。(5)接着,通过tem,获得随机选择切片的表面(树脂片的截面)的0.5μm2以上的范围的图像(tem图像)。图像的获取倍率设为20万倍。
[0018]
在得到的tem图像中,如图1所示,作为连续相,观察到环氧树脂固化物区域(以下也称为“树脂区域”),作为分散在树脂区域内的分散相,观察到聚合物粒子群(c)的各粒子的区域(以下也称为“粒子区域”)。另外,在tem图像中,如图1所示,粒子区域中的聚有机硅氧烷(a1)区域(以下,也称为“聚有机硅氧烷(a1)域”)用明对比度确认,乙烯基聚合物(a2)或乙烯基聚合物(b)区域(以下也称为“乙烯基聚合物域”。)用暗对比度确认。另外,图1“tem像”的图像是对后述的实施例中制造的聚合物粒子群(c-1)取得的tem图像。
[0019]
如图2所示,可以使用图像分析软件(例如imagej)从得到的tem图像中分别提取粒子区域、聚有机硅氧烷(a1)域及乙烯基聚合物域。具体地,对于tem图像,通过线轮廓测量提取粒子区域的对比度。在线轮廓测量中,可以将一个粒子区域中的最大费雷特径设为长径,将最小费雷特径设为短径,通过其长径与短径的交叉点(以下,也称为“中央点”),以切断粒径的方式画线。在最大费雷特径和最小费雷特径为相同值的情况下,可以任意地确定表示相同费雷特径的多条线,从它们的交点求出中央点。另外,以线的两端不与邻接的粒子区域相接的方式画线。将画出的线的长度设为粒子区域的直径l。另外,虽然可以在满足上述规定的地方任意地画线,但优选在一个粒子区域中画出多条线,将表示粒子区域的直径l与通过后述的方法求出的聚有机硅氧烷(a1)区域的最大长度m之比(以下也称为“m/l值”)为最大值的线用于在该粒子区域中的线轮廓。多条画线时,首先任意地画出第一条线,然后画出与得到的第一条线在中央点所成的角(这里所成的角是指90
°
以下的角)为30
°
以上、60
°
以下的第二条线,进一步画出与第二条线在中央点垂直的第三条线,对总计三条线进行评价,选择表示最大的m/l值的线。但是,在不能画出多条满足上述规定的线的情况下,可以采用对不足三条的线进行评价而得到的m/l值。
[0020]
此时,由于符合下述的粒子区域不能判别为1个粒子,因此不选择作为测定线轮廓的粒子区域。(i)在图像边缘截断的粒子区域。(ii)尺寸小于平均粒径的80%的粒子区域。(iii)在3个方向以上存在相邻的粒子区域,各粒子区域的边界不清楚的粒子区域。测定图像中可确认的所有剩余的粒子区域,对合计50个以上的粒子区域进行线轮廓测定。在一张tem像中无法测定50个以上不符合上述(i)~(iii)的粒子区域的情况下,根据在不同视场中取得的多张tem像进行测定并合计。
上述(ii)中的平均粒径是在图像中可确认的粒子区域的等效圆直径的平均值。
[0021]
然后,将在粒子区域中画出的线的从一端到另一端的距离设为横轴(x轴),将线上的对比度(gray value)作为y值绘制为纵轴(y轴),求出一个粒子区域中y值的最大值和最大值的75%的值(设为ya值)。表示大于ya值的y值的部位规定为hgv部位。这里,由于颜色越亮(淡)y值越大,聚有机硅氧烷(a1)域用明对比度确认,乙烯基聚合物域用暗对比度确认,因此能够判别hgv部位与聚有机硅氧烷(a1)域。将hgv部位中、一个粒子区域内的具有最大连续长度的部位定义为该粒子区域中的聚有机硅氧烷(a1)的最大区域,将该hgv部位的连续长度设为区域的最大长度m。在此,hgv部位的连续长度是指在灰度值(gray value)超过ya值的峰值中,与以ya值画的线(图2中用虚线表示)的交点间距离。
[0022]
在进行了线轮廓测定的粒子区域中,满足上述式(1)的粒子区域的个数设为z1,不满足上述式(1)的粒子区域的个数设为z2,根据下述式(2)计算z值。z值={z1/(z1+z2)}
×
100
···
(2)
[0023]
图2中,“tem像”是对于在后述实施例中制造的聚合物(c-1)所取得的tem像,在imagej中进行了线轮廓测定的图像。图2中,“解析例”是将对聚合物(c-1)使用imagej进行线轮廓测定的结果图表化并进行分析的例子。在“解析例”中,为了简便地比较不同粒径的粒子区域,示出了将线的长度(直径l)转换为1的例子。在后述图3~16中也同样。
[0024]
在上述海岛结构中,乙烯基聚合物域的平均直径优选为50nm以下,更优选为30nm以下,特别优选为20nm以下。如果乙烯基聚合物域的平均直径为50nm以下,则成形体的冲击强度更优异。乙烯基聚合物域的平均直径的下限没有特别限定,例如为2nm。
[0025]
在上述海岛结构中,从成形体的冲击强度的观点出发,优选在聚有机硅氧烷(a1)域中含有多个乙烯基聚合物域。进一步,乙烯基聚合物域的平均直径优选为50nm以下,更优选为30nm以下。乙烯基聚合物(a2)域的平均直径的下限没有特别限定,例如为2nm。
[0026]
乙烯基聚合物域可以仅是乙烯基聚合物(a2)的区域,也可以是乙烯基聚合物(a2)的区域和乙烯基聚合物(b)的区域这两者。当乙烯基聚合物域仅为乙烯基聚合物(a2)的区域时,乙烯基聚合物域的平均直径为乙烯基聚合物(a2)区域的平均直径。
[0027]
乙烯基聚合物域的平均直径可以通过各种方法确认,例如可以从上述tem图像确认。具体而言,可以按照以下步骤进行确认。首先,通过上述步骤得到tem图像。接着,从得到的tem图像中,使用图像解析软件(例如商品名“image-pro(注册商标)plus”、日本roper公司制),分别提取粒子区域、聚有机硅氧烷(a1)域和乙烯基聚合物域。具体而言,对于tem像,在进行背景亮度不均的平坦化及去噪之后,通过以周围的树脂区域未被选择的最大亮度为界点进行二值化,能够分别提取粒子区域、聚有机硅氧烷(a1)域及乙烯基聚合物域。从面积大的粒子区域中选择确认的粒子区域中30%以上且100个以上作为测定对
象。此时选择的粒子区域的比率是指选择的粒子区域的个数相对于图像中可确认的等效圆直径10nm以上的粒子区域的总个数的比率。对于所选择的测定对象的各粒子区域,在该区域内存在的乙烯基聚合物域中,选择等效圆直径10nm以上的乙烯基聚合物域,将这些乙烯基聚合物域的等效圆直径的平均值作为平均直径。可以将对所有测定对象的粒子区域求出的平均直径的平均值作为乙烯基聚合物域的平均直径。
[0028]
聚有机硅氧烷(a1)相对于聚合物粒子群(c)100质量%的比率优选为50质量%以下,更优选为40质量%以下,进一步优选为30质量%以下,进一步更优选为20质量%以下,特别优选为10质量%以下,最优选为6质量%以下,另一方面,优选为1质量%以上。如果聚有机硅氧烷(a1)的比率为所述下限值以上,则成形体的冲击强度更优异,如果为所述上限值以下,则成形体的着色外观更优异。
[0029]
聚合物(a)相对于聚合物粒子群(c)100质量%的比率优选为60质量%以上,更优选为65质量%以上,另一方面,优选为95质量%以下,更优选为90质量%以下,进一步优选为85质量%以下。如果聚合物(a)的含量为上述下限值以上,则成形体的冲击强度更优异,如果为上述上限值以下,则聚合物粒子群(c)在热塑性树脂中的分散性更优异,得到的成形体的外观更优异。聚合物(a)相对于聚合物粒子群(c)100质量%的比率例如可以为60~95质量%,可以为60~90质量%,可以为65~90质量%,可以为65~85质量%。
[0030]
乙烯基聚合物(b)相对于聚合物粒子群(c)100质量%的比率优选为5质量%以上,更优选为10质量%以上,进一步优选为15量%以上,另一方面,优选为40质量%以下,更优选为35质量%以下。如果乙烯基聚合物(b)含量为上述下限值以上,则聚合物粒子群(c)在热塑性树脂中的分散性更优异,得到的成形体的外观更优异,如果为上述上限值以下,则成形体的冲击强度更优异。乙烯基聚合物(b)相对于聚合物粒子群(c)100质量%的比率例如可以为5~40质量%,可以为10~40质量%,可以为10~35质量%,可以为15~35质量%。
[0031]
典型地,聚合物粒子群(c)的各粒子中的一部分不溶于四氢呋喃(以下也称为“thf”)。换言之,聚合物粒子群(c)的各粒子中的一部分可溶于thf。以下,将聚合物粒子群(c)各粒子的不溶于thf的部分称为“thf不溶成分”,将可溶于thf的部分称为“thf可溶成分”。
[0032]
thf不溶成分相对于聚合物粒子群(c)100质量%的比率优选为80质量%以上,更优选为85质量%以上,进一步优选为90量%以上,特别优选为93质量%以上,另一方面,优选小于100质量%,更优选为99质量%以下。如果thf不溶成分的比率为上述下限值以上,则聚合物粒子群(c)在热塑性树脂中的分散性更优异,得到的成形体的冲击强度及外观更优异。如果thf不溶成分的比率小于100质量%,则冲击强度与显色性的平衡更优异,如果为99质量%以下,则添加到热塑性树脂中时的熔融流动性更优异。thf不溶成分相对于聚合物粒子群(c)100质量%的比率可以为80质量%以上小于100质量%,可以为85质量%以上小于100质量%,可以为90质量%以上99质量%以下,可以为93质量%以上99质量%以下。
[0033]
thf不溶成分通过进行以下(1-1)~(1-5)的操作来测定。
(1-1)向thf 50ml(44.5g)中加入0.5g试样(聚合物粒子群(c)),调制混合溶液,在25℃静置8小时后,用搅拌器搅拌30分钟,使thf可溶成分溶解。(1-2)将所述混合溶液放入测定质量的离心管中,用离心分离机(16000rpm,4小时)对含有thf不溶成分和thf可溶成分的液体进行离心分离。(1-3)除去上清液后,重新加入thf搅拌后,再次与上述(1-2)同样地进行离心分离,清洗thf不溶成分。(1-4)重复上述(1-3)2次后,除去上清液。将残留有thf不溶成分的离心管浸渍在温水浴中(80℃、8小时),使thf挥发后,在65℃下真空干燥6小时,得到干燥试样(附着在离心管上的thf不溶成分)。(1-5)测定得到的干燥试样的质量(thf不溶成分+离心管),通过下式计算thf不溶成分的比率w
ais
(%)。w
ais
=(w
c1-w
as
)/wt
×
100wt:测定所用的聚合物粒子群(c)的质量w
as
:离心管的质量w
c1
:thf不溶成分的质量(包含离心管的质量)
[0034]
从使聚合物粒子群(c)中的thf不溶成分的比率在上述范围内的观点出发,优选乙烯基聚合物(a2)充分交联,聚有机硅氧烷(a1)与乙烯基聚合物(a2)共价键合。另外,优选乙烯基聚合物(b)与聚合物(a)共价键合。
[0035]
聚合物粒子群(c)的thf可溶成分的重均分子量优选为2万以上,更优选为5万以上,另一方面,优选为50万以下,更优选为35万以下,进一步优选为20万以下,特别优选为15万以下。如果thf可溶成分的重均分子量为2万以上50万以下,则聚合物粒子群(c)在热塑性树脂中的分散性优异,得到的成形体的冲击强度及外观优异。其中,如果thf可溶成分的重均分子量为5万以上15万以下,则聚合物粒子群(c)在热塑性树脂中的分散性特别良好,在包括混炼水平低的广泛的成形条件下,得到的成形体的冲击强度及外观也优异。聚合物粒子群(c)的thf可溶成分的重均分子量可以为2万以上50万以下,可以为2万以上35万以下,可以为5万以上20万以下,可以为5万以上15万以下。
[0036]
thf可溶成分的重均分子量通过进行以下(2-1)~(2-3)的操作来测定。(2-1)从通过测定上述thf不溶成分的比率而采集的含有thf可溶成分的液体中,使用旋转蒸发器减压蒸馏除去thf,得到thf可溶成分。(2-2)将上述(2-1)中得到的thf可溶成分再次溶解在thf中,使试样浓度为0.1~0.3质量%,得到thf可溶成分的thf溶液。(2-3)对上述(2-2)中得到的thf可溶成分的thf溶液进行凝胶渗透色谱(gpc)测定,由标准聚苯乙烯的定量曲线求出重均分子量(mw)。gpc测定条件如后述的实施例所述。
[0037]
thf可溶成分的重均分子量可以通过在使构成乙烯基聚合物(b)的乙烯基单体成分(b)聚合时的引发剂量、还原剂量、聚合温度和链转移剂的使用来调整。例如,通过由引发剂量、还原剂量的增量或聚合温度的高温化等引起的自由基产生的增进、或由链转移剂的添加及添加量的增量引起的链转移反应的促进,降低thf可溶成分的重均分子量。
[0038]
聚合物粒子群(c)的数均粒径优选为10nm以上,更优选为30nm以上,进一步优选为50nm以上,特别优选为70nm以上,另一方面,优选为800nm以下,更优选为500nm以下,更优选200nm以下,特别优选150nm以下,特别优选130nm以下,最优选100nm以下。如果数均粒径为上述下限值以上,则成形体的冲击强度更优异,如果为上述上限值以下,则成形体的着色外观更优异。聚合物粒子群(c)的数均粒径例如可以为10~800nm,可以为30~500nm,可以为50~200nm,可以为50~150nm,可以为70~130nm,可以为70~100nm。
[0039]
聚合物粒子群(c)的数均粒径的测定方法没有特别限定,例如通过以下的方法进行测定。将聚合物粒子群(c)的胶乳用去离子水稀释至浓度约3%作为试样,使用毛细管方式的粒度分布计(美国matec公司制造的chdf2000型粒度分布计)测定个数基准的粒度分布,将中位直径作为数均粒径。
[0040]
粒度分布的测定可以在matec公司推荐的下述标准条件下进行。卡盒(cartridge):专用的粒子分离用毛细管式卡盒(商品名:c-202)载液:专用载液(商品名;2xgr500)载液的液性:中性载液的流速:1.4ml/分钟载液的压力:约4,000psi(2,600kpa)测定温度:35℃试样使用量:0.1ml此外,作为标准粒径物质,可使用美国duke公司制的粒径已知的单分散聚苯乙烯且在40~800nm的粒径范围内的12种粒子。
[0041]
聚合物粒子群(c)的数均粒径例如在通过乳液聚合制造聚合物粒子群(c)时,可以通过乳化剂的量来调整。
[0042]
(聚有机硅氧烷(a1))聚有机硅氧烷(a1)是含有有机硅氧烷单元的聚合物。聚有机硅氧烷(a1)可以通过聚合含有有机硅氧烷的有机硅氧烷混合物而得到。有机硅氧烷混合物还可包含根据需要使用的成分。作为根据需要使用成分,可以举出选自由硅氧烷系交联剂、硅氧烷系交叉剂和具有末端封端基的硅氧烷低聚物构成的组中的至少一种。
[0043]
作为有机硅氧烷,可以举出链状有机硅氧烷、烷氧基硅烷化合物、环状有机硅氧烷等,均可以使用。其中,优选烷氧基硅烷化合物、环状有机硅氧烷,特别地,由于聚合稳定性高、聚合速度快,因此优选环状有机硅氧烷。
[0044]
作为烷氧基硅烷化合物,优选2官能性烷氧基硅烷化合物,例如可以举出二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、二乙氧基二乙基硅烷、二丙氧基二甲基硅烷、二苯基二甲氧基硅烷、二苯基二乙氧基硅烷、甲基苯基二甲氧基硅烷、甲基苯基二乙氧基硅烷等。它们可以单独使用1种,或者将2种以上组合使用。
[0045]
作为环状有机硅氧烷,优选3~7元环的物质,例如,可举出六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、十二甲基环六硅氧烷、三甲基三苯基环三硅氧烷、四
甲基四苯基环四硅氧烷、八苯基环四硅氧烷等。它们可单独使用1种,也可并用2种以上。这些之中,从易于控制粒径分布出发,优选八甲基环四硅氧烷。
[0046]
作为有机硅氧烷,从得到可以使成形体冲击强度更高的聚合物粒子群(c)的观点出发,优选选自由环状二甲基硅氧烷及2官能性二烷基硅烷化合物构成的组中的至少一种。
[0047]
环状二甲基硅氧烷是在硅原子上具有2个甲基的环状硅氧烷,可以举出六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、十二甲基环己硅氧烷等。它们可以单独使用1种,或者将2种以上组合使用。
[0048]
2官能性二烷基硅烷化合物是指,在硅原子上具有烷氧基和烷基各2个的硅烷化合物,可以举出二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、二乙氧基二乙基硅烷、二丙氧基二甲基硅烷等。它们可以单独使用1种,或者将2种以上组合使用。
[0049]
作为硅氧烷系交联剂,优选具有硅氧烷基的交联剂。作为硅氧烷系交联剂,例如可以举出三甲氧基甲基硅烷、三乙氧基苯基硅烷、四甲氧基硅烷、四乙氧基硅烷、四正丙氧基硅烷、四丁氧基硅烷等3官能性或4官能性硅烷系交联剂。其中,优选4官能性交联剂,更优选四乙氧基硅烷。
[0050]
硅氧烷系交联剂相对于有机硅氧烷混合物100质量%的比率优选为10质量%以下,更优选为5质量%以下,也可以为0质量%。如果硅氧烷系交联剂含有率为10质量%以下,则可以得到能够使成形体的冲击强度更良好的聚合物粒子群(c)。
[0051]
硅氧烷系交叉剂具有硅氧基(-si-o-),并且具有能够与乙烯基单体聚合的官能团。作为硅氧烷系交叉剂,例如可以举出下述式(i)表示的硅氧烷。r-si(r1)n(or2)
(3-n)
ꢀꢀꢀꢀꢀ
(i)式(i)中,r1表示甲基、乙基、丙基或苯基。r2表示烃基等有机基团,例如优选甲基、乙基、丙基或苯基。n表示0、1或2。r表示下述式(i-1)~(i-4)中任一种所示的官能团。ch2=c(r3)-coo-(ch2)
p
‑ꢀꢀꢀ
(i-1)ch2=c(r4)-c6h4‑ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(i-2)ch2=ch
‑ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(i-3)hs-(ch2)
p
‑ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(i-4)其中r3和r4各自独立地表示氢原子或甲基,p表示1~6的整数。
[0052]
作为式(i-1)表示的官能团,例如可以举出甲基丙烯酰氧基烷基。作为具有该基团的硅氧烷,例如可以举出以下物质。β-甲基丙烯酰氧基乙基二甲氧基甲基硅烷、γ-甲基丙烯酰氧基丙基甲氧基二甲基硅烷、γ-甲基丙烯酰氧基丙基二甲氧基甲基硅烷、γ-甲基丙烯酰氧基丙基三甲氧基硅烷、γ-甲基丙烯酰氧基丙基乙氧基二乙基硅烷、γ-甲基丙烯酰氧基丙基二乙氧基甲基硅烷、δ-甲基丙烯酰氧基丁基二乙氧基甲基硅烷等。
[0053]
作为式(i-2)表示的官能团,例如可以举出乙烯基苯基。具有该基团的硅氧烷,例如可举出乙烯基苯基乙基二甲氧基硅烷。作为具有式(i-3)所示官能团的硅氧烷,例如可以举出乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷。
[0054]
作为式(i-4)所示的官能团,可以举出巯基烷基。作为具有该基团的硅氧烷,例如可以举出以下物质。γ-巯基丙基二甲氧基甲基硅烷、γ-巯基丙基甲氧基二甲基硅烷、γ-巯基丙基二乙氧基甲基硅烷、γ-巯基丙基乙氧基二甲基硅烷、γ-巯基丙基三甲氧
基硅烷等。
[0055]
硅氧烷系交叉剂可以单独使用1种,或者将2种以上组合使用。作为硅氧烷系交叉剂,出于聚有机硅氧烷(a1)与乙烯基聚合物(a2)复合化时容易形成海岛结构的角度,优选3-甲基丙烯酰氧基丙基甲基二甲氧基硅烷。
[0056]
硅氧烷系交叉剂相对于有机硅氧烷混合物100质量%的比率优选为0.05质量%以上,更优选为0.1质量%以上,进一步优选为0.5质量%以上,另一方面,优选为20质量%以下,更优选为10质量%以下,进一步优选为5质量%以下。如果硅氧烷系接枝交叉剂的比率在上述上限值及上述下限值的范围内,则能够充分形成聚有机硅氧烷(a1)与乙烯基聚合物(a2)的共价键,能够得到冲击强度良好的聚合物粒子群(c)。硅氧烷系交叉剂相对于有机硅氧烷混合物100质量%的比率例如可以为0.05~20质量%,可以为0.1~10质量%,可以为0.5~5质量%。
[0057]
聚有机硅氧烷(a1)的数均粒径优选为1nm以上,更优选为10nm以上,进一步优选为30nm以上,另一方面,优选为500nm以下,更优选为300nm以下,更优选150nm以下,特别优选100nm以下,特别优选80nm以下,最优选60nm以下。如果聚有机硅氧烷(a1)的数均粒径在上述上限值及上述下限值的范围内,则容易将聚合物粒子群(c)的数均粒径调整到上述优选的上限值及下限值的范围内。聚有机硅氧烷(a1)的数均粒径例如可以为1~500nm,可以为1~300nm,可以为10~150nm,可以为10~100nm,可以为30~80nm,可以为30~60nm。
[0058]
由聚有机硅氧烷(a1)的质量平均粒径(nm)/数均粒径(nm)表示的比(以下也称为“dw/dn”。)优选为1.0以上,另一方面,优选为1.7。如果dw/dn在上述上限值及上述下限值范围内,则含有聚合物粒子群(c)和热塑性树脂的树脂组合物的着色性更优异,使用了树脂组合物的成形体的着色外观更优异。
[0059]
聚有机硅氧烷(a1)的数均粒径(dn)的测定方法与上述聚有机硅氧烷(a1)的数均粒径的测定方法相同。聚有机硅氧烷(a1)的质量平均粒径(dw)的测定方法除了测定质量基准的粒度分布代替个数基准的粒度分布以外,与上述聚有机硅氧烷(a1)的数均粒径的测定方法相同。即,将聚有机硅氧烷(a1)的胶乳用去离子水稀释至浓度约3%作为试样,使用毛细管方式的粒度分布计(美国matec公司制造的chdf2000型粒度分布计)测定质量基准的粒度分布,将中位直径作为质量平均粒径。
[0060]
<聚有机硅氧烷(a1)的制造方法>作为聚有机硅氧烷(a1)的制造方法没有特别限制,例如可以采用以下的制造方法。首先,将含有有机硅氧烷、根据需要的硅氧烷系交联剂、根据需要的硅氧烷系交叉剂、及根据需要具有封端基团的硅氧烷低聚物的有机硅氧烷混合物,通过乳化剂和水进行乳化而制备成乳液,在该乳液中,在酸催化剂的存在下使有机硅氧烷混合物在高温下聚合,然后用碱性物质中和酸催化剂,得到聚有机硅氧烷胶乳的制造方法。另外,在以下制造方法的说明中,对使用“有机硅氧烷混合物”作为聚合用原料的情况进行了说明,但对于使用“有机硅氧烷”的情况也可以使用同样的制造工艺。
[0061]
在该制造方法中,作为乳液的配制方法,可以举出使用以高速旋转带来的剪切力
进行微粒化的均质搅拌机的方法、使用以高压发生器的喷出力进行微粒化的均质机等通过高速搅拌进行混合的方法等。这些之中,由于可使聚有机硅氧烷胶乳的粒径分布变狭窄,因而使用均质机的方法是优选的方法。
[0062]
作为聚合时的酸催化剂的混合方法,可举出(1)将有机硅氧烷混合物、乳化剂和水连同酸催化剂一举添加,进行混合的方法;(2)向有机硅氧烷混合物的乳液中,一举添加酸催化剂水溶液的方法;(3)以一定的速度将有机硅氧烷混合物的乳液滴加到高温的酸催化剂水溶液中,进行混合的方法等。其中,由于容易控制聚有机硅氧烷粒径,因此优选将有机硅氧烷混合物的乳液以一定速度滴加到高温的酸催化剂水溶液中进行混合的方法。
[0063]
聚合温度优选为50℃以上,更优选为70℃以上。聚合温度的上限例如为100℃。在将有机硅氧烷混合物的乳液以一定速度滴加到高温的酸催化剂水溶液中进行聚合时,聚合时间通常为2小时以上,优选为5小时以上。
[0064]
进一步地,由于在30℃以下的温度下会进行硅烷醇之间的交联反应,因此为提高聚有机硅氧烷的交联密度,也可以在50℃以上的高温下使其聚合后,将生成的胶乳在30℃以下的温度下保持5小时至100小时左右。
[0065]
有机硅氧烷混合物的聚合反应中,可以用氢氧化钠、氢氧化钾、氨水溶液等碱性物质将包含胶乳的反应体系中和至ph6以上8以下,使聚合反应终止。
[0066]
作为上述制造方法中所使用的乳化剂,只要可将有机硅氧烷混合物进行乳化则没有特别限制,优选阴离子系乳化剂或非离子系乳化剂。作为阴离子系乳化剂,例如可举出烷基苯磺酸钠、烷基二苯醚二磺酸钠、烷基硫酸钠、聚氧乙烯烷基硫酸钠、聚氧乙烯壬基苯基醚硫酸钠等。作为非离子系乳化剂,例如可举出以下物质。聚氧乙烯烷基醚、聚氧乙烯亚烷基烷基醚、聚氧乙烯二苯乙烯化苯基醚、聚氧乙烯三苄基苯基醚、聚氧乙烯聚氧丙烯二醇等。这些乳化剂可单独使用1种,或组合使用2种以上。
[0067]
相对于100质量份有机硅氧烷混合物,乳化剂的使用量优选为0.05质量份以上,更优选为0.1质量份以上,另一方面,优选为20质量份以下,更优选为10质量份以下。根据乳化剂的用量,可以将聚有机硅氧烷胶乳的粒径调整为所希望的值。如果乳化剂的使用量为上述下限值以上,则可以提高有机硅氧烷混合物乳液的乳化稳定性。乳化剂使用量为上述上限值以下时,成形体的耐热变色性及表面外观更优异。
[0068]
作为用于有机硅氧烷混合物的聚合的酸催化剂,可举出脂肪族磺酸、脂肪族取代苯磺酸、脂肪族取代萘磺酸等磺酸类和硫酸、盐酸、硝酸等无机酸类。这些酸催化剂可单独使用1种,或可组合使用2种以上。这些之中,若使用硫酸、盐酸、硝酸等无机酸,则可以使聚有机硅氧烷胶乳的粒径分布变狭窄,进一步地,可以抑制聚有机硅氧烷胶乳中因乳化剂成分所引起的不良情况(成形品的耐热分解性降低以及外观不良)的产生。
[0069]
相对于有机硅氧烷100质量份,酸催化剂的使用量优选为0.005质量份以上40质量份以下。如果酸催化剂的使用量为0.005质量份以上,则可以在短时间内使有机硅氧烷混合物聚合。如果酸催化剂使用量为40质量份以下,则成形体的耐热变色性及表面外观更优异。
[0070]
另外,由于酸催化剂使用量成为决定聚有机硅氧烷(a1)粒径的因素,因此为了得到后述粒径的聚有机硅氧烷(a1),更优选使酸催化剂的使用量相对于100质量份的有机硅氧烷为1质量份以上30质量份以下。
[0071]
在提高机械稳定性的目的下,可以根据需要在通过上述方法得到的聚有机硅氧烷胶乳中添加乳化剂。作为乳化剂,优选与上述例示相同的阴离子系乳化剂、非离子系乳化剂。
[0072]
(乙烯基聚合物(a2))乙烯基聚合物(a2)是乙烯基单体成分(a2)聚合而成的聚合物,由基于乙烯基单体的单元构成。构成乙烯基聚合物(a2)的乙烯基单体成分(a2)由一种以上的乙烯基单体构成。
[0073]
从成形体的冲击强度的观点出发,乙烯基单体成分(a2)优选为单官能(甲基)丙烯酸酯单体(以下也称为“单体(a2-1)”。)。乙烯基单体成分(a2)除了含有单体(a2-1)以外,还可以含有由不同于能够与单体(a2-1)共聚的单体(a2-1)的其他单官能单体(以下也记为“单体(a2-2)”)以及能够与单体(a2-1)共聚的多官能单体(以下也记为“单体(a2-3)”)构成的组中的至少一种。从成形体的冲击强度的观点及将聚合物粒子群(c)中的thf不溶成分的比率设为上述范围内的观点出发,乙烯基单体成分(a2)优选含有单体(a2-1)和单体(a2-3)。
[0074]
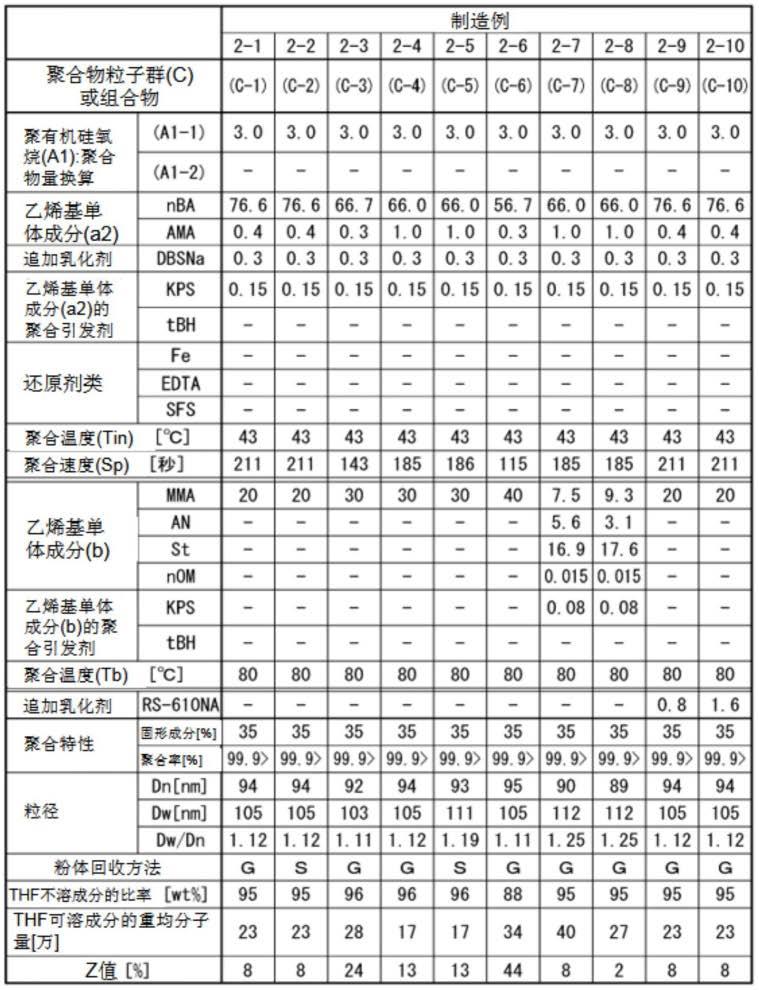
作为单体(a2-1),可以举出例如丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丙酯、丙烯酸正丁酯、丙烯酸2-乙基己酯、丙烯酸正辛酯等丙烯酸烷基酯;甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸正丙酯、甲基丙烯酸正丁酯、甲基丙烯酸异丁酯、甲基丙烯酸叔丁酯、甲基丙烯酸己酯、甲基丙烯酸2-乙基己酯、甲基丙烯酸月桂酯、甲基丙烯酸十三烷基酯、甲基丙烯酸硬脂酯等甲基丙烯酸烷基酯。其中,从成形体冲击强度变得更好出发,单体(a2-1)优选含有选自由丙烯酸乙酯、丙烯酸正丁酯、丙烯酸2-乙基己酯和丙烯酸正辛酯构成的组中的至少一种以上的单体,更优选含有丙烯酸正丁酯。这些单体(a2-1)可以单独使用1种,或者将2种以上组合使用。
[0075]
作为单体(a2-2),例如可以举出苯乙烯、α-甲基苯乙烯等芳香族乙烯基单体、丙烯腈、甲基丙烯腈等氰化乙烯基单体、(甲基)丙烯酰基改性硅酮等各种乙烯基单体。这些单体(a2-2)可以单独使用1种,或者将2种以上组合使用。
[0076]
作为单体(a2-3),例如可以举出乙二醇二甲基丙烯酸酯、丙二醇二甲基丙烯酸酯、1,3-丁二醇二甲基丙烯酸酯、1,4-丁二醇二甲基丙烯酸酯、乙二醇二丙烯酸酯、丙二醇二丙烯酸酯、1,3-丁二醇二丙烯酸酯、1,4-丁二醇二丙烯酸酯、1,6-己二醇二丙烯酸酯、二乙烯基苯、多官能(甲基)丙烯酰基改性硅酮、甲基丙烯酸烯丙酯、三烯丙基氰脲酸酯、三烯丙基异氰脲酸酯等、偏苯三酸三烯丙酯等。其中,由于成形体的冲击强度变得更好,因此单体(a2-3)优选选自由甲基丙烯酸烯丙酯、三烯丙基氰脲酸酯和三烯丙基异氰脲酸酯中的至少一种,更优选甲基丙烯酸烯丙酯。这些单体(a2-3)可以单独使用1种,或者将2种以上组合使用。
[0077]
单体(a2-1)相对于乙烯基单体成分(a2)100质量%的比率,从成形体的冲击强度的观点出发,优选为60质量%以上,更优选为70质量%以上,进一步优选为80质量%以上,特别优选为90质量%以上。单体(a2-1)相对于乙烯基单体成分(a2)100质量%的比率可以为100质量%,但优
选为99.9质量%以下。
[0078]
从成形体的冲击强度的观点出发,单体(a2-2)相对于乙烯基单体成分(a2)100质量%的比例优选为40质量%以下,更优选为30质量%以下,进一步优选为20质量%以下,特别优选为10质量%以下,也可以是0质量%。
[0079]
单体(a2-3)相对于乙烯基单体成分(a2)100质量%的比率优选为0.1质量%以上4质量%以下。从进一步提高成形体的冲击强度的观点出发,单体(a2-3)相对于乙烯基单体成分(a2)100质量%的比率,更优选为0.1质量%以上2质量%以下,进一步优选为0.1质量%以上1质量%以下。从进一步提高成形体的着色外观的观点出发,单体(a2-3)相对于乙烯基单体成分(a2)100质量%的比率,更优选为0.5质量%以上4质量%以下,进一步优选为1质量%以上4质量%以下。从成形时的熔融流动性的观点和取得成形体的着色外观及冲击强度的平衡的观点出发,单体(a2-3)相对于乙烯基单体成分(a2)100质量%的比率,更优选为0.3质量%以上3质量%以下,进一步优选为0.5质量%以上2.5质量%以下。
[0080]
(聚合物(a))聚合物(a)包含聚有机硅氧烷(a1)和乙烯基聚合物(a2)。从成形体的冲击强度的观点出发,聚合物(a)中,由聚有机硅氧烷(a1)/乙烯基聚合物(a2)表示的质量比(以下也称为“a1/a2”。)优选为1/99~60/40,更优选为1/99~40/60,进一步优选为2/98~30/70。
[0081]
《聚合物(a)的制造方法》聚合物(a)的制造方法没有特别限定,但出于成形体的冲击强度更优异的角度,优选在含有聚有机硅氧烷(a1)的胶乳的存在下,使构成乙烯基聚合物(a2)的乙烯基单体成分(a2)聚合的方法。
[0082]
在含有聚有机硅氧烷(a1)的胶乳的存在下使乙烯基单体成分(a2)聚合的方法没有特别限定,可举出(i)在含有聚有机硅氧烷(a1)的胶乳中滴加乙烯基单体成分(a2)进行聚合的方法;(ii)将乙烯基单体成分(a2)的一部分在不开始聚合的条件下添加至含有聚有机硅氧烷(a1)的胶乳中,含浸于聚有机硅氧烷(a1)的粒子中后,开始聚合,然后滴加或一并添加乙烯基单体成分(a2)的剩余部分进行聚合的方法;(iii)在不开始聚合条件下将乙烯基单体成分(a2)的总量添加到含有聚有机硅氧烷(a1)的胶乳中,含浸于聚有机硅氧烷(a1)的粒子中后,进行聚合的方法。
[0083]
作为聚合物(a)制造方法,在上述中,由于成形体的冲击强度更优异,因此优选在不开始聚合的条件下,将乙烯基单体成分(a2)的总量添加到含有聚有机硅氧烷(a1)的胶乳中,含浸在聚有机硅氧烷(a1)的粒子中后聚合方法。
[0084]
作为聚合物(a)的制造方法,特别优选具有以下工序i~iv的制造方法。根据该制造方法,具有将聚有机硅氧烷(a1)作为海成分、将乙烯基聚合物(a2)作为岛成分的海岛结构,在聚有机硅氧烷(a1)区域中具有多个乙烯基聚合物(a2)区域,容易得到能够实现z值小于60%的聚合物粒子群(c)的聚合物(a)。
[0085]
工序i:
在任意条件下制造聚有机硅氧烷(a1)的胶乳。聚有机硅氧烷(a1)的胶乳可以通过上述方法制造。此时,聚有机硅氧烷(a1)优选由有机硅氧烷和硅氧烷系交叉剂构成。
[0086]
工序ii:在乙烯基单体成分(a2)不开始聚合的条件下,将乙烯基单体成分(a2)的总量和自由基聚合引发剂添加至工序i中得到的聚有机硅氧烷(a1)的胶乳中,使其含浸于聚有机硅氧烷(a1)的粒子中。此时,通过添加全部量的乙烯基单体成分(a2),成形体的冲击强度更优异。
[0087]
在乙烯基单体成分(a2)含有多种乙烯基单体的情况下,例如含有单体(a2-1)和由选自单体(a2-2)和单体(a2-3)构成的组中的至少一种的情况下,这些单体的添加方法没有特别限定。例如,可以同时添加单体(a2-1)和单体(a2-2)和/或单体(a2-3),也可以分别添加单体(a2-1)及单体(a2-2)和/或单体(a2-3)。乙烯基单体成分(a2)含有单体(a2-3)时,从得到适当交联结构的观点出发,优选与单体(a2-1)和/或单体(a2-2)混合添加。
[0088]
作为自由基聚合引发剂,没有特别限定,例如可以举出偶氮化合物、过氧化物、二卤化物等。它们可以单独使用1种,或者将2种以上组合使用。
[0089]
作为偶氮系引发剂,例如可举出如下的物质。2,2
’‑
偶氮双异丁腈、二甲基-2,2
’‑
偶氮双(2-甲基丙酸酯)、2,2
’‑
偶氮双(2,4-二甲基戊腈)、2,2
’‑
偶氮双(2-丁腈)等油溶性偶氮系引发剂;4,4
’‑
偶氮双(4-氰基戊酸)、2,2
’‑
偶氮双[n-(2-羧基甲基)-2-甲基丙基脒]水合物、2,2
’‑
偶氮双-(n,n
’‑
二亚甲基异丁基脒)二盐酸盐、2,2
’‑
偶氮双[2-(2-咪唑啉-2-基)丙烷]二盐酸盐等水溶性偶氮系引发剂等。它们可以单独使用1种,或者将2种以上组合使用。
[0090]
使用偶氮化合物作为自由基聚合引发剂时,相对于待聚合的单体的合计100质量%,偶氮化合物的使用量优选为0.01质量%以上,另一方面,优选为3质量%以下,更优选为1质量%以下,进一步优选为0.5质量%以下。通过使偶氮化合物使用量在所述上限值及所述下限值的范围内,可以抑制聚合速度过度提高,容易构筑所述海岛结构。
[0091]
作为过氧化物,例如可以举出以下的过氧化物。过氧化氢、过硫酸钾、过硫酸氨等无机过氧化物;过氧化氢二异丙苯、过氧化氢对薄荷烷、过氧化氢异丙苯、叔丁基过氧化氢、过氧化丁二酸、过氧化新癸酸叔丁酯、过氧化新庚酸叔丁酯、过氧化新戊酸叔丁酯、过氧化-2-乙基己酸-1,1,3,3-四甲基丁酯、过氧化-2-乙基己酸叔丁酯等有机过氧化物等。它们可以单独使用1种,或者将2种以上组合使用。其中,出于乳液聚合时处理变得容易,10小时半衰期温度优选在25℃~105℃之间。
[0092]
使用过氧化物作为自由基聚合引发剂时,过氧化物使用量相对于待聚合的单体的合计100质量%优选为0.01质量%以上,另一方面,优选为1质量%以下,更优选为0.5质量%以下,进一步优选为0.2质量%以下。通过使过氧化物使用量在所述上限值及所述下限值的范围内,能够抑制聚合速度过度提高,容易构筑所述海岛结构。
[0093]
作为自由基聚合引发剂,出于聚合速度容易控制,成形体的冲击强度更优异的角度,优选过氧化物。
[0094]
在使用过氧化物作为自由基聚合引发剂情况下,为了促进过氧化物的分解,可以
并用还原剂。
[0095]
作为还原剂,可以举出亚硫酸盐、亚硫酸氢盐、碱金属亚硫酸氢盐、丙酮亚硫酸氢盐、碱金属亚硫酸盐、焦亚硫酸盐及其盐等硫化合物;硫代硫酸盐、亚磺酸、羟基烷基亚磺酸、羟基甲基亚磺酸及2-羟基-2-磺基乙酸、甲脒亚磺酸、丙基亚磺酸、异丙基亚磺酸、苯基亚磺酸等有机硫化合物;甲醛次硫酸及其盐、羟胺、羟胺硫酸氢盐、羟铵盐、多胺、二甲基苯胺等还原性氮化合物;山梨糖、果糖、葡萄糖、乳糖、无水葡萄糖等还原糖类;抗坏血酸和异抗坏血酸等烯二醇等。作为“盐”,可举出例如钠离子、钾离子、铵离子和锌离子。
[0096]
另外,存在于周期表的第3族~第11族之间的过渡金属的硫酸盐、硝酸盐、乙酸盐、碳酸盐及氯化物也可用作还原剂。作为过渡金属,可以举出第3族的ce等、第4族的ti等、第5族的v等、第6族的cr、mo等、第7族的mn等、第8族的fe等、第9族的co等、第10族的ni等、第11族的cu、ag等。
[0097]
作为还原剂,基于工业上容易得到,成形体的耐热变色性及表面外观更优异的角度,优选选自由甲醛次硫酸钠、l(+)-酒石酸、二亚硫酸钠、异抗坏血酸钠及l-抗坏血酸、硫酸亚铁构成的组中的至少一种。
[0098]
还原剂的使用量优选为乙烯基单体成分(a2)的聚合中使用的过氧化物的2.0摩尔当量以下,更优选为1.0摩尔当量以下,进一步优选为0.6摩尔当量以下,还可以为0摩尔当量。通过使还原剂使用量为所述上限值以下,能够抑制聚合速度过度提高,容易构筑所述海岛结构。
[0099]
使用过渡金属盐作为还原剂时,为了提高其反应性,可以并用螯合剂。作为螯合剂,可以使用含有能够与对象过渡金属原子形成配位键的2个以上的供电子原子的化合物,可举出例如乙二胺四乙酸、羟乙基乙二胺三乙酸、硝基三乙酸、柠檬酸、酒石酸、葡糖酸、5-磺基水杨酸、乙二胺、二亚乙基三胺、三亚乙基四胺、三氨基三乙胺、三乙醇胺、n-羟乙基乙二胺、草酸钠及它们的金属盐等。其中,出于聚合稳定性优异的角度,优选乙二胺四乙酸及其金属盐。
[0100]
从聚合反应性控制方面出发,螯合剂的用量相对于还原剂优选为0.5摩尔当量以上,更优选为1.0摩尔当量以上,另一方面,优选为5.0摩尔当量以下,更优选为2.5摩尔当量以下。
[0101]
在步骤ii中,根据需要,可以向聚有机硅氧烷(a1)的胶乳中添加水系介质。作为水系介质,可以举出水、以及水与有机溶剂的混合介质。作为混合介质中的有机溶剂,只要可以与水混合即可,例如可以举出甲醇、乙醇。
[0102]
在步骤ii中,根据需要,可以向聚有机硅氧烷(a1)的胶乳中添加乳化剂。作为乳化剂,没有特别限制,可以使用与在上述聚有机硅氧烷(a1)的制造中使用的乳化剂相同的乳化剂。其中,优选阴离子系乳化剂或非离子系乳化剂。另外,也可以在工序ii中不添加乳化剂,仅用聚有机硅氧烷(a1)胶乳中所含的乳化剂进行聚合。
[0103]
当乙烯基单体成分(a2)聚合时,可以使用链转移剂。作为链转移剂,可以举出正十二烷基硫醇、叔十二烷基硫醇、正辛基硫醇、正十四烷基硫醇、正己基硫醇、正丁基硫醇等硫醇类;四氯化碳、溴化乙烯等卤素化合物;α-甲基苯乙烯二聚体等。这些链转移剂可以单独使用1种,也可以并用2种以上。
[0104]
相对于100质量%的乙烯基单体成分(a2),链转移剂的使用量优选为1.0质量%以下,也可以为0质量%。通过使链转移剂使用量为1.0质量%以下,可以抑制聚合物粒子群(c)中thf不溶成分比率的降低,成形体的冲击强度更优异。
[0105]
工序iii:对于在工序ii中添加了乙烯基单体成分(a2)和自由基聚合引发剂的聚有机硅氧烷(a1)的胶乳,在满足下述式(a)的条件下,使乙烯基单体成分(a2)开始聚合。t
10
》(t
in
+10)
···
(a)在此,t
10
表示所使用的自由基聚合引发剂的10小时半衰期温度,t
in
表示使乙烯基单体成分(a2)开始聚合的温度。组合使用多个自由基聚合引发剂时,使用表示最低10小时半衰期温度的自由基聚合引发剂的t
10
值。
[0106]
满足式(a)换言之即通过使乙烯基单体成分(a2)开始聚合的温度t
in
(以下称为聚合温度(t
in
))为比自由基聚合引发剂的10小时半衰期温度t
10
低10℃以上的温度,能够抑制聚合速度,容易构筑上述聚有机硅氧烷(a1)和乙烯基聚合物(a2)复合的海岛结构,可以得到能够使得到的成形体的冲击强度更好的聚合物粒子群(c)。聚合温度(t
in
)优选为比自由基聚合引发剂的10小时半衰期温度t
10
低15℃以上的温度,更优选为低20℃以上的温度。聚合温度(t
in
)的下限值没有特别限定,从聚合开始的稳定性的观点出发,优选不比自由基聚合引发剂的10小时半衰期温度t
10
低50℃以上。聚合时间根据聚合温度(t
in
)而不同,例如为0.1~30小时。
[0107]
10小时半衰期温度t
10
是指使用的自由基聚合引发剂的50摩尔%在10小时内热分解时的温度。例如,关于在下述式(b)和下述式(c)中代入自由基聚合引发剂转化率x=50[%]、时间t=36000[s](=10小时)、气体常数r=8.314[j/kmol]、频率因子a及活化能e代入文献值或计算值,由此可以计算自由基聚合引发剂的10小时半衰期温度t
10
。作为10小时半衰期温度t
10
,也可以使用文献值。
[0108]
x=100
×
exp(-kdt)
···
(b)kd=a
×
exp(-e/rt)
···
(c)(x[%]:转化率、kd[1/s]:反应速度、t[s]:时间、a[1/s]:频率因子、e[j/mol]:活化能、r[j/kmol]:气体常数、t[k]:温度)例如,过硫酸钾的10小时半衰期温度为67℃,叔丁基过氧化氢(商品名perbutylh69、日本油脂公司制)的10小时半衰期温度为167℃。
[0109]
工序iv:在步骤iii中使聚合开始后的聚合行为中,乙烯基单体成分(a2)聚合所达到的体系内的最高温度设为tp(℃),体系内的温度达到t
in
+1(℃)之后、达到t
in
+{(t
p-t
in
)/3}(℃)所需的时间设为s
p
(秒)(以下称为聚合速度(s
p
))时,以满足以下式(d)的方式进行聚合。s
p
≥80
···
(d)
[0110]
通过使聚合速度(s
p
)为80秒以上,容易构筑上述聚有机硅氧烷(a1)和乙烯基聚合物(a2)复合的海岛结构,可以得到能够使得到的成形体的冲击强度更良好的聚合物粒子群(c)。聚合速度(s
p
)更优选为100秒以上,进一步优选为120秒以上,特别优选为150秒以上。聚合速度(s
p
)除了上述自由基聚合引发剂种类、量、聚合开始温度t
in
以外,还可以
通过聚合时的慢热系统进行调整。
[0111]
(乙烯基聚合物(b))乙烯基聚合物(b)是乙烯基单体成分(b)聚合而成的聚合物,由基于乙烯基单体的单元构成。构成乙烯基聚合物(b)的乙烯基单体成分(b)由一种以上的乙烯基单体构成。作为构成乙烯基单体成分(b)的乙烯基单体,没有特别限定,例如可以举出(甲基)丙烯酸酯单体、芳香族乙烯基单体、氰化乙烯基单体等各种乙烯基单体。
[0112]
作为(甲基)丙烯酸酯单体,可以举出甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸正丁酯、甲基丙烯酸异丁酯等甲基丙烯酸烷基酯;丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丁酯等丙烯酸烷基酯等。作为芳香族乙烯基单体,可以举出苯乙烯、烷基取代苯乙烯(对甲基苯乙烯、间甲基苯乙烯、邻甲基苯乙烯、2,4-二甲基苯乙烯、2,5-二甲基苯乙烯、3,4-二甲基苯乙烯、3,5-二甲基苯乙烯、对乙基苯乙烯、间乙基苯乙烯、邻乙基苯乙烯等)、烷基取代异丙烯基苯(异丙烯基苯(α-甲基苯乙烯)、异丙烯基甲苯、异丙烯基乙基苯、异丙烯基丙基苯、异丙烯基丁基苯、异丙烯基戊基苯、异丙烯基己基苯、异丙烯基辛基苯等)、1,1-二苯基乙烯等。其中,基于能够抑制碎料产生的角度,优选苯乙烯和α-甲基苯乙烯。作为氰化乙烯基单体,可以举出丙烯腈、甲基丙烯腈等。它们可以单独使用1种,或者将2种以上组合使用。
[0113]
从成形体的耐候性更优异的观点出发,乙烯基单体成分(b)优选含有选自由(甲基)丙烯酸酯单体和芳香族乙烯基单体构成的组中的至少一种。相对于乙烯基单体成分(b)100质量%,(甲基)丙烯酸酯单体和芳香族乙烯基单体合计的比率优选为50质量%以上。
[0114]
乙烯基单体成分(b)在(甲基)丙烯酸酯单体和芳香族乙烯基单体中,基于成形体的耐候性特别优异的角度,更优选含有甲基丙烯酸甲酯和苯乙烯中的任一者或双者。相对于乙烯基单体成分(b)100质量%,甲基丙烯酸甲酯和苯乙烯合计的比率优选为50质量%以上。
[0115]
乙烯基单体成分(b)从聚合物粒子群(c)在热塑性树脂中的分散性及成形体的耐候性更优异的角度出发,优选含有(甲基)丙烯酸酯单体。相对于乙烯基单体成分(b)100质量%,(甲基)丙烯酸酯单体的比率优选为50质量%以上。
[0116]
乙烯基单体成分(b)在(甲基)丙烯酸酯单体中,基于聚合物粒子群(c)在热塑性树脂中的分散性及成形体的耐候性特别优异的角度,更优选含有甲基丙烯酸甲酯。相对于乙烯基单体成分(b)100质量%,甲基丙烯酸甲酯的比率更优选为50质量%以上。
[0117]
乙烯基聚合物(b)的玻璃化转变温度(以下也称为“tg”。)优选70℃以上,更优选80℃以上,进一步优选90℃以上,另一方面,优选105℃以下。如果乙烯基聚合物(b)的tg为上述下限值以上,则聚合物粒子群(c)的粉体特性(粉体的流动性或粒径)良好。乙烯基聚合物(b)tg可以根据构成乙烯基单体成分(b)的乙烯基单体的种类和比率来调整。
[0118]
乙烯基聚合物(b)的tg由fox式求得。此时,构成乙烯基单体成分(b)的乙烯基单体的均聚物的tg例如可以使用“聚合物手册(polymer handbook)”(wiley interscience公司/1999年)中记载的值。该文献中没有记载的乙烯基单体均聚物的tg可以使用bicerano方法“聚合物性能预测(prediction of polymer properties)”(marcel dekker公司/2002年)算出。
[0119]
(聚合物粒子群(c)的制造方法)聚合物粒子群(c)例如可以通过在聚合物(a)的存在下使乙烯基单体成分(b)聚合(接枝聚合)的方法来制造。由此,得到在聚合物(a)上接枝了乙烯基聚合物(b)的一部分或全部的聚合物。
[0120]
作为聚合物粒子群(c)的制造方法,优选在聚合物(a)的胶乳中添加乙烯基单体成分(b),在胶乳中使乙烯基单体成分(b)聚合的方法。如上所述,聚合物(a)的胶乳优选通过在含有聚有机硅氧烷(a1)的胶乳的存在下使乙烯基单体成分(a2)聚合来制造。
[0121]
使乙烯基单体成分(b)聚合的温度(以下,也称为“聚合温度(tb)”。)没有特别限定,可以适用惯用条件,例如可以举出45~95℃、聚合时间为0.1~10小时的条件。
[0122]
向聚合物(a)的胶乳中添加乙烯基单体成分(b)的方法没有特别限定,但基于抑制碎料的产生,聚合物(a)和乙烯基单体成分(b)的接枝化率良好的角度,优选滴加添加。此时,既可以连续滴加乙烯基单体成分(b)的总量,也可以在其间设置不滴加乙烯基单体成分(b)的保持时间而分多次滴加。在乙烯基单体成分(b)由多种乙烯基单体构成的情况下,连续滴加乙烯基单体成分(b)的总量的方法没有特别限定,可以举出连续添加相同组成的混合物的方法、如机械进料聚合那样连续地使组成变化的同时添加的方法等。在乙烯基单体成分(b)由多种乙烯基单体构成情况下,作为在其间设置保持时间而分多次滴加添加的方法,可以举出将相同组成的混合物分多次添加的方法、将各成分单体和/或不同组成的混合物分多次添加的方法。
[0123]
乙烯基单体成分(b)含有(甲基)丙烯酸酯单体、芳香族乙烯基单体和氰化乙烯基单体时,优选使(甲基)丙烯酸酯单体聚合,接着使芳香族乙烯基单体和氰化乙烯基单体聚合。通过用该方法聚合,聚合后进行粉体回收工序而得到的聚合物粒子群(c)的粉体特性(粉体的流动性或粒径)良好。
[0124]
乙烯基单体成分(b)通过与基于聚有机硅氧烷(a1)中所含硅氧烷系交叉剂的单元和/或基于乙烯基聚合物(a2)中所含的单体(a2-3)的单元化学键合,能够形成与聚合物(a)的接枝聚合物。
[0125]
为了提高该接枝化的效率,在添加乙烯基单体成分(b)之前,例如可以使乙二醇二甲基丙烯酸酯、丙二醇二甲基丙烯酸酯、1,3-丁二醇二甲基丙烯酸酯、1,4-丁二醇二甲基丙烯酸酯、乙二醇二丙烯酸酯、丙二醇二丙烯酸酯、1,3-丁二醇二丙烯酸酯、1,4-丁二醇二丙烯酸酯、1,6-己二醇二丙烯酸酯、二乙烯基苯、多官能(甲基)丙烯酰基改性硅酮、甲基丙烯酸烯丙酯、三烯丙基氰脲酸酯、三烯丙基异氰脲酸酯等、偏苯三酸三烯丙酯等多官能单体预先聚合。
[0126]
聚合乙烯基单体成分(b)时使用的乳化剂没有特别限制,可以使用与聚有机硅氧烷(a1)的制造和/或乙烯基聚合物(a2)的制造中使用的乳化剂相同的乳化剂,但优选阴离
子系乳化剂或非离子系乳化剂。另外,在使乙烯基单体成分(b)聚合时不追加特殊的乳化剂,而仅用乙烯基聚合物(a2)胶乳中所含乳化剂进行聚合。
[0127]
聚有机硅氧烷(a1)制造、乙烯基聚合物(a2)的制造及乙烯基单体成分(b)聚合时使用的乳化剂的总量,相对于形成聚合物粒子群(c)的所有单体的合计100质量份,优选为0.05质量份以上,更优选为0.1质量份以上,另一方面,优选为10质量份以下,更优选为5质量份以下。根据乳化剂的总量,可以将聚合物粒子群(c)胶乳的粒径调整为所希望的值。如果乳化剂的总量为上述下限值以上,则可以充分提高聚有机硅氧烷(a1)胶乳、聚合物(a)胶乳、聚合物(c)胶乳各自的稳定性。如果乳化剂总量为上述上限值以下,则能够充分降低聚合物粒子群(c)粉体中残留的乳化剂的量,能够抑制使用了含有聚合物粒子群(c)和热塑性树脂的树脂组合物的成形体的耐热分解性及表面外观的降低。
[0128]
在聚合乙烯基单体成分(b)时,也可以以thf可溶成分的调整、分子量的调整等为目的使用链转移剂。作为链转移剂,可以举出正十二烷基硫醇、叔十二烷基硫醇、正辛基硫醇、正十四烷基硫醇、正己基硫醇、正丁基硫醇等硫醇类;四氯化碳、溴化乙烯等卤素化合物;α-甲基苯乙烯二聚体等。这些链转移剂可以单独使用1种,也可以并用2种以上。
[0129]
链转移剂的使用量相对于乙烯基单体成分(b)100质量%优选为2.0质量%以下,也可以为0质量%。通过使链转移剂使用量为2.0质量%以下,聚合物粒子群(c)的thf不溶成分的比率的降低被抑制,成形体的冲击强度更优异。
[0130]
也可以将乙烯基单体成分(b)聚合后,从得到的聚合物粒子群(c)的胶乳中将聚合物粒子群(c)作为粉体回收。将聚合物粒子群(c)作为粉体回收时,可以使用喷雾干燥法等直接干燥法或凝聚法。在凝聚法中,通过絮凝后的清洗工序,可以降低聚合时使用的乳化剂及其絮凝盐、引发剂等所得粉体中所含的聚合助剂残留物。另一方面,在直接干燥法中,可以使聚合时添加的助剂类大致残留在得到的粉体中。在将聚合物粒子群(c)添加到热塑性树脂中时,为了形成优选的残留状态,可以适当选择这些粉体回收法。
[0131]
喷雾干燥法是将聚合物粒子群(c)胶乳以微小液滴状喷雾到干燥机中,将干燥用的加热气体与其接触而进行干燥的方法。作为产生微小液滴的方法,例如可以举出旋转圆盘型式、压力喷嘴式、双流体喷嘴式、加压双流体喷嘴式。干燥机的容量可以是从实验室使用的小规模容量到工业使用的大规模容量中的任意一种。干燥用加热气体的温度优选为200℃以下,更优选为120~180℃。也可以将单独制备的两种以上的接枝共聚物的胶乳一起喷雾干燥。进一步,为了提高喷雾干燥时的粘连、体积密度等粉末特性,也可以在聚合物粒子群(c)胶乳中添加二氧化硅等任意成分进行喷雾干燥。
[0132]
凝聚法是对聚合物粒子群(c)胶乳进行絮凝,将聚合物粒子群(c)分离、回收并干燥的方法。首先,在溶解有凝聚剂的热水中投入聚合物(c)胶乳,进行盐析并凝聚,由此分离聚合物粒子群(c)。接着,对分离出的湿润状的聚合物粒子群(c)进行脱水等,回收水分量降低的聚合物粒子群(c)。回收的聚合物粒子群(c)使用压榨脱水机或热风干燥机进行干燥。
[0133]
作为凝聚剂,可以举出氯化铝、硫酸铝、硫酸钠、硫酸镁、硝酸钠、乙酸钙等无机盐;硫酸等酸等,特别优选乙酸钙。这些凝聚剂可以单独使用1种,或者将2种以上组合使用。
[0134]
上述凝聚剂通常作为水溶液使用。从使聚合物粒子群(c)稳定地凝聚、回收的观点
出发,凝聚剂水溶液的浓度优选为0.1质量%以上,特别优选为1质量%以上。另外,从减少回收的聚合物粒子群(c)中残留的凝聚剂的量来防止成形体的成形外观降低的观点出发,凝聚剂水溶液的浓度优选为20质量%以下,特别优选为15质量%以下。凝聚剂水溶液量没有特别限定,优选相对于聚合物粒子群(c)胶乳100质量份为10质量份以上、500质量份以下。
[0135]
使聚合物粒子群(c)胶乳与凝聚剂水溶液接触的方法没有特别限定,通常可以举出下述方法。(1)搅拌凝聚剂水溶液的同时在其中连续添加胶乳并保持一定时间的方法,(2)将凝聚剂水溶液和胶乳以一定的比例连续地注入带搅拌机的容器内并使其接触,从容器中连续地抽出含有絮凝的聚合物和水的混合物的方法。使胶乳与凝聚剂水溶液接触时的温度没有特别限定,优选为30℃以上、100℃以下。接触时间没有特别限定。
[0136]
絮凝后的聚合物粒子群(c)用聚合物粒子群(c)的1~100质量倍左右的水进行清洗,过滤。过滤后的湿润状的聚合物粒子群(c)使用流动干燥机或压榨脱水机等进行干燥。干燥温度、干燥时间可以根据得到的聚合物粒子群(c)适当决定。另外,也可以不回收从压榨脱水机或挤出机排出聚合物粒子群(c),直接送到制造树脂组合物的挤出机或成形机,与热塑性树脂混合而得到成形体。
[0137]
〔组合物〕本发明的一个实施方式涉及的组合物(以下也称为“本组合物”)包含选自由聚合物粒子群(c)、磷酸化合物及其碱金属盐构成的组中的至少一种成分(以下也称作“成分(d)”)。成分(d)使含有聚合物粒子群(c)的树脂组合物可塑化,形成时的树脂组合物的流动性提高。另外,能抑制树脂组合物分子量的降低、提高成形稳定性。
[0138]
作为成分(d)中的磷酸化合物,可以举出聚氧化烯烷基醚磷酸等烷基磷酸;聚氧化烯烷基苯基醚磷酸等烷基芳基磷酸等。在聚氧化烯烷基苯基醚磷酸和聚氧化烯烷基醚磷酸中,作为聚氧化烯基,可以举出聚氧乙烯基等,优选聚氧乙烯基。聚氧乙烯基中的氧乙烯单元的单元数例如为2~14,优选为2~10,更优选为2~8,进一步优选为2~6。烷基的碳原子数例如为1~20,优选为5~18,更优选为7~16,进一步优选为10~16。作为碱金属盐,可以举出钠盐、钾盐等。
[0139]
作为成分(d),从容易调整后述的磷原子含量的观点出发,优选磷酸化合物的碱金属盐,更优选烷基磷酸的碱金属盐和烷基芳基磷酸的碱金属盐。其中,从成形时树脂组合物的流动性和成形稳定性的观点出发,优选聚氧化烯烷基苯基醚磷酸的碱金属盐、聚氧化烯烷基醚磷酸的碱金属盐,更优选聚氧化烯烷基醚磷酸的碱金属盐。作为聚氧化烯烷基苯基醚磷酸的碱金属盐,优选聚氧乙烯烷基苯基醚磷酸的碱金属盐。作为聚氧化烯烷基醚磷酸的碱金属盐,优选聚氧乙烯烷基醚磷酸的碱金属盐。其中,优选聚氧乙烯烷基醚磷酸的碱金属盐。这些化合物可以单独使用1种,或者将2种以上组合使用。
[0140]
本组合物中的成分(d)的含量考虑相对于聚合物粒子群(c)和成分(d)的合计100质量%的成分(d)中所含的磷原子的比率(以下也称为“磷含量”)而设定。从成形时树脂组合物的流动性和成形稳定性的观点出发,磷含量优选为10质量ppm以上,更优选为50质量ppm以上,进一步优选为100质量ppm以上,特别优选为200质量ppm以上,最优选300质量ppm以上。磷原子含量上限没有特别限定,例如为2000质量ppm以下,优选为1500质量ppm以下。
[0141]
所述组合物可以进一步包含除成分(d)以外的其它乳化剂。作为其他乳化剂,没有特别限制,例如可以使用与在聚合物粒子群(c)的制造中使用的乳化剂相同的乳化剂。聚合物粒子群(c)和成分(d)的合计比率相对于本组合物100质量%优选为90质量%以上,更优选为95质量%以上,进一步优选为98质量%以上。
[0142]
本组合物的一个优选方式是从胶乳中回收粉体,所述乳胶包含聚合物粒子群(c)和乳化剂,且乳化剂的至少一部分为所述磷酸化合物的碱金属盐。该组合物例如可以如下而制造:在聚合物粒子群(c)胶乳(也可以含有其他乳化剂)中添加磷酸化合物的碱金属盐,进行粉体回收,或者在聚合物粒子群(c)的聚合过程(聚合物(a)或乙烯基聚合物(b)的聚合时)中添加磷酸化合物的碱金属盐,并回收粉体。作为粉体回收方法,可以举出与从上述聚合物粒子群(c)胶乳中以粉体的形式回收聚合物粒子群(c)的方法相同的方法。但是,本组合物不限于此。例如,也可以是将聚合物粒子群(c)粉体和成分(d)混合而成的物质。
[0143]
〔树脂组合物〕本发明的一个方式的树脂组合物(以下,也称为“本树脂组合物”)包含聚合物粒子群(c)和热塑性树脂(以下也称为“热塑性树脂(e)”)。本发明的树脂组合物可以含有本组合物替代聚合物粒子群(c)。此时,本树脂组合物含有聚合物粒子群(c)、成分(d)和热塑性树脂(e)。
[0144]
作为热塑性树脂(e),没有特别限定,例如可以举出工程塑料(芳香族聚碳酸酯等)、苯乙烯系树脂、聚酯树脂、烯烃系树脂(聚乙烯等)、热塑性弹性体、生物降解性树脂、卤素系树脂(氯乙烯树脂等)、丙烯酸系树脂等。
[0145]
作为工程塑料,可以没有特别限制地使用公知的各种热塑性工程塑料。作为工程塑料,可以示例为聚苯醚、聚碳酸酯、聚酯系聚合物(聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯等)、间规聚苯乙烯、尼龙系聚合物(6-尼龙、6,6-尼龙等)、聚芳酯、聚苯硫醚、聚醚酮、聚醚醚酮、聚砜、聚醚砜、聚酰胺酰亚胺、聚醚酰亚胺、聚缩醛。
[0146]
另外,耐热性高度优异、需要熔融流动性的耐热abs等特殊的苯乙烯系树脂或耐热丙烯酸系树脂等也可以作为本发明中的工程塑料例示。其中,在更需要显示强度性能的情况下,更优选芳香族聚碳酸酯或聚对苯二甲酸丁二醇酯。作为芳香族聚碳酸酯,例如可以举出4,4
’‑
二羟基二苯基-2,2-丙烷(即双酚a)系聚碳酸酯等4,4
’‑
二氧基二芳基烷烃系聚碳酸酯。
[0147]
作为烯烃系树脂,可以举出高密度聚乙烯、中密度聚乙烯、低密度聚乙烯、乙烯和其他α-烯烃的共聚物;聚丙烯、丙烯和其他α-烯烃的共聚物;聚丁烯、聚-4-甲基-1-戊烯等。
[0148]
作为热塑性弹性体,可以举出苯乙烯系弹性体、聚氨酯系弹性体、聚烯烃系弹性
体、聚酰胺系弹性体、氟系弹性体、氯化pe系弹性体、丙烯酸系弹性体等。
[0149]
作为苯乙烯系弹性体,可以举出苯乙烯-丁二烯-苯乙烯共聚物(sbs)、苯乙烯-异戊二烯-苯乙烯共聚物(sis)、苯乙烯-乙烯
·
丁烯共聚物(seb)、苯乙烯-乙烯
·
丙烯共聚物(sep)、苯乙烯-乙烯
·
丁烯-苯乙烯共聚物(sebs)、苯乙烯-乙烯
·
丙烯-苯乙烯共聚物(seps)、苯乙烯-乙烯
·
乙烯
·
丙烯-苯乙烯共聚物(seeps)、苯乙烯-丁二烯
·
丁烯-苯乙烯共聚物(苯乙烯-丁二烯-苯乙烯共聚物的部分氢化物:sbbs)、苯乙烯-异戊二烯-苯乙烯共聚物的部分氢化物、苯乙烯-异戊二烯
·
丁二烯-苯乙烯共聚物的部分氢化物等。
“‑”
表示形成由
“‑”
连接的单元的单体共聚而成者,“·”表示共聚后通过氢化等随机改性而存在。
[0150]
作为聚氨酯弹性体,可以举出高分子二醇与有机二异氰酸酯与扩链剂的反应产物。作为高分子二醇,可以举出聚酯二醇、聚醚二醇、聚酯醚二醇、聚碳酸酯二醇、聚酯聚碳酸酯二醇等。作为有机二异氰酸酯,可以举出4,4
’‑
二苯基甲烷二异氰酸酯、甲苯二异氰酸酯、对苯二异氰酸酯、苯二甲基二异氰酸酯、萘二异氰酸酯、氢化4,4
’‑
二苯基甲烷二异氰酸酯(4,4
’‑
二环己基甲烷二异氰酸酯)、异佛尔酮二异氰酸酯、六亚甲基二异氰酸酯等。在这些有机二异氰酸酯中,优选4,4
’‑
二苯基甲烷二异氰酸酯。作为扩链剂,可以举出乙二醇、二乙二醇、1,4-丁二醇、1,5-戊二醇、2-甲基-1,3-丙二醇、1,6-己二醇、新戊二醇、1,9-壬二醇、环己烷二醇、1,4-双(β-羟基乙氧基)苯等。
[0151]
作为聚烯烃系弹性体,可以举出乙烯-丙烯橡胶、乙烯-丙烯-二烯橡胶、乙烯-乙酸乙烯酯共聚物、丁基橡胶、丁二烯橡胶、丙烯-丁烯共聚物、乙烯-丙烯酸酯共聚物等。
[0152]
作为苯乙烯系树脂,可以举出聚苯乙烯、丙烯腈-苯乙烯共聚物、丙烯腈-苯乙烯-α-甲基苯乙烯共聚物、丙烯腈-甲基丙烯酸甲酯-苯乙烯-α-甲基苯乙烯共聚物、abs树脂、as树脂、mabs树脂、mbs树脂、aas树脂、aes树脂、丙烯腈-丁二烯-苯乙烯-α-甲基苯乙烯共聚物、丙烯腈-甲基丙烯酸甲酯-丁二烯-苯乙烯-α-甲基苯乙烯共聚物、苯乙烯-马来酸酐共聚物、苯乙烯-马来酰亚胺共聚物、苯乙烯-n-取代马来酰亚胺共聚物、丙烯腈-苯乙烯-n-取代马来酰亚胺共聚物、丙烯腈-丁二烯-苯乙烯-β-异丙烯基萘共聚物及丙烯腈-甲基丙烯酸甲酯-丁二烯-苯乙烯-α-甲基苯乙烯-马来酰亚胺共聚物等。
[0153]
聚酯树脂是多元酸和多元醇的聚合物,以具有热塑性为条件没有特别限定。作为多元酸,可以举出对苯二甲酸、萘二羧酸、环己基二羧酸及其酯等。多元醇包括乙二醇、丙二醇、丁二醇、戊二醇、新戊二醇、己二醇、辛二醇、癸二醇、环己烷二甲醇、对苯二酚、双酚a、2,2-双(4-羟基乙氧基苯基)丙烷、1,4-二羟甲基四溴苯、四溴双酚a双(2-羟基乙基)醚(tba-eo)等。聚酯系树脂可以是均聚物、共聚物或这两种以上的混合物。作为聚酯系树脂,也可以使用eastman chemical公司制的商品名“petg”等的市售品。
[0154]
作为生物降解性树脂,可以举出微生物系聚合物、化学合成死聚合物、天然物系聚合物等。作为微生物系聚合物,可以举出聚羟基丁酸酯/戊酸酯(phb/v)等生物聚酯、细菌纤维素、微生物多糖(普鲁兰、凝胶多糖(curdlan)等)等。
作为化学合成系聚合物,可以举出脂肪族聚酯(聚己内酯、聚丁二酸丁二醇酯、聚丁二酸乙二醇酯、聚乙醇酸、聚乳酸等)、聚乙烯醇、聚氨基酸类(pmlg等)等。作为天然物系聚合物,可以举出壳聚糖、纤维素、淀粉、乙酸纤维素等。
[0155]
作为卤素系树脂,例如可以举出氯乙烯均聚物、以80质量%以上的比例含有氯乙烯的共聚物、高氯化聚氯乙烯等氯乙烯树脂。作为共聚物的成分,除了氯乙烯以外,还可以例示乙烯、乙酸乙烯酯、甲基丙烯酸甲酯和丙烯酸丁酯等单亚乙烯化合物。在共聚物中,这些化合物以其总量计可以以20质量%以下的比例含有。作为卤素系树脂,除了氯乙烯树脂以外,还可以举出氟化聚合物、溴化聚合物、碘化聚合物等。
[0156]
作为丙烯酸系树脂,例如可以举出甲基丙烯酸甲酯与可共聚的乙烯基单体聚合而成的共聚物等。作为可共聚乙烯基单体,可以举出丙烯酸甲酯、丙烯酸乙酯、丙烯酸异丙酯、丙烯酸正丁酯、丙烯酸2-乙基己酯等丙烯酸烷基酯、甲基丙烯酸乙酯、甲基丙烯酸丙酯、甲基丙烯酸正丁酯等甲基丙烯酸烷基酯、苯乙烯、α-甲基苯乙烯、甲基苯乙烯等芳香族乙烯基化合物等。
[0157]
聚苯醚、聚碳酸酯、聚对苯二甲酸乙二醇酯及聚对苯二甲酸丁二醇酯等聚酯系树脂,间规聚苯乙烯、6-尼龙及6,6-尼龙等聚酰胺系树脂,聚芳酯、聚苯硫醚、聚醚酮、聚醚醚酮、聚砜、聚醚砜、聚酰胺酰亚胺、聚醚酰亚胺、聚缩醛等工程塑料与其他热塑性树脂的聚合物合金也包含在本发明的热塑性树脂(e)的范围内。
[0158]
这些热塑性树脂(e)可以单独使用1种,或者将2种以上组合使用。基于在工业上容易获得,成形体的冲击强度和着色性的平衡优异的角度,热塑性树脂(e)优选含有选自由芳香族聚碳酸酯、聚甲基丙烯酸甲酯、苯乙烯-丙烯腈共聚物、聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚氯乙烯、聚苯硫醚和聚缩醛构成的组中至少一种,更优选含有选自由聚甲基丙烯酸甲酯和苯乙烯-丙烯腈共聚物构成的组中的至少一种。
[0159]
本发明的树脂组合物除上述以外,在不损害本发明目的的范围内,还可以含有公知的各种添加剂。作为添加剂,可以举出阻燃剂(磷系、溴系、硅酮系、有机金属盐系等)、防滴落剂(例如氟化聚烯烃、硅酮及芳族聚酰胺纤维)、润滑剂(例如硬脂酸镁等长链脂肪酸金属盐等)、脱模剂(例如季戊四醇四硬脂酸酯等)、成核剂、抗静电剂、稳定剂(例如酚系稳定剂、硫系稳定剂、磷系稳定剂、紫外线吸收剂、胺系光稳定剂等)、填充材料(氧化钛、滑石、云母、高岭土、碳酸钙、玻璃片等)、增塑剂、强化剂(例如玻璃纤维、碳纤维等)、色素及颜料等。
[0160]
酚系稳定剂是具有酚性羟基的稳定剂,其中,可以优选使用与酚性羟基结合的芳香环的碳原子相邻的1个或2个碳原子被碳原子数为4以上的取代基取代的受阻酚系抗氧化剂。此时,碳原子数为4以上取代基可以通过碳-碳键与芳香环的碳原子结合,也可以介由碳以外的原子结合。
[0161]
作为酚系稳定剂,例如可以举出对环己基苯酚、3-叔丁基-4-甲氧基苯酚、4,4
’‑
异亚丙基二苯酚、1,1-双(4-羟基苯基)环己烷等非受阻酚系抗氧化剂、2-叔丁基-4-甲氧基苯酚、2,6-二叔丁基对甲酚、2,4,6-三叔丁基苯酚、4-羟基甲基-2,6-二叔丁基苯酚、苯乙烯化苯酚、2,5-二叔丁基对苯二酚、十八烷基-3-(3,5-二叔丁基-4-羟基苯基)丙酸酯、三甘醇双
[3-(3-叔丁基-5-甲基-4-羟基苯基)丙酸酯]、1,6-己二醇双[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、季戊四醇四亲[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、2,2
’‑
亚甲基双(4-甲基-6-叔丁基苯酚)、2,2
’‑
亚甲基双(6-叔丁基-4-乙基苯酚)、2,2,2
’‑
亚甲基双[4-甲基-6-(1,3,5-三甲基己基)苯酚]、4,4
’‑
亚甲基双(2,6-二叔丁基苯酚)、4,4
’‑
亚丁基二(3-甲基-6-叔丁基苯酚)、2,6-双(2-羟基-3-叔丁基-5-甲基苄基)-4-甲基苯酚、1,1,3-三[2-甲基-4-羟基-5-叔丁基苯基]丁烷、1,3,5-三甲基-2,4,6-三[3,5-二-叔丁基-4-羟基苄基]苯、三(3,5-二叔丁基-4-羟基苄基)异氰脲酸酯、三[3-(3,5-二叔丁基-4-羟基苯基)丙氧基乙基]异氰脲酸酯、4,4
’‑
硫代双(3-甲基-6-叔丁基苯酚)、2,2
’‑
硫代双(4-甲基-6-叔丁基苯酚)、4,4
’‑
硫代双(2-甲基-6-叔丁基苯酚)、硫代双(β-萘酚)等受阻酚系抗氧化剂。特别是受阻酚系抗氧化剂,由于其自身容易成为稳定的自由基,适合作为自由基捕获剂使用。这些酚系稳定剂可以单独使用或两种以上组合使用。
[0162]
相对于本树脂组合物100质量%,酚系稳定剂的含量优选为0.001质量%以上,更优选为0.003质量%以上,另一方面,优选为2质量%以下,更优选为1质量%以下。如果酚系稳定剂含量为上述下限值以上,则抗氧化效果优异,如果为上述上限值以下,则能够进一步抑制树脂组合物的氧化热稳定性和熔融混炼时的树脂分解。
[0163]
硫系稳定剂是具有硫原子但不具有酚性羟基的稳定剂,通过作为热塑性树脂劣化产生的氢过氧化物的分解剂发挥作用,具有改善树脂组合物的耐热老化性、提高色调、拉伸强度、伸长率等保持率的效果。硫系稳定剂也可以单独使用,但通过与上述酚系稳定剂组合使用,可以进一步改善长期热稳定性。
[0164]
作为硫系稳定剂,例如可以举出硫代二丙酸双十二烷基酯、硫代二丙酸双十四烷基酯、硫代二丙酸双十八烷基酯、季戊四醇四(3-十二烷基硫代丙酸酯)、硫代双(n-苯基-β-萘胺)、2-巯基苯并噻唑、2-巯基苯并咪唑、一硫化四甲基秋兰姆、二硫化四甲基秋兰姆、二丁基二硫代氨基甲酸镍、异丙基黄原酸镍、三月桂基三硫代亚磷酸酯等。特别地,具有硫醚结构的硫醚稳定剂可以适当地用于从氧化的物质中接收氧进行还原。这些硫系稳定剂可以单独使用或两种以上组合使用。
[0165]
相对于本树脂组合物100质量%,硫系稳定剂的含量优选为0.001质量%以上,更优选为0.003质量%以上,另一方面,优选为2质量%以下,更优选为1质量%以下。如果硫系稳定剂含量为上述下限值以上,则热稳定化效果优异,如果为上述上限值以下,则能够进一步抑制树脂组合物熔融混炼时的树脂分解。
[0166]
磷系稳定剂是指具有磷原子的稳定剂,是具有p(or)3结构的亚磷酸酯化合物。这里,r是烷基、亚烷基、芳基、亚芳基等,3个r可以相同也可以不同,2个r可以形成环结构。此外,一个分子中可以具有多个p(or)3结构。磷系稳定剂通过作为热塑性树脂的劣化产生的氢过氧化物的分解剂发挥作用,具有改善树脂组合物的耐热老化性、提高色调、拉伸强度、伸长率等保持率的效果。磷系稳定剂也可以单独使用,但通过与所述酚系稳定剂组合使用,特别地能够改善长期热稳定性,且能够抑制来自酚系稳定剂的黄变。
[0167]
作为亚磷酸酯化合物,例如可以举出三芳基亚磷酸酯(三苯基亚磷酸酯、三甲苯基亚磷酸酯、三(二甲苯基)亚磷酸酯、三萘基亚磷酸酯等)、二芳基烷基亚磷酸酯(二苯基异辛基亚磷酸酯、二苯基癸基亚磷酸酯等二芳基c1-18烷基亚磷酸酯等)、芳基二烷基亚磷酸酯(苯基二异辛基亚磷酸酯等芳基c1-18二烷基亚磷酸酯等)、三烷基亚磷酸酯(三甲基亚磷酸
酯、三乙基亚磷酸酯、三正丁基亚磷酸酯、三异辛基亚磷酸酯、十三烷基亚磷酸酯、三异癸基亚磷酸酯等三c1-18烷基亚磷酸酯等)、二烷基亚磷酸酯(二月桂基亚磷酸酯等二c1-18烷基亚磷酸酯等)、含有烷基芳基单元的亚磷酸酯[三(2,4-叔丁基苯基)亚磷酸酯、三(壬基苯基)亚磷酸酯、三(二壬基苯基)亚磷酸酯、二壬基苯基-o-联苯亚磷酸酯等三(c1-18烷基-芳基)亚磷酸酯、2,2-亚甲基双(4,6-二叔丁基苯基)辛基亚磷酸酯等)、脂肪族羧酸亚磷酸酯(三硬脂基亚磷酸酯等c1-18脂肪族羧酸亚磷酸酯等)、含有环氧烷单元的亚磷酸酯(聚二丙二醇壬基苯基亚磷酸酯、四苯基二丙二醇亚磷酸酯等)、含有环式新戊烷单元的亚磷酸酯[双十八烷基季戊四醇双亚磷酸酯、双(2,4-二叔丁基苯基)季戊四醇双亚磷酸酯、双(2,6-二叔丁基-4-甲基苯基)季戊四醇双亚磷酸酯等]、二亚磷酸酯类(二异癸基季戊四醇二亚磷酸酯、双十二烷基季戊四醇二亚磷酸酯、4,4
’‑
亚异丙二苯基双十二烷基二亚磷酸酯等)、三亚磷酸酯类[七聚二丙二醇三亚磷酸酯、六
·
十三烷基-1,1,3-三(3-叔丁基-6-甲基-4-氧基苯基)-3-甲基丙烷三亚磷酸酯等]等。这些亚磷酸酯化合物可以单独使用或两种以上组合使用。
[0168]
其中,作为热塑性树脂(e),使用聚碳酸酯树脂或聚酯树脂、聚酰胺树脂、聚缩醛树脂等,在聚合物结构单元中使用含有碳酸酯键或酯键、酰胺键、缩醛键的树脂时,从抑制这些热塑性树脂的耐湿热分解性的降低等观点出发,优选使用耐水解性高的磷系稳定剂。其中,优选一分子中的磷原子数为1,且各酯部的碳原子数(在多个酯部形成环的情况下,将位于酯键所夹区域的总碳原子数作为各碳原子数)为8以上的亚磷酸酯化合物,或者优选在一分子中具有多个磷原子且在各磷元素之间的酯部的碳原子数为8以上的亚磷酸酯化合物,例如可举出三c6-18烷基亚磷酸酯(三异癸基亚磷酸酯等)、含有支链c3-6烷基(叔丁基等)的亚磷酸酯[三(2,4-叔丁基苯基)亚磷酸酯、2,2-亚甲基双(4,6-二-叔丁基苯基)辛基亚磷酸酯等]、四烷基(c12-15)-4,4
’‑
亚异丙基二苯基二亚磷酸酯等。
[0169]
相对于本树脂组合物100质量%,磷系稳定剂的含量优选为0.001质量%以上,更优选为0.003质量%以上,另一方面,优选为2质量%以下,更优选为1质量%以下。如果磷系稳定剂含量为所述下限值以上,则热稳定化效果更优异,如果为所述上限值以下,则能够进一步抑制树脂组合物在熔融混炼时的树脂分解和耐湿热分解性的降低。
[0170]
在本树脂组合物中,在并用酚系稳定剂和硫系稳定剂和/或磷系稳定剂时,硫系稳定剂和磷系稳定剂的总添加质量相对于酚系稳定剂的添加质量优选为0.1倍以上,进一步优选为0.2倍以上,另一方面优选5倍以下,更优选3倍以下。如果硫系稳定剂和磷系稳定剂含量为上述下限值以上,则耐热老化性的提高效果更优异,如果为上述上限值以下,则能够进一步抑制树脂组合物在熔融混炼时的树脂分解。
[0171]
在本树脂组合物中,含有酚系稳定剂和硫系稳定剂和/或磷系稳定剂时,相对于树脂组合物100质量%,稳定剂的含量的合计优选为2质量%以下。通过使稳定剂含量的合计为2质量%以下,可以在熔融混炼时进一步抑制树脂的分解。
[0172]
作为色素或颜料,若为无机系颜料,例如可以举出氧化铁、群青、氧化钛、炭黑等。若为有机系颜料,例如可以举出酞菁系或蒽醌系蓝色颜料、苝系或喹吖啶酮系红色颜料、异吲哚啉酮系黄色颜料等。另外,作为特殊颜料,可以举出荧光颜料、金属粉颜料、珍珠颜料等。作为染料,可以举出苯胺黑类、紫环酮类、蒽醌类染料。这些色素和颜料根据所要求的颜色,市场上有各种等级,可以使用。它们可以单独使用1种,或者将2种以上组合使用。
[0173]
聚合物粒子群(c)或本组合物相对于本树脂组合物100质量%的比率优选为0.5质量%以上,更优选为1质量%以上,进一步优选为2质量%以上,另一方面,优选为60质量%以下,更优选为50质量%以下。如果聚合物粒子群(c)或本组合物比率为上述下限值以上,则得到的成形体的冲击强度更优异,如果为上述上限值以下,则能够抑制树脂组合物的流动性或耐热变形温度的降低。
[0174]
热塑性树脂(e)相对于本树脂组合物100质量%的比率优选为40质量%以上,更优选为50质量%以上,另一方面,优选为99.5质量%以下,更优选为99质量%以下,进一步优选为98质量%以下。如果热塑性树脂(e)的比率为上述下限值以上,则能够抑制树脂组合物的流动性或耐热变形温度的降低,如果为上述上限值以下,则得到的成形体的冲击强度更优异。
[0175]
(树脂组合物的制造方法)树脂组合物可以通过将聚合物粒子群(c)或本组合物、热塑性树脂(e)和根据需要的添加剂混合来制造。作为各材料的混合方法可以举出公知的混合方法,没有特别限定。例如,可以举出用转筒、v型搅拌机、超级混合器、诺塔混合机、班伯里密炼机、混炼辊、挤出机等进行混合、混炼的方法。作为本发明的树脂组合物的制造方法的一个例子,可以举出使用挤出机将聚合物粒子群(c)或本组合物、粒料状热塑性树脂(e)和根据需要的添加剂混合,挤出成股线状,用旋转式切割器等切成粒料状的方法。通过该方法,可以得到粒料状的树脂组合物。
[0176]
〔成形体〕本发明的一个方式的成形体(以下,也称为“本成形体”)包含聚合物粒子群(c)。本成形体还可以含有成分(d)。本成形体还可以含有热塑性树脂(e)。本成形体优选由上述本树脂组合物构成。
[0177]
本成形体例如可以通过成形聚合物粒子群(c)、本组合物或本树脂组合物来制造。作为成形方法,可以举出通常热塑性树脂组合物的成形中使用的成形法,例如注射成形法、挤出成形法、吹塑成形法、压延成形法等。
[0178]
本成形体可以作为汽车领域、oa机器领域、家电、电气
·
电子领域、建筑领域、生活
·
化妆品领域、医用品领域等各种材料在工业上广泛利用。更具体地说,可以作为电子设备等的框体、各种部件、被覆材料、汽车结构部件、汽车内装部件、光反射板、建筑物结构部件、门窗隔扇等使用。更具体地说,可以作为电脑框体、手机框体、便携信息终端框体、便携游戏机框体、打印机、复印机等内装
·
外装部件、导电体被覆材料、汽车内装
·
外装部件、建筑物外装材料、树脂窗框部件、地板材料、配管部件等使用。
[0179]
本发明的其他方式如下所示。〔1〕一种含聚有机硅氧烷的聚合物,是由含有聚有机硅氧烷(a1)和第一乙烯基聚合物(a2)的聚合物(a)和第二乙烯基聚合物(b)构成的含聚有机硅氧烷的聚合物,使所述含聚有机硅氧烷的聚合物分散在液体的环氧树脂中使其固化制成树脂片,用透射型电子显微镜观察其截面时,所述聚合物(a)具有将所述聚有机硅氧烷(a1)作为海成分、将所述第一乙烯基聚合物(a2)作为岛成分的海岛结构,将所述含聚有机硅氧烷聚合
物的粒子的直径设为l,将所述粒子中所含的所述聚有机硅氧烷(a1)区域的最大长度设为m时,相对于全部所述粒子的个数,满足下述式(1)的粒子的个数的比率小于60%。m/l》0.1
…
(1)〔2〕根据〔1〕所述的含聚有机硅氧烷的聚合物,相对于所述含聚有机硅氧烷的聚合物100质量%,所述聚有机硅氧烷(a1)的比率为1质量%以上50质量%以下。〔3〕根据〔1〕或〔2〕所述的含聚有机硅氧烷的聚合物,相对于所述含聚有机硅氧烷的聚合物100质量%,所述聚有机硅氧烷(a1)的比率为1质量%以上10质量%以下。〔4〕根据〔1〕~〔3〕中任一项所述的含聚有机硅氧烷的聚合物,数均粒径为10nm以上150nm以下。〔5〕根据〔1〕~〔4〕中任一项所述的含聚有机硅氧烷的聚合物,所述含聚有机硅氧烷的聚合物的一部分不溶于四氢呋喃,相对于所述含聚有机硅氧烷的聚合物粒子群100质量%,不溶于四氢呋喃的所述含聚有机硅氧烷的聚合物的比率为80质量%以上小于100质量%。〔6〕根据〔1〕~〔5〕中任一项所述的含聚有机硅氧烷的聚合物,所述含聚有机硅氧烷的聚合物的一部分可溶于四氢呋喃,可溶于四氢呋喃的所述含聚有机硅氧烷的聚合物的重均分子量为2万以上50万以下。〔7〕根据〔1〕~〔6〕中任一项所述的含聚有机硅氧烷的聚合物,在所述海岛结构中,在所述聚有机硅氧烷(a1)区域中含有多个所述第一乙烯基聚合物(a2)区域。〔8〕根据〔1〕~〔7〕中任一项所述的含聚有机硅氧烷的聚合物,使所述含聚有机硅氧烷的聚合物分散在液体的环氧树脂中使其固化制成树脂片,用透射型电子显微镜观察其截面时,所述含聚有机硅氧烷的聚合物具有将所述聚有机硅氧烷(a1)作为海成分、将所述第一乙烯基聚合物(a2)作为第一岛成分、将所述第二乙烯基聚合物(b)作为第二岛成分的海岛结构。〔9〕根据〔1〕~〔8〕中任一项所述的含聚有机硅氧烷的聚合物,相对于所述含聚有机硅氧烷的聚合物100质量%,所述聚合物(a)的比率为60质量%以上95质量%以下。〔10〕根据〔1〕~〔9〕中任一项所述的含聚有机硅氧烷的聚合物,构成所述第一乙烯基聚合物(a2)的乙烯基单体成分(a2)含有(甲基)丙烯酸酯单体。〔11〕根据〔1〕~〔10〕中任一项所述的含聚有机硅氧烷的聚合物,构成所述第二乙烯基聚合物(b)的乙烯基单体成分(b)含有选自由(甲基)丙烯酸酯单体及芳香族乙烯基单体构成的组中的至少一种,相对于所述乙烯基单体成分(b)100质量%,所述(甲基)丙烯酸酯单体及所述芳香族乙烯基单体的合计比率为50质量%以上。〔12〕根据〔1〕~〔11〕中任一项所述的含聚有机硅氧烷的聚合物,构成所述第二乙烯基聚合物(b)的乙烯基单体成分(b)含有甲基丙烯酸甲酯,相对于所述乙烯基单体成分(b)100质量%,所述甲基丙烯酸甲酯的比率为50质量%以上。〔13〕根据〔1〕~〔12〕中任一项所述的含聚有机硅氧烷的聚合物,所述聚合物(a)是在含有所述聚有机硅氧烷(a1)的胶乳的存在下,由构成所述第一乙烯基聚合物(a2)的乙烯基单体成分(a2)聚合而成的聚合物。
〔14〕一种组合物,含有〔1〕~〔13〕中任一项所述的含聚有机硅氧烷的聚合物,和选自由磷酸化合物及其碱金属盐构成的组中的至少一种成分。〔15〕根据〔14〕所述组合物,所述磷酸化合物的碱金属盐是选自由烷基磷酸的碱金属盐和烷基芳基磷酸的碱金属盐构成的组中的至少一种。〔16〕根据〔14〕或〔15〕所述组合物,其中,所述磷酸化合物的碱金属盐是聚氧化烯烷基醚磷酸的碱金属盐。〔17〕根据〔14〕~〔16〕中任一项所述组合物,相对于所述含聚有机硅氧烷的聚合物和选自由所述磷酸化合物及其碱金属盐构成的组中的至少一种成分的合计100质量%,所述成分中所含的磷原子的比率为100质量ppm以上。〔18〕一种树脂组合物,含有〔1〕~〔13〕中任一项所述的含聚有机硅氧烷的聚合物和热塑性树脂。〔19〕一种树脂组合物,含有〔14〕~〔17〕中任一项所述的组合物和热塑性树脂。〔20〕根据〔18〕或〔19〕所述树脂组合物,所述热塑性树脂含有选自由芳香族聚碳酸酯、聚甲基丙烯酸甲酯、苯乙烯-丙烯腈共聚物、聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚氯乙烯、聚苯硫醚及聚缩醛构成的组中至少一种。〔21〕一种成形体,含有〔1〕~〔13〕中任一项所述的含聚有机硅氧烷的聚合物。[实施例]
[0180]
以下,通过制造例及实施例更详细地说明本发明。制造例1-1~1-2、2-1~2-20是聚有机硅氧烷(a1)、聚合物(a)、聚合物粒子群(c)及组合物的制造例。另外,“份”表示“质量份”,“%”表示“质量%”,“ppm”表示“质量ppm”。另外,下述记载了各种测定方法。
[0181]
[固形成分的测定]将质量w1的聚有机硅氧烷胶乳用180℃的热风干燥机干燥30分钟,测定干燥后的残渣的质量w2,通过下式(e)算出固形成分[%]。固形成分[%]=w2/w1
×
100
···
(e)
[0182]
[粒径的测定]将“聚有机硅氧烷(a1)胶乳”或“聚合物粒子群(c)胶乳”用去离子水稀释至固形成分浓度约3%作为试样,使用上述美国matec公司制造的chdf2000型粒度分布计,使用下述条件,测定数均粒径dn和质量平均粒径dw。卡盒(cartridge):专用的粒子分离用毛细管式卡盒(商品名:c-202)载液:专用载液(商品名;2xgr500)载液的液性:中性载液的流速:1.4ml/分钟载液的压力:约4,000psi(2,600kpa)测定温度:35℃试样使用量:0.1ml
[0183]
[粉体回收方法]聚合物粒子群(c)的粉体回收通过下述的絮凝法(在表1及2中记载为g)或喷雾回收法(在表1及2中记载为s)中的任一种进行。絮凝法:将630份的乙酸钙浓度为0.8%的水溶液加热至50℃,进行搅拌同时向该
水溶液中缓慢滴加聚合物粒子群(c)胶乳使其凝聚。将得到的聚合物粒子群(c)过滤、清洗、脱水后,使其干燥,得到聚合物粒子群(c)的粉体。喷雾回收法:将聚合物粒子群(c)胶乳在下述处理条件下使用雾化器式喷雾干燥机(大川原化工机公司制、l8喷雾干燥器)进行喷雾干燥处理,得到聚合物粒子群(c)的粉体。<喷雾干燥处理条件>喷雾方式:转盘式圆盘转速:25000rpm热风温度入口温度:130℃、出口温度:60℃
[0184]
[thf不溶成分的测定]对于聚合物粒子群(c),按照以下方法进行thf不溶成分的测定。(1-1)向thf 50ml(44.5g)中加入0.5g试样,调制混合溶液,在25℃静置8小时后,用搅拌器搅拌30分钟,使thf可溶成分溶解。(1-2)将所述混合溶液放入测定了质量的离心管中,用离心分离机(16000rpm,4小时)对含有thf不溶成分和thf可溶成分的液体进行离心分离。(1-3)分离含有thf可溶成分的上清液后,再加入thf搅拌,再次与上述(1-2)同样地进行离心分离,清洗thf不溶成分。(1-4)重复上述(1-3)2次后,除去上清液。将残留有thf不溶成分的离心管浸渍在温水浴中(80℃、8小时),使thf挥发后,在65℃下真空干燥6小时,得到干燥试样(附着在离心管上的thf不溶成分)。(1-5)测定得到的干燥试样的质量(thf不溶成分+离心管),通过下式(f)计算thf不溶成分的比率w
ais
(%)。w
ais
=(w
c1-w
as
)/wt
×
100
···
(f)wt:测定所用的聚合物粒子群(c)的质量w
as
:离心管的质量w
c1
:thf不溶成分的质量(包含离心管的质量)
[0185]
[thf可溶成分的重均分子量的测定]thf可溶成分的重均分子量通过进行以下(2-1)~(2-3)的操作来测定。(2-1)从上述“thf不溶成分的测定”中采集的含有thf可溶成分的液体中,使用旋转蒸发器减压蒸馏除去thf,得到thf可溶成分。(2-2)将上述(2-1)中得到的thf可溶成分再次溶解在thf中,使试样浓度为:0.1~0.3%,得到thf可溶成分的thf溶液。(2-3)对上述(2-2)中得到的thf可溶成分的thf溶液进行凝胶渗透色谱(gpc)测定,由标准聚苯乙烯的标准曲线求出重均分子量(mw)。gpc测定条件如后述的实施例所述。装置:东曹公司制造的“hlc8220”、色谱柱:东曹公司制造的“tskgel supermultiporehz-h”(内径4.6mm
×
长15cm
×
2根、排除极限4
×
107(推定)洗脱液:thf
洗脱液流量:0.35ml/分钟测定温度:40℃试样注入量:10μl
[0186]
[tem图像的获取和图像分析]将聚合物粒子群(c)取入聚乙烯胶囊中,注入液态环氧树脂(epiform(注册商标)r-2100、h-105、somar公司制造),进行搅拌。在25℃下放置12小时,使上述环氧树脂固化。使用超薄切片机leica em uc7(leica microsystems公司制)对得到的树脂片切面,进行修整。将得到的树脂片用四氧化锇水溶液(23℃、12小时)进行染色,然后使用四氧化钌水溶液(23℃、5小时)进行染色。从染色后的树脂片,在切削温度23℃、切削速度0.4mm/秒、薄片厚度50nm的条件下切出切片,回收到带支撑膜的铜栅上。
[0187]
通过tem(日立公司制、h-7600),在加速电压为80kv、倍率为20万倍的条件下,观察回收的切片表面上随机选择的0.5μm2以上的范围,取得tem像。在得到的tem图像中,观察到环氧树脂固化物区域(树脂区域)和分散在该区域内的粒子区域。另外,在粒子区域中,聚有机硅氧烷(a1)部分用明对比度确认,乙烯基聚合物部分用暗对比度确认。
[0188]
在1张tem观察图像中观察的粒子区域中,除去符合下述(i)~(iii)的粒子区域。(i)在图像边缘截断的粒子区域。(ii)尺寸小于平均粒径的80%的粒子区域。(iii)在3个方向以上存在相邻的粒子区域,各粒子区域的边界不清楚的粒子区域。
[0189]
对于剩余的粒子区域中80%以上且50个以上的粒子区域,按照以下的顺序进行对比度的线轮廓测定,计算z值,求出它们的平均值。此时选择的粒子区域的比率是指选择的粒子区域的个数相对于图像中可确认的粒子区域的全部个数的比例。
[0190]
根据得到的tem图像,使用图像分析软件(imagej)计算粒子区域的直径l和聚有机硅氧烷(a1)区域的最大长度m。如上所述,粒子区域的直径l以通过连接一个粒子区域的长径和短径的中央点并切断粒子直径的方式进行线轮廓来求出。如上所述,最大长度m是对于由线轮廓获得的1个粒子区域中的对比度值(gray value),由在连续表示最大值的75%以上的值的部位中,具有最大连续长度的部位求出。根据得到的直径l及最大长度m,将满足下述式(1)的粒子区域的个数规定为z1,将不满足的粒子区域的个数规定为z2,根据下述式(2)计算z值[%]。m/l>0.1
···
(1)z值〔%〕={z1/(z1+z2)}
×
100
···
(2)。
[0191]
《制造例1-1》(聚有机硅氧烷(a1-1)的制造)将98份环状有机硅氧烷混合物(信越有机硅公司制,产品名:dmc,3~6元环的环状有机硅氧烷的混合物)及2份3-甲基丙烯酰氧基丙基甲基二甲氧基硅烷(信越有机硅公司制、产品名:kbm-502)混合得到100份有机硅氧烷混合物。将在300份的去离子水中溶解了0.7份十二烷基苯磺酸钠(dbsna,花王公司制、产品名:neopelex g-15,固形成分换算)的水溶液添加到上述混合物中,用均质搅拌机以10,000rpm搅拌5分钟后在20mpa的压力下通过
均质机两次,得到稳定的预混合乳液。接着,在具备冷却冷凝器的容量5升的可分离烧瓶内,在去离子水90份中放入溶解了15份十二烷基苯磺酸(dbsh、花王公司制、产品名:neopelex gs)的水溶液后,将该水溶液加热到温度80℃,接着将上述乳液在240分钟内连续投入进行聚合反应后,冷却至25℃,添加5%氢氧化钠水溶液,将反应液中和至ph7.0,得到聚有机硅氧烷胶乳(a1-1)。聚有机硅氧烷胶乳(a1-1)的固形成分含量为20%。另外,该胶乳的由毛细管粒度分布计得到的数均粒径(dn)为26nm,质量平均粒径(dw)为35nm,dw/dn为1.35。
[0192]
《制造例1-2》(聚有机硅氧烷(a1-2)的制造)将98份环状有机硅氧烷混合物(信越有机硅公司制、产品名:dmc、3~6元环的环状有机硅氧烷的混合物)和2份3-甲基丙烯酰氧基丙基甲基二甲氧基硅烷(kbm-502)混合,得到100份有机硅氧烷混合物。将0.7份十二烷基苯磺酸钠(dbsna)溶解在350份去离子水中的水溶液添加到所述混合物中,用均质搅拌机在10,000rpm下搅拌5分钟后,在20mpa的压力下通过两次均质机,得到稳定的预混合乳液。
[0193]
接着,在具备冷却冷凝器的容量5升的可分离烧瓶内,在40份去离子水中放入溶解了4份十二烷基苯磺酸(dbsh)的水溶液后,将该水溶液加热至温度80℃,接着,将上述乳液在240分钟内连续投入,使其聚合反应,然后冷却至25℃,添加5%氢氧化钠水溶液,将反应液中和至ph7.0,得到聚有机硅氧烷胶乳(a1-2)。
[0194]
聚有机硅氧烷胶乳(a1-2)的固形成分为18质量%。另外,该胶乳的由毛细管粒度分布计得到的数均粒径(dn)为67nm,质量平均粒径(dw)为83nm,dw/dn为1.24。
[0195]
《制造例2-1》(聚合物粒子群(c-1)的制造)将在制造例1-1中得到的聚有机硅氧烷胶乳(a1-1)18份(换算成聚合物为3.0份)采集到容量为5升的可分离烧瓶内,添加170份去离子水并混合。接着,在该可分离烧瓶内添加丙烯酸正丁酯(nba)76.6份、甲基丙烯酸烯丙酯(ama)0.4份、十二烷基苯磺酸钠(dbsna)0.3份,通入氮气流进行烧瓶内气氛气体的氮气置换,将液温升温至43℃,搅拌1小时。添加0.15份过硫酸钾(kps),开始自由基聚合,搅拌10小时确认到聚合发热峰后,冷却至25℃保持15小时,由此使聚合结束,得到复合橡胶胶乳。
[0196]
将该复合橡胶胶乳升温至80℃,以0.6份/分钟的速度向该胶乳中滴加甲基丙烯酸甲酯(mma)20份,开始接枝聚合反应。滴加结束后,在温度80℃下保持1小时后,冷却至25℃,得到聚合物粒子群(c-1)胶乳。该胶乳的固形成分为35%,聚合率为99.9%以上。该聚合率是从复合橡胶的制造到接枝聚合的全部工序中使用的单体成分的聚合率。该胶乳的由毛细管粒度分布计得到的数均粒径(dn)为94nm,质量平均粒径(dw)为105nm,dw/dn为1.12。
[0197]
接着,通过上述絮凝法,得到聚合物粒子群(c-1)的粉体。聚合物粒子群(c-1)的thf不溶成分的比率为95%。另外,thf可溶成分的重均分子量为23万。
[0198]
如图2所示,聚合物粒子群(c-1)具有以聚有机硅氧烷(a1)为海成分、以乙烯基聚合物(a2)为岛成分的海岛结构。另外,z值为8%。
[0199]
《制造例2-2》
(聚合物粒子群(c-2)的制造)将制造例2-1中得到的聚合物粒子群(c-1)胶乳通过上述喷雾干燥法进行粉体回收,由此得到聚合物粒子群(c-2)。由于同一聚合后的胶乳,因此粒径、thf不溶成分的比率、thf可溶成分的重均分子量及z值与聚合物粒子群(c-1)相同。
[0200]
《制造例2-3~2-6》(聚合物粒子群(c-3)~(c-6)的制造)除了将使用的单体的量及粉体回收方法如表1所述进行变更以外,与制造例2-1同样地,得到聚合物粒子群(c-3)~(c-6)。通过絮凝法得到聚合物粉体时的凝聚温度配合得到的粉体性状在50~85℃之间适当变更。
[0201]
《制造例2-7》(聚合物粒子群(c-7)的制造)将在制造例1-1中得到的聚有机硅氧烷胶乳(a1-1)18份(换算成聚合物为3.0份)采集到容量为5升的可分离烧瓶内,添加170份去离子水并混合。接着,在该可分离烧瓶内添加丙烯酸正丁酯(nba)66.0份、甲基丙烯酸烯丙酯(ama)1.0份、十二烷基苯磺酸钠(dbsna)0.3份,通入氮气流进行烧瓶内气氛气体的氮气置换,将液温升温至43℃,搅拌1小时。添加0.15份过硫酸钾(kps),开始自由基聚合,搅拌10小时确认到聚合发热峰后,冷却至25℃保持15小时,由此使聚合结束,得到复合橡胶胶乳。
[0202]
将该复合橡胶胶乳升温至80℃,以0.6份/分钟的速度将7.5份甲基丙烯酸甲酯(mma)滴加到该胶乳中,开始接枝聚合反应。滴加结束后,在温度80℃下保持1小时。然后,添加0.04份过硫酸钾(kps)搅拌15分钟后,以0.3份/分钟的速度将5.6份丙烯腈(an)、16.9份苯乙烯(st)、0.015份正辛基硫醇(nom)的混合溶液滴加到该胶乳中,再次开始接枝聚合反应。滴加结束后,在80℃的温度下保持2小时,添加0.04份过硫酸钾(kps),再搅拌2小时后,冷却至25℃,得到聚合物粒子群(c-7)胶乳。该胶乳的固形成分为35%,聚合率为99.9%以上。该聚合率是从复合橡胶的制造到接枝聚合的全部工序中使用的单体成分的聚合率。该胶乳的由毛细管粒度分布计得到的数均粒径(dn)为90nm,质量平均粒径(dw)为112nm,dw/dn为1.25。
[0203]
接着,通过上述絮凝法,得到聚合物粒子群(c-7)的粉体。聚合物粒子群(c-7)的thf不溶成分的比率为95质量%。另外,thf可溶成分的重均分子量为40万。
[0204]
如图6所示,聚合物粒子群(c-7)具有将聚有机硅氧烷(a1)作为海成分、将乙烯基聚合物(a2)作为岛成分的海岛结构。另外,z值为8%。
[0205]
《制造例2-8》(聚合物粒子群(c-8)的制造)除了将所使用单体的量如表1所述那样变更以外,与制造例2-7同样地,得到聚合物粒子群(c-8)的粉体。
[0206]
《制造例2-9、2-10》(组合物(c-9)、(c-10)的制造)在制造例2-1中得到聚合物粒子群(c-1)胶乳中,作为成分(d)添加记载于表1中的量的十三烷基氧乙烯磷酸钠(rs-610na,东邦化学工业公司制、产品名:phosphanol(注册商标)rs-610na,固形成分换算、烷基为十三烷基的聚氧乙烯烷基磷酸盐、聚氧乙烯基中的氧
乙烯单元的单元数:6),得到组合物(c-9)胶乳和组合物(c-10)胶乳。
[0207]
然后,通过上述絮凝法将各胶乳粉体化,得到组合物(c-9)的粉体和组合物(c-10)的粉体。在组合物(c-9)和(c-10)中,聚合物粒子群(c-1)的thf不溶成分的比率为95%,thf可溶成分的重均分子量为23万,z值为8%。相对于聚合物粒子群(c-1)和追加乳化剂的合计质量,磷原子的含量在组合物(c-9)中为230ppm,在组合物(c-10)中为490ppm。另外,制造例2-1中得到聚合物(c-1)的磷原子含量小于16ppm。
[0208]
《制造例2-11、2-12》(聚合物粒子群(c-11)、(c-12)的制造)除了将使用的聚有机硅氧烷胶乳、单体及乳化剂的种类及量如表2所述那样变更以外,与制造例2-1同样地,得到聚合物粒子群(c-11)、(c-12)的粉体。用于得到各粉体的絮凝温度配合得到的粉体性状在55~95℃间适当变更。
[0209]
《制造例2-13~2-15》(聚合物粒子群(c-13)~(c-15)的制造)使用的聚有机硅氧烷胶乳、单体及乳化剂的种类及量如表2所述进行了变更,除此之外,与制造例2-7同样地,得到聚合物粒子群(c-13)~(c-15)的粉体。用于得到各粉体的絮凝温度配合得到的粉体性状在55~75℃间适当变更。
[0210]
《制造例2-16》(聚合物粒子群(c-16)的制造)将丙烯酸正丁酯(nba)5.0份、甲基丙烯酸烯丙酯(ama)0.03份、十二烷基苯磺酸钠(dbsna)0.3份及去离子水160份装入容量为5升的可分离烧瓶内,通过使氮气流流通的同时搅拌1小时,进行烧瓶内气氛气体的氮气置换。将液温升温至80℃后,添加0.05份过硫酸钾(kps),搅拌180分钟后,冷却至25℃,静置18小时。接着,添加丙烯酸正丁酯(nba)74.6份、甲基丙烯酸烯丙酯(ama)0.37份及十二烷基苯磺酸钠(dbsna)0.4份,使氮气流流通的同时在25℃下搅拌90分钟后,将液温升温至43℃,搅拌1小时。添加0.15份过硫酸钾(kps),开始自由基聚合,搅拌10小时确认到聚合发热峰后,冷却至25℃保持15小时,由此使聚合结束,得到复合橡胶胶乳。
[0211]
将该复合橡胶胶乳升温至80℃,以0.6份/分钟的速度向该胶乳中滴加甲基丙烯酸甲酯(mma)20份,开始接枝聚合反应。滴加结束后,在温度80℃下保持1小时后,冷却至25℃,得到聚合物粒子群(c-16)胶乳。该胶乳的固形成分为35%,聚合率为99.9%以上。该聚合率是从丙烯酸橡胶的制造到接枝聚合的全部工序中使用的单体成分的聚合率。该胶乳的由毛细管粒度分布计得到的数均粒径(dn)为88nm,质量平均粒径(dw)为94nm,dw/dn为1.07。
[0212]
接着,通过上述絮凝法进行粉体化,得到聚合物粒子群(c-16)的粉体。聚合物粒子群(c-16)的thf不溶成分的比率为96%。另外,thf可溶成分的重均分子量为17万。另外,聚合物粒子群(c-16)是不含有聚有机硅氧烷(a1),仅为乙烯基聚合物(a2)的聚合物,因此不
进行tem观察。
[0213]
《制造例2-17》(聚合物粒子群(c-17)的制造)除了如表2所述变更所使用的单体的量以外,与制造例2-1同样地得到聚合物粒子群(c-17)胶乳。该胶乳的固形成分为35%,聚合率为99.9%以上。另外,由毛细管粒度分布计得到的数均粒径(dn)为98nm,质量平均粒径(dw)为108nm,dw/dn为1.11。
[0214]
接着,通过上述絮凝法进行粉体化,得到聚合物粒子群(c-17)的粉体。聚合物粒子群(c-17)的thf不溶成分的比率为79%。另外,thf可溶成分的重均分子量为41万。另外,z值为68%。
[0215]
《制造例2-18》(聚合物粒子群(c-18)的制造)将在制造例1-1中得到的聚有机硅氧烷胶乳(a1-1)18份(换算成聚合物为3.0份)采集到容量为5升的可分离烧瓶内,添加去离子水220份并混合。接着,在该可分离烧瓶内添加丙烯酸正丁酯(nba)76.6份、甲基丙烯酸烯丙酯(ama)0.4份、十二烷基苯磺酸钠(dbsna)0.3份,通过使氮气流流通进行烧瓶内气氛气体的氮气置换,将液温升温至65℃并搅拌1小时。
[0216]
添加0.25份过硫酸钾(kps),开始自由基聚合。然后,将液温升温至80℃,保持1小时使聚合结束,得到复合橡胶胶乳。
[0217]
将该胶乳的液温维持在80℃的同时,将20份甲基丙烯酸甲酯(mma)以0.6份/分钟的速度滴加到该胶乳中,开始接枝聚合反应。滴加结束后,在温度80℃下保持1小时后,冷却至25度,得到聚合物粒子群(c-18)胶乳。
[0218]
胶乳的固形成分为30质量%,聚合率为99.9%以上。另外,由毛细管粒度分布计得到的数均粒径(dn)为98nm,质量平均粒径(dw)为113nm,dw/dn为1.16。
[0219]
接着,将630份乙酸钙浓度为0.8质量%的水溶液加热到70℃,搅拌的同时将得到的接枝共聚物胶乳缓慢地滴加在该水溶液中并使其凝聚。将得到的接枝共聚物过滤、清洗、脱水后进行干燥,得到聚合物粒子群(c-18)的粉体。聚合物粒子群(c-18)的thf不溶成分率为93质量%。另外,thf可溶成分的重均分子量为19万。另外,z值为66%。
[0220]
《制造例2-19》(聚合物粒子群(c-19)的制造)将在制造例1-1中得到的聚有机硅氧烷胶乳(a1-1)18份(换算成聚合物为3.0份)采集到容量为5升的可分离烧瓶内,添加去离子水155份并混合。接着,在该可分离烧瓶内添加丙烯酸正丁酯(nba)76.6份、甲基丙烯酸烯丙酯(ama)0.4份、叔丁基过氧化氢(t-bh)0.15份、十二烷基苯磺酸钠(dbsna)0.3份,通过使氮气流流通进行烧瓶内气氛气体的氮气置换,使液温升温至50℃并搅拌1小时。
[0221]
一次性添加溶解有0.0005份硫酸亚铁
·
七水合物(fe)、0.0015份乙二胺四乙酸二钠盐
·
二水合物(edta)、0.2份甲醛次硫酸钠(sfs)的去离子水5份,开始自由基聚合。然后,保持1小时使聚合结束,得到复合橡胶胶乳。
[0222]
在将该胶乳的液温维持在50℃的同时,将20份甲基丙烯酸甲酯(mma)和0.05份叔
丁基过氧化氢(t-bh)的混合液以0.6份/分钟的速度滴加到该胶乳中,开始接枝聚合反应。滴加结束后,在温度50℃下保持1小时后,冷却至25度,得到聚合物粒子群(c-19)胶乳。
[0223]
胶乳的固形成分为35质量%,聚合率为99.9%以上。另外,该胶乳的由毛细管粒度分布计得到的数均粒径(dn)为96nm,质量平均粒径(dw)为106nm,dw/dn为1.11。
[0224]
接着,将630份乙酸钙浓度为0.8质量%的水溶液加热到70℃,搅拌的同时将得到的接枝共聚物胶乳缓慢地滴加在该水溶液中并使其凝聚。将得到的接枝共聚物过滤、清洗、脱水后进行干燥,得到聚合物粒子群(c-19)的粉体。聚合物粒子群(c-19)的thf不溶成分率为93质量%。另外,thf可溶成分的重均分子量为17万。另外,z值为96%。
[0225]
《制造例2-20》(聚合物粒子群(c-20)的制造)除了如表2的记载变更所使用的单体的种类和量以外,与制造例2-19同样地得到聚合物粒子群(c-20)的粉体。聚合物粒子群(c-20)的thf不溶成分率为85质量%。另外,thf可溶成分的重均分子量为15万。另外,z值为68%。
[0226]
[表1]
[0227]
[表2]
[0228]
《实施例1~14、比较例1~11》将上述制造例中得到的聚合物、苯乙烯-丙烯腈共聚物、炭黑(cb、三菱化学公司制、产品名:#960b)及硬脂酸镁(mgst、nacalai tesk公司制)以表3~6的比率配合,得到混合物。将该混合物供给到加热到筒体温度240℃的脱挥式双螺杆挤出机(东芝机械公司制、tem-35b(商品名))中进行混炼,制作各树脂组合物粒料。
[0229]
在此,作为苯乙烯-丙烯腈共聚物,使用以下的共聚物。san-1:techno umg公司制,产品名:sr-56b(an含有率33%)
san-2:techno umg公司制,产品名:ap-h(an含有率27%)san-3:techno umg公司制,产品名称:ap-a(an含有率30%)an含量是丙烯腈单元相对于苯乙烯-丙烯腈共聚物100%的质量比例
[0230]
将各粒料在下述条件下注塑成形,制作评价用的试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:240℃、模具温度:80℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm试验片b:长度100mm
×
宽度50mm
×
厚度2mm
[0231]
[夏比冲击强度]对试验片a,依据iso179-1刻划出a型缺口,在23℃和-30℃下测定夏比冲击强度。可以说数值越高,耐冲击性越好,越优选。
[0232]
[显色性试验]作为着色外观的指标,使用分光色差计(日本电色工业公司制、se7700(商品名)),在c光源、2度视野的条件下,用反射光测定法测定试验片b的明度(基于l
*
、jis z8781-4)。在本技术实施例中,由于着色为黑色,因此可以说数值越低,着色外观(漆黑性)越好,越优选。
[0233]
[耐候性试验]使用与上述显色性试验相同的成形体(平板),在下述照射条件下进行耐候性试验。“照射条件”试验装置:daipla wintes公司制、daipla
·
metal weather、型号kw-r5tp-a,5槽内温度:设定为50℃紫外线照射条件:设定65mw/cm2用(ushio电机公司制、照度计uit-101/uvd-365pd、测定波长330~390nm、峰值灵敏度波长365nm)进行测定滤波器:kf-1滤波器(透射波长范围:295nm~780nm)试验气氛:仅照射20小时,黑暗4小时(24小时/1循环)试验时间:96小时(4个循环)
[0234]
使用分光色差计(日本电色工业公司制造,se7700(商品名)),在c光源、2度视野条件下,用反射光测定法测定耐候性试验前后的黄色度(yi、基于jis k7105)、明度(l
*
)及色度(a
*
、b
*
)(基于jis z8781-4)。
[0235]
耐候性试验前的黄色度(yi1)为初始黄色度(yi)。黄色度变化(δyi)由耐候性试验前的黄色度(yi1)及耐候性试验后的黄色度(yi2)通过下述式(g)算出。此外,色差(δe
*ab
)由耐候性试验前的明度(l*1)和色度(a*1、b
*1
)以及耐候性试验后的明度(l
*2
)和色度(a
*2
、b
*2
)通过下述式(h)算出。式(g):δyi=yi
2-yi1式(h):δe
*ab
=((l
*2-l
*1
)+(a
*2-a
*1
)+(b
*2-b
*1
))
0.5
初始黄色度越低,可以说成形时的耐热变色性越好,越优选。另外,可以说黄色度
变化及色差越小,耐候性越好,越优选。
[0236]
[表3]
[0237]
实施例1~6的冲击强度及着色外观优异。在比较例1中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例2中,由于使用了不含聚有机硅氧烷的丙烯酸系聚合物,因此冲击强度为低位。在比较例3~5中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度及着色外观为低位。
[0238]
[表4]
[0239]
实施例7~12的冲击强度及着色外观优异。在比较例6中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度及着色外观为低位。
[0240]
[表5]
[0241]
实施例13的冲击强度和着色外观优异。在比较例8、9中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度及着色外观为低位。
[0242]
[表6]
[0243]
实施例14的冲击强度和耐候性优异。在比较例10、11中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度、着色外观、耐热变色性及耐候性为低位。
[0244]
《实施例15~25、比较例12~19》将上述制造例中得到的各含聚有机硅氧烷的聚合物、甲基丙烯酸树脂(pmma)及各种有机染料以表7及表8的比率配合,得到混合物。将该混合物供给到加热到筒体温度250℃
的脱挥式双螺杆挤出机(东芝机械公司制、tem-35b(商品名)中进行混炼,制作各树脂组合物粒料。
[0245]
在此,作为甲基丙烯酸树脂(pmma),使用acrypet vh001(商品名,含有90%以上甲基丙烯酸甲酯单元,三菱化学公司制)。另外,作为着色剂使用以下物质。(od-1):有机染料、三菱化学公司制、diaresin green(
ダイアレジングリーン
)c(od-2):有机染料、三菱化学公司制、diaresin red(
ダイアレジンレッド
)a(od-3):有机染料、三菱化学公司制、diaresin blue(
ダイアレジンブルー
)gcb:炭黑、三菱化学公司制、#960b
[0246]
将树脂组合物粒料在下述条件下注塑成形,制作评价用试验片。注塑成形机:东芝机械公司制注射成型机、ec20pnii(商品名)料筒温度:250℃、模具温度:60℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm试验片c:长度50mm
×
宽度50mm
×
厚度3mm
[0247]
[夏比冲击强度]对试验片a,依据iso179-1刻划出a型切口,在23℃和-30℃下测定夏比冲击强度。可以说数值越高,耐冲击性越好,越优选。
[0248]
[显色性试验]作为树脂组合物的着色外观的指标,按照下述方法评价明度(l*)。使用分光色差仪(商品名:sd7000,日本电色工业公司制),以sce方式对上述试验片c进行色相测定,根据iso11664-4求出l
*
。在本技术实施例中,由于着色为黑色,因此可以说数值越低,着色外观(漆黑性)越好,越优选。
[0249]
[表7]
[0250]
实施例15~18的冲击强度及着色外观优异。
在比较例12中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例13~15中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度及着色外观的平衡处于低位。
[0251]
[表8]
[0252]
实施例19~25中,冲击强度及着色外观优异。在比较例16中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例17~19中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度及着色外观为低位。
[0253]
《实施例26~31、比较例20~34》将所述制造例中得到的各含聚有机硅氧烷的聚合物、各种助剂及各种热塑性树脂以表9~12的比率配合,得到混合物。将该混合物供给到脱挥式双螺杆挤出机(池贝公司制、pcm-30(商品名)中进行混炼,制作各树脂组合物粒料。
[0254]
在此,作为热塑性树脂使用以下的热塑性树脂。pc:聚碳酸酯树脂(iupilon s-2000f,三菱工程塑料公司制,粘度平均分子量24,000)。san-2:苯乙烯-丙烯腈共聚物(ap-h、techno-umg公司制)。pet:聚对苯二甲酸乙二醇酯树脂(trn8550ff,帝人公司制)。pcgf-1:添加玻璃纤维的聚碳酸酯树脂(iupilon gs 2020mr2、三菱工程塑料公司制、添加玻璃纤维20质量%)。
[0255]
将树脂组合物粒料注塑成形,制作评价用试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm试验片d:长度127mm
×
宽度12.7mm
×
厚度1.6mm此时,挤出条件、注塑成形条件如下,鉴于成形树脂的流动性而适当变更。(实施例26、比较例20~22)挤出筒体温度280℃,注塑料筒温度280℃,模具温度80℃。(实施例27~28、比较例23~26)
挤出筒体温度260℃、注塑料筒温度260℃、模具温度80℃。(实施例29~30、比较例27~30)挤出筒体温度280℃,注塑料筒温度300℃,模具温度70℃。(实施例31、比较例31~34)挤出筒体温度300℃,注塑料筒温度300℃,模具温度90℃。
[0256]
[夏比冲击强度]对于试验片a,基于iso179-1刻划出a型缺口,测定夏比冲击强度。可以说数值越高,耐冲击性越好,越优选。另外,在本实施例中,在一部分冲击试验中存在耐冲击性高、试验片不完全断裂的情况。在这种情况下,由于不能计算出正确的冲击值,因此在表中标记为n.b。
[0257]
[表9]
[0258]
实施例26中,冲击强度优异。在比较例20中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例21~22中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度为低位。
[0259]
[表10]
[0260]
实施例27~28中,冲击强度优异。在比较例23中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例24~26中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度
为低位。
[0261]
[表11]
[0262]
实施例29~30中,冲击强度优异。在比较例27中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例28~30中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度为低位。
[0263]
[表12]
[0264]
实施例31中,冲击强度优异。在比较例31中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例32~34中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度为低位。
[0265]
《实施例32~33、比较例35~36》将上述制造例中得到的各含聚有机硅氧烷的聚合物、各种助剂及聚缩醛树脂(pom)以表13的比率配合,得到混合物。将该混合物供给到加热至筒体温度180℃的脱挥式双螺杆挤出机(池贝公司制、pcm-30(商品名)中进行混炼,制作各树脂组合物粒料。
[0266]
在此,作为聚缩醛树脂(pom),使用了hostaform c9021(商品名,塞拉尼斯日本公司制)。
另外,作为各种助剂,使用以下的助剂。st-ca:硬脂酸钙(硬脂酸钙gf-200、日油公司制)。irg245:苯酚类抗氧化剂(irganox 245、汽巴日本公司制)。
[0267]
将树脂组合物粒料在下述条件下注塑成型,制作评价用试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:205℃、模具温度:70℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm
[0268]
[夏比冲击强度]对于试验片a,依据iso179-1刻划出a型切口,在23℃和-30℃下测定夏比冲击强度。可以说数值越高,耐冲击性越好,越优选。
[0269]
[表13]
[0270]
实施例32~33中,冲击强度优异。在比较例35中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例36中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度为低位。
[0271]
《实施例34~35、比较例37~38》将上述制造例中得到的各含聚有机硅氧烷的聚合物及聚氯乙烯树脂以表14的比率配合,得到混合物。在此,作为聚氯乙烯树脂,使用了将氯乙烯树脂(产品名:tk-1000、信越化学工业公司制、平均聚合度1050)100份、ca/zn系复合稳定剂(产品名:ht-547a、日东化成工业公司制)3份、碳酸钙(产品名:μ-powder 3s、备北粉化工业公司制)6份、氧化钛(产品名:tipaque r830、石原产业公司制)3份、甘油脂肪酸酯(产品名:loxiol gh-4、意慕利油脂化学日本公司制)0.5份、高分子复合酯(产品名:loxiol vpn963、意慕利油脂化学日本公司制)0.2份、高分子复合酯(产品名:loxiol g70s、意慕利油脂化学日本公司制)0.3份、聚乙烯蜡(产品名:loxiol vpn233、意慕利油脂化学日本公司制)0.2份、丙烯酸系加工助剂(产品名:甲基
丁烯p-570a、三菱化学公司制)供给亨舍尔混合机均匀混合得到的粉体。
[0272]
将上述氯乙烯树脂和各含聚有机硅氧烷的聚合物的混合物,使用介电加热式8英寸试验辊(关西辊公司制),在190℃、辊间隔0.4mm的条件下熔融混炼5分钟,得到片状成形体。使用热压机(shoji公司制),在190℃、15mpa的条件下对得到的片状成形体进行5分钟的热压,得到厚度4mm的成形体。从得到的成形体切出以下的试验片,用于评价。试验片a:长度80mm
×
宽度10mm
×
厚度4mm。
[0273]
[夏比冲击强度]对试验片a,依据iso179-1刻划出a型切口,在23℃和-10℃下测定夏比冲击强度。可以说数值越高,耐冲击性越好,越优选。另外,在本实施例中,在一部分冲击试验中存在耐冲击性高、试验片不完全断裂的情况。在这种情况下,由于不能计算出正确的冲击值,因此在表中标记为n.b。
[0274]
[表14]
[0275]
实施例34~35中,冲击强度优异。在比较例37中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例38中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度为低位。
[0276]
《实施例36、比较例39~40》将上述制造例中得到的各含聚有机硅氧烷的聚合物及聚酰胺树脂(pa)以表15的比率配合,得到混合物。将该混合物在80℃下干燥12小时后,供给到加热至筒体温度250℃的脱挥式双螺杆挤出机(池贝公司制、pcm-30(商品名)中进行混炼,制作各树脂组合物粒料。
[0277]
在此,作为聚酰胺树脂(pa),使用ube尼龙1022b(商品名、聚酰胺6、宇部兴产公司制)。
[0278]
将树脂组合物粒料在下述条件下注塑成型,制作评价用试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:250℃、模具温度:80℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm
[0279]
[夏比冲击强度]
对试验片a,依据iso179-1刻划出a型切口,在23℃和-40℃下测定绝干条件下的夏比冲击强度。可以说数值越高,耐冲击性越好,越优选。
[0280]
[表15]
[0281]
实施例36中,冲击强度优异。在比较例39中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例40中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度为低位。
[0282]
《实施例37、比较例41~42》将上述制造例中得到的各含聚有机硅氧烷聚合物及聚乳酸树脂(pla)以表16的比率配合,得到混合物。将该混合物供给到加热到筒体温度200℃的脱挥式双螺杆挤出机(池贝公司制、pcm-30(商品名)中进行混炼,制作各树脂组合物粒料。
[0283]
在此,作为聚乳酸树脂(pla),使用了ingeo生物聚合物2003d(商品名、natureworks llc公司制)。
[0284]
将树脂组合物粒料在下述条件下注塑成形,制作评价用试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:200℃、模具温度:30℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm
[0285]
[夏比冲击强度]对试验片a,依据iso179-1刻划出a型切口,在23℃下测定夏比冲击强度。在有退火的条件下,对试验片a依据iso179-1刻划出a型切口,在100℃的烘箱内静置3小时后,冷却至23℃,测定夏比冲击强度。均为数值越高,耐冲击性越好,越优选。
[0286]
[表16]
[0287]
实施例37中,冲击强度优异。在比较例41中,由于不含聚合物粒子群(c),因此冲击强度为极低。在比较例42中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度为低位。
[0288]
《实施例38~39、比较例43~44》将上述制造例2-10中得到组合物及聚合物粒子群(c-1)、聚对苯二甲酸丁二醇酯(pbt、novaduran 5010r5、三菱工程塑料公司制)及聚碳酸酯(pc、iupilon s-2000f、三菱工程塑料公司制、粘度平均分子量24,000)按表17的比率配合,得到混合物。将该混合物供给到加热到筒体温度260℃的脱挥式双螺杆挤出机(池贝公司制、pcm-30(商品名))中进行混炼,制作各树脂组合物粒料。
[0289]
将各粒料在下述条件下注塑成形,制作评价用试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:260℃、模具温度:60℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm
[0290]
对试验片a,依据iso179-1刻划出a型切口,在23℃和-30℃下测定夏比冲击强度。可以说数值越高,耐冲击性越好,越优选。
[0291]
[熔融流动性]对各粒料在下述条件下连续注塑成形15次,评价其螺旋流动长度(sfl)。15次中最大sfl和最小sfl和它们的差值δsfl(最大sfl-最小sfl)如表15所示。数值越高,熔融流动性越高,越优选。另外,最小和最大的差越小,成形稳定性越好,越优选。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:300℃、模具温度:70℃、注塑速度:20mm/sec、注塑压力:50mpa
[0292]
[表17]
[0293]
实施例38的冲击强度、熔融流动性和成形稳定性优异。实施例39的冲击强度和熔融流动性优异。但是,由于不含成分(d),因此成形稳定性比实施例41低。在比较例43中,由于不使用聚合物粒子群(c),因此冲击强度为低位。在比较例44中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度、熔融流动性及成形稳定性为低位。
[0294]
《实施例40~42、比较例45~46》将上述制造例2-9~2-10中得到的各组合物、聚合物粒子群(c-1)、聚碳酸酯(pc、iupilon s-2000f、三菱工程塑料公司制、粘度平均分子量24,000)和聚对苯二甲酸乙二醇酯(pet、trn8550ff、帝人公司制)按表18的比例配合,得到混合物。将该混合物供给到加热至筒体温度280℃的脱挥式双螺杆挤出机(池贝公司制、pcm-30(商品名)中进行混炼,制作各树脂组合物粒料。
[0295]
将各粒料在下述条件下注塑成形,制作评价用试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:300℃、模具温度:70℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm
[0296]
[夏比冲击强度]对试验片a,依据iso179-1刻划出a型切口,在23℃和-30℃下测定夏比冲击强度。数值越高,耐冲击性越好,越优选。
[0297]
[熔融流动性]对各粒料在下述条件下连续注塑成形15次,评价其螺旋流动长度(sfl)。15次中最大sfl和最小sfl和它们的差值δsfl(最大sfl-最小sfl)如表15所示。数值越高,熔融流动性越高,越优选。另外,最小和最大的差越小,成形稳定性越好,越优选。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:300℃、模具温度:70℃、注塑速度:20mm/sec、注塑压力:50mpa
[0298]
[表18]
[0299]
实施例40~41的冲击强度、熔融流动性及成形稳定性优异。实施例42的冲击强度和熔融流动性优异。但是,由于不含成分(d),因此成形稳定性比实施例40~41低。在比较例45中,由于不使用聚合物粒子群(c),因此冲击强度和流动性为低位。在比较例46中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度、熔融流动性及成形稳定性为低位。
[0300]
《实施例43~44、比较例47~48》将上述制造例2-10中得到组合物及聚合物(c-1)和玻璃纤维添加聚碳酸酯树脂(pcgf-2、iupilon gsh2030r2、三菱工程塑料公司制、添加30质量%的玻璃纤维)和聚对苯二甲酸乙二醇酯(pet、trn8550ff、帝人公司制)以表19的比例配合,得到混合物。将该混合物供给到加热到筒体温度300℃的脱挥式双螺杆挤出机(池贝公司制、pcm-30(商品名)中进行混炼,制作各树脂组合物粒料。
[0301]
将各粒料在下述条件下注塑成形,制作评价用试验片。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:300℃、模具温度:70℃试验片规格:试验片a:长度80mm
×
宽度10mm
×
厚度4mm
[0302]
[夏比冲击强度]对试验片a,依据iso179-1刻划出a型切口,在23℃和-30℃下测定夏比冲击强度。数值越高,耐冲击性越好,越优选。
[0303]
[熔融流动性]对各粒料在下述条件下连续注塑成形15次,评价其螺旋流动长度(sfl)。15次中最大sfl和最小sfl和它们的差值δsfl(最大sfl-最小sfl)如表15所示。数值越高,熔融流动性越高,越优选。另外,最小和最大的差越小,成形稳定性越好,越优选。注塑成形机:住友重机械工业公司制、se100du(商品名)料筒温度:300℃、模具温度:70℃、注塑速度:20mm/sec、注塑压力:50mpa
[0304]
[表19]
[0305]
实施例43的冲击强度、熔融流动性和成形稳定性优异。实施例44的冲击强度和熔融流动性优异。但是,由于不含成分(d),因此成形稳定性比实施例43低。在比较例47中,由于不使用聚合物粒子群(c),因此冲击强度和流动性为低位。在比较例48中,由于使用z值为60%以上的聚合物粒子群(c),因此冲击强度、熔融流动性及成形稳定性为低位。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1