可用于治疗糖尿病的6-甲氧基-3,4-二氢-1H-异喹啉化合物的制作方法
可用于治疗糖尿病的6-甲氧基-3,4-二氢-1h-异喹啉化合物
1.本发明涉及6-甲氧基-3,4-二氢-1h-异喹啉化合物、胰高血糖素样肽-1受体激动剂、葡萄糖依赖性的促胰岛素多肽(gip)激动剂、胰高血糖素激动剂和所述化合物用于治疗ii型糖尿病的治疗用途。
2.变构调节剂是通过改变配体结合环境而远程改变配体与其受体的相互作用的药剂。这种类型的调节的一个例子是,当调节剂与变构(二级)位点的结合产生受体蛋白中的构象变化时,所述构象变化被传递到配体的正构(初级)结合位点。如果调节剂促进或增强配体与正构结合位点的相互作用,就说变构效应的性质是正的。变构效应可以用于正构结合位点中的配体结合的表征和结晶。
3.提供的化合物是胰高血糖素样肽-1(glp-1)受体的正变构调节剂(pam)。提供的化合物是葡萄糖依赖性的促胰岛素多肽(gip)受体的正变构调节剂(pam)。提供的化合物是胰高血糖素受体的正变构调节剂(pam)。glp-1是由肠的肠道内分泌l-细胞分泌的肠降血糖素家族的肽激素的一个成员。glp-1以葡萄糖依赖性的方式诱导胰岛素从β细胞的释放。glp-1激动剂已被批准用于治疗ii型糖尿病;但是,存在对替代性glp-1激动剂治疗以及结合表征的需要。存在对gip结合表征的需求。
4.在一个实施方案中,本发明提供了新颖的化合物,其为glp-1r和/或gipr和/或胰高血糖素受体的有效正变构调节剂。
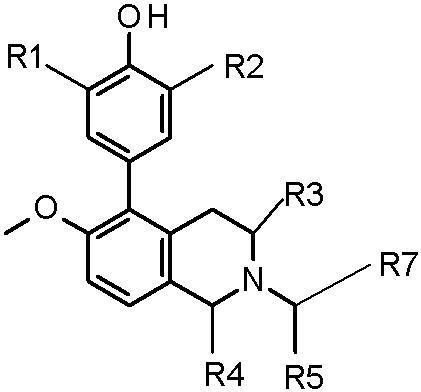
5.在一个实施方案中,本发明提供了下式的化合物或其药学上可接受的盐:
[0006][0007]
其中
[0008]
r1选自下组:f和h;
[0009]
r2选自下组:ch3、ch3chch3和-nhch2ch(ch3)2;
[0010]
r3选自下组:h和-ch2och3;
[0011]
r4选自下组:h和ch3;
[0012]
r5选自下组:h和ch3;
[0013]
r6选自下组:h和oh;
[0014]
r7选自下组:
[0015]
在一个实施方案中,本发明提供了下式的化合物或其药学上可接受的盐:
[0016][0017]
其中
[0018]
r1选自下组:f和h;
[0019]
r2选自下组:ch3和-nhch2ch(ch3)2;
[0020]
r3选自下组:h和-ch2och3;
[0021]
r4选自下组:h和ch3;
[0022]
r5选自下组:h和ch3;
[0023]
r6选自下组:h和oh。
[0024]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r4是ch3。
[0025]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r5是ch3。
[0026]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r6是h。
[0027]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r1是f。
[0028]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r3是h。
[0029]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r4是ch3、r5是ch3且r6是h。
[0030]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r4是ch3、r5是ch3、r6是h、r1是f且r3是h。
[0031]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r2是nhch2ch(ch3)2。
[0032]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐,其中r2是ch3。
[0033]
在一个实施方案中是式i的化合物或其药学上可接受的盐,其中所述化合物选自下组:
[0034][0035]
在一个实施方案中是下式的化合物:
[0036]
或其药学上可接受的盐。
[0037]
在一个实施方案中是下式的化合物:
[0038][0039]
或其药学上可接受的盐。
[0040]
在一个实施方案中是下式的化合物:
[0041][0042]
或其药学上可接受的盐。
[0043]
在一个实施方案中是式i的化合物,其中所述化合物是盐酸盐。
[0044]
在一个实施方案中是式ii的化合物,其中所述化合物是
[0045]
或其药学上可接受的盐。
[0046]
在一个实施方案中是一种药物组合物,其包含式i或式ii的化合物或其药学上可接受的盐和至少一种药学上可接受的载体、稀释剂或赋形剂。
[0047]
在一个实施方案中是一种用于治疗哺乳动物中的ii型糖尿病的方法,所述方法包括给所述哺乳动物施用有效量的式i或式ii的化合物或其药学上可接受的盐。
[0048]
在一个实施方案中是用于疗法中的式i的化合物或其药学上可接受的盐。
[0049]
在一个实施方案中是用于治疗ii型糖尿病的式i或式ii的化合物或其药学上可接受的盐。
[0050]
在一个实施方案中是式i或式ii的化合物或其药学上可接受的盐在制备药物中的用途,所述药物用于治疗ii型糖尿病。
[0051]
式ii和式i包括所有单独对映异构体、及其混合物、以及外消旋体及其药学上可接受的盐。
[0052]
在另一个实施方案中,本发明也提供了一种药学上可接受的组合物,其包含式i或式ii的化合物或其药学上可接受的盐以及至少一种药学上可接受的载体、稀释剂或赋形剂。在一个优选的实施方案中,药学上可接受的组合物被配制用于口服施用。
[0053]
在另一个实施方案中,本发明提供了一种治疗哺乳动物中的ii型糖尿病的方法,所述方法包括给需要治疗的哺乳动物施用药学上可接受的组合物,其包含有效量的式i的化合物或其药学上可接受的盐以及至少一种药学上可接受的载体、稀释剂或赋形剂。优选地,所述哺乳动物是人。
[0054]
在另一个实施方案中,本发明也提供了一种用于治疗哺乳动物中的ii型糖尿病的方法,所述方法包括给需要治疗的哺乳动物施用有效量的根据式i或式ii的化合物或其药学上可接受的盐。
[0055]
式i的化合物可以与其它化合物组合使用,所述其它化合物用于治疗式i或式ii的化合物对其有用的疾病或病症,包括ii型糖尿病。
[0056]
在另一个实施方案中,本发明也提供了一种降低哺乳动物中的血糖水平的方法,
所述方法包括给需要治疗的哺乳动物施用有效量的根据式ii或式i的化合物或其药学上可接受的盐。
[0057]
在另一个实施方案中,本发明也提供了一种治疗哺乳动物中的高血糖症的方法,所述方法包括给需要治疗的哺乳动物施用有效量的根据式i的化合物或其药学上可接受的盐。
[0058]
在一个实施方案中,本发明提供了用于疗法中的根据式i的化合物或其药学上可接受的盐。
[0059]
在另一个实施方案中,本发明也提供了用于治疗哺乳动物中的ii型糖尿病的根据式i的化合物或其药学上可接受的盐。
[0060]
在一个实施方案中,本发明提供了根据式i的化合物或其药学上可接受的盐用于制备药物的用途,所述药物用于治疗哺乳动物中的ii型糖尿病。
[0061]
本文中使用的术语“药学上可接受的盐”表示被认为对于临床和/或兽医学应用而言可接受的本发明的化合物的盐。药学上可接受的盐的例子和制备它们的常用方法可以参见“handbook of pharmaceutical salts:properties,selection and use”p.stahl,等人,第2次修订版,wiley-vch,2011和s.m.berge,等人,“pharmaceutical salts”,journal of pharmaceutical sciences,1977,66(1),1-19。
[0062]
药物组合物及其制备方法的例子可以参见“remington:the science and practice of pharmacy”,loyd,v.,等人编,第22版,mack publishing co.,2012。在一个实施方案中,药物组合物可以被配制用于口服施用。优选地,药物组合物被配制为片剂、胶囊或溶液。所述片剂、胶囊或溶液可以包括有效治疗需要治疗的患者的量的本发明的化合物。
[0063]
术语“有效量”表示本发明的化合物或其药学上可接受的盐的量或剂量,其在向患者单剂量或多剂量施用后,在诊断或治疗的患者中提供期望的效果。作为本领域技术人员的主治医师通过使用常规技术和通过观察在类似情况下得到的结果,可以容易地确定有效量。在确定化合物的有效量或剂量时考虑的因素包括:将施用化合物还是其盐;如果使用的话,其它药剂的共同施用;哺乳动物的物种;它的大小、年龄和一般健康;所述障碍的涉及程度或严重程度;个体哺乳动物的应答;施用模式;施用的制品的生物利用度特征;选择的给药方案;和其它有关的情况。本发明的化合物在落在约0.01至约15mg/kg体重的范围内的每日剂量是有效的。
[0064]
本文中使用的术语“治疗”或“处理”(
″
treating
″
、“to treat”或
″
treatment
″
)表示降低、减轻或逆转现有症状、障碍或病症的进展或严重程度,诸如高血糖症,其可以包括增加胰岛素分泌。
[0065]
本文中使用的术语“患者”表示哺乳动物。优选地,所述患者是人。
[0066]
本发明的化合物可以被配制为药物组合物,其通过使所述化合物生物可利用的任何途径施用。优选地,这样的组合物用于口服施用。这样的药物组合物及其制备方法是本领域众所周知的(参见,例如,remington,j.p.,“remington:the science and practice of pharmacy”,l.v.allen,编,第22版,pharmaceutical press,2012)。
[0067]
式i的化合物及其药学上可接受的盐可用于本发明的治疗用途中,其中某些构型是优选的。
[0068]
尽管一个实施方案考虑了所有单独对映异构体、其混合物和外消旋体,但式ic的
化合物或其药学上可接受的盐是特别优选的。在一个实施方案中,式ib的化合物或其药学上可接受的盐是特别优选的。在一个实施方案中,式ia的化合物或其药学上可接受的盐是特别优选的。
[0069]
通过方法诸如选择性结晶技术、手性色谱(参见例如,j.jacques,等人,“enantiomers,racemates,and resolutions”,john wiley and sons,inc.,1981,以及e.l.eliel和s.h.wilen,“stereochemistry of organic compounds”,wiley-interscience,1994)或超临界流体色谱(sfc)(参见例如,t.a.berger;“supercritical fluid chromatography primer,”agilent technologies,2015年7月),本领域普通技术人员可以在本发明的化合物的合成中的任何方便点分离或拆分单独对映异构体。
[0070]
例如,通过使本发明的化合物的适当中性形式与适当的药学上可接受的酸或碱在合适的溶剂中在本领域众所周知的标准条件下反应(参见,例如,bastin,r.j.,等人;org.process.res.dev.,4,427-435,2000和berge,s.m.,等人;j.pharm.sci.,66,1-19,1977),可以形成本发明的化合物的药学上可接受的盐。
[0071]
根据daub g.h.,等人,“the use of acronyms in organic chemistry”aldrichimica acta,1984,17(1),6-23定义本文中使用的某些缩写。某些缩写定义如下:“acn”表示乙腈;“atp”表示腺苷三磷酸;“boc”表示叔丁氧基羰基;“bsa”表示牛血清白蛋白;“camp”表示环状腺苷-3’,5
’‑
单磷酸;“dcm”表示二氯甲烷(dichloromethane或methylene chloride);“dipea”表示n,n-二异丙基乙胺;“dmf”表示n,n-二甲基甲酰胺;“dmso”表示二甲基亚砜;“ec
50”表示与预定义的阳性对照化合物(绝对ec
50
)相比,产生目标活性的50%应答的药剂的浓度;“es/ms”表示电喷雾质谱;“etoac”表示乙酸乙酯;“hatu”表示1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐;“hek”表示人胚胎肾;“hepes”表示4-(2-羟基乙基)-1-哌嗪乙磺酸;“h”分别表示几小时或1小时;“meoh”表示甲醇(methanol或methyl alcohol);“min”表示1分钟或几分钟;“mtbe”表示甲基叔丁基醚;“pd(dppf)cl2·
dcm”表示1,1
’‑
双(二苯基膦基)二茂铁-二氯化钯(ii)二氯甲烷复合物;“rt”表示室温;“scx”表示强阳离子交换;“sfc”表示超临界流体色谱;且“thf”表示四氢呋喃。
[0072]
本发明的化合物可以通过多种程序制备,其中一些在下面的制备和实施例中进行了举例说明。通过常规方法,包括萃取、蒸发、沉淀、色谱、过滤、研磨和结晶,可以回收以下每个步骤的产物。通过方法诸如选择性结晶技术或手性色谱(参见例如,j.jacques,等人,“enantiomers,racemates,and resolutions”,john wiley and sons,inc.,1981,以及e.l.eliel和s.h.wilen,“stereochemistry of organic compounds”,wiley-interscience,1994),可以在合成中的任何方便点分离或拆分单独异构体、对映异构体和非对映异构体。并非限制本发明范围,提供以下制备和实施例以进一步举例说明本发明。
[0073]
方案1
[0074][0075]
方案1显示了通过两个途径合成式i的化合物。在第一个途径中,在步骤1a中,将关键中间体1在有机溶剂中使用hcl去保护,随后在ki和有机碱存在下在升高的温度用烷基氯2烷基化以产生中间体3。在步骤2a中,中间体3然后经历一锅miyaura硼基化/suzuki偶联:使用双戊酰二硼、pd(dppf)cl2·
dcm和乙酸钾在升高的温度,芳基卤4被转化成硼酸酯5,此后将中间体3和磷酸三钾水溶液加入反应中,产生式i的化合物。或者,在类似的miyaura硼基化条件下在单独的步骤中从芳基卤4制备硼酸酯5,并然后在步骤2a中,它经历与中间体3的suzuki偶联以产生式i的化合物。
[0076]
在第二个途径中,在步骤1b中,中间体1经历与硼酸酯5的suzuki偶联,其中在升高的温度在2-甲基-2-丁醇中使用第2代xphos预催化剂和磷酸三钾水溶液,这也除去boc保护基团,产生胺中间体6。在步骤2b中用三乙酰氧基硼氢化钠对中间体6和羰基中间体7的还原胺化产生式i的化合物。
[0077]
在hp1200液相色谱系统上进行lc-es/ms。在与hplc接口的质量选择性检测器(mass selective detector)四极质谱仪上进行电喷雾质谱测量(以正和/或负模式获取),所述hplc可以具有或没有elsd。lc-es/ms条件(低ph):柱:nx c18 2.0
×
50mm3.0mm,梯度:在1.5min内5-95%b,然后在95%b保持0.5min;柱温:50℃+/-10℃;流速:1.2ml/min;1ml注射体积;溶剂a:含有0.1%hcooh的去离子水;溶剂b:含有0.1%甲酸的acn;波长200-400nm和212-216nm。如果hplc配备elsd,则设置为45℃蒸发器温度,40℃雾化器温度,和1.6slm气体流速。替代性lc-ms条件(高ph):柱:watersc18柱2.1
×
50mm,3.5mm;梯度:在1.5min内5-95%b,然后在95%b保持0.50min;柱温:50℃+/-10℃;流速:1.2ml/min;1μl注射体积;溶剂a:10mm nh4hco
3 ph 9;溶剂b:acn;波长:200-400nm和212-216nm;如果具有elsd:45℃蒸发器温度,40℃雾化器温度,和1.60slm气体流速。
[0078]
制备1
[0079]
n-[2-(3-甲氧基苯基)乙基]乙酰胺
[0080][0081]
将3-甲氧基苯乙胺(48g,308mmol)溶解在dcm(384ml)中,加入三乙胺(64.4ml,462mmol)并冷却至0-5℃。逐滴加入乙酰氯(24.1ml,339mmol)并在环境温度搅拌1h。加入水并分离各层。将有机层用0.5m hcl和饱和nahco3洗涤,经mgso4干燥,过滤并在真空中浓缩以得到作为黄色油的标题化合物(59g,99%)。es/ms m/z:194[m+h]
+
。
[0082]
制备2
[0083]
6-甲氧基-1-甲基-3,4-二氢异喹啉
[0084]
将n-[2-(3-甲氧基苯基)乙基]乙酰胺(5g,26mmol)溶解在acn(74ml)中。加入磷酰氯(2.9ml,31.0mmol)并将反应物在75℃加热3h。将混合物冷却至环境温度,倒入k3po4水溶液(2m,200ml)中并搅拌30min。用etoac萃取两次。将有机层合并并用饱和nacl水溶液洗涤,经mgso4干燥,过滤并在真空中浓缩以得到作为棕色油的标题化合物(4.6g,91%)。es/ms m/z:176[m+h]
+
。
[0085]
制备3
[0086]
(1r)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0087][0088]
将6-甲氧基-1-甲基-3,4-二氢异喹啉(4.6g,26mmol)溶解在acn(90ml)中。加入甲酸/三乙胺5:2复合物(17ml,40mmol),随后加入(s,s)-n-(对甲苯磺酰基)-1,2-二苯基乙烷二胺(氯)(对伞花烃)钌(ii)(86mg,0.13mmol)。在温和氮气流下在环境温度搅拌17h。用etoac稀释,用k2co3水溶液(3m)和饱和nacl水溶液洗涤两次。将有机物经na2so4干燥,过滤并在真空中浓缩。将粗制物质在水(2ml)中搅拌。加入在水(2ml)中的二碳酸二叔丁酯(0.2g,0.9mmol)并剧烈搅拌2.5h。加入khso4水溶液(1m溶液,20ml),搅拌2分钟并分离各层。将有机层用k2co3水溶液(3m)和饱和nacl水溶液洗涤。将有机物经mgso4干燥,过滤并在真空中浓缩。将粗产物溶解在dcm中并在上浓缩,然后使用0-50%的在己烷类中的etoac的梯度通过硅胶色谱纯化以得到作为无色油的标题化合物(6.26g,82%)。es/ms m/z:222[m+h-t-bu]
+
。
[0089]
制备4
[0090]
(1r)-5-溴-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0091][0092]
将(1r)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯(60g,216mmol)和乙酸钠(17.4ml,324mmol)溶解在乙酸(300ml)中并搅拌30min。加入n-溴代琥珀酰亚胺(42.3g,238mmol)并在环境温度搅拌1h。加入水并用己烷萃取两次。将有机层合并并用饱和
nahco3水溶液洗涤。将有机物经mgso4干燥,过滤并在真空中浓缩。将粗产物通过快速色谱纯化,用10-15%的在己烷中的etoac的梯度洗脱。将纯化的物质与己烷(40ml)一起研磨并搅拌过夜。过滤固体以得到作为白色固体的标题化合物(21.5g,28%),通过分析型手性sfc(柱:ad 10
×
46cm,5μm;流动相:等度1∶9(异丙醇+0.2%异丙胺):co2;流速:5ml/min;柱温:40℃;出口压强:150巴;保留时间:0.79min(期望的异构体),0.97min)确定为98%ee。es/ms m/z:300,302[m+h]
+
。
[0093]
制备5
[0094]
(1r)-5-溴-6-甲氧基-1-甲基-1,2,3,4-四氢异喹啉;盐酸盐
[0095][0096]
将(1r)-5-溴-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯(7.4g,21mmol)溶解在dcm(100ml)中并加入氯化氢(4m在1,4-二氧杂环己烷中,26ml,104mmol)。在环境温度搅拌17h。过滤并用etoac洗涤固体。将固体在高真空下干燥以得到作为白色固体的标题化合物(5.93g,95%)。es/ms m/z:256,258[m+h]
+
。
[0097]
制备6
[0098]
(1r)-5-溴-2-[1-(4-异丙基苯基)乙基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉(异构体1和异构体2)
[0099][0100]
在可重新密封的试管中将外消旋的1-(1-氯乙基)-4-异丙基-苯(2g,8.76mmol)、n,n-二异丙基乙胺(2ml,11.5mmol)和碘化钾(1.3g,7.8mmol)溶解在无水n,n-二甲基乙酰胺(100ml)中,并在环境温度搅拌10min。一次性加入(1r)-5-溴-6-甲氧基-1-甲基-1,2,3,4-四氢异喹啉;盐酸盐(2g,6.83mmol)。给管形瓶加盖并在60℃搅拌96h。冷却至环境温度并在水/饱和nahco3(1∶1)和etoac之间分配。将水层用etoac萃取两次。将有机层合并,用水和饱和nacl水溶液洗涤,经na2so4干燥,过滤并在真空中浓缩。将粗制物质通过硅胶上的快速色谱纯化,用17%的在dcm中的etoac洗脱以得到作为淡棕色固体的标题化合物的非对映异构体的混合物(2g,67%)。es/ms m/z:402,404[m+h]
+
。
[0101]
将所述非对映异构体混合物与另一批如上制备的非对映异构体混合物(1.5g)合并。将非对映异构体混合物(3.5g,8.2mmol)溶解在4:1meoh/dcm(100ml)中。将所述混合物通过手性sfc(柱:ig(25
×
2cm,5μm);流动相:在co2中的等度10%(含有0.2%二甲基乙胺的乙醇);温度:40℃;流速:65g/min;出口压强:120巴)分离以得到作为白色固体的异构体1(第一洗脱,1.08g,33%)和异构体2(第二洗脱,0.91,28%)。
[0102]
分析型手性sfc(柱:chiralpak(10cm
×
4.6mm,5μm);流动相:在co2中的5至25%(含有0.2%异丙胺的乙醇);温度:40℃;流速:4ml/min;出口压强:120巴)显示异构体1
(保留时间1.4min)是95%de且异构体2(保留时间1.6min)是>98%de。
[0103]
制备7
[0104]
4-溴-2-氟-6-(异丁基氨基)苯酚
[0105][0106]
将2-氨基-4-溴-6-氟苯酚(700mg,3.40mmol)和2-甲基丙醛(0.5ml,5mmol)溶解在thf(5.7ml)中。加入乙酸(3.4ml,59mmol)和甲醇(34ml)并在0℃搅拌2min。加入2-甲基吡啶硼烷复合物(640mg,5.08mmol)并在60℃搅拌17h。在减压下除去溶剂并将残余物在scx柱上纯化,将化合物用nh3在meoh中的溶液(2m)洗脱。将所述物质使用0-30%的在己烷类中的etoac的梯度通过硅胶色谱重新纯化,以得到作为粘性油的标题化合物(380mg,43%)。es/ms m/z:260,262[m-h]
+
。
[0107]
实施例1
[0108]
2-氟-6-(异丁基氨基)-4-[(1r)-2-[1-(4-异丙基苯基)乙基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-5-基]苯酚;二盐酸盐
[0109][0110]
混合4-溴-2-氟-6-(异丁基氨基)苯酚(200mg,0.76mmol)和无水1,4-二氧杂环己烷(2ml)。用氮气在混合物中鼓泡15min并加入双戊酰二硼(240mg,0.9263mmol)、pd(dppf)cl2·
dcm(32mg,0.038mmol)和乙酸钾(116mg,1.15mmol)。用氮气在混合物中鼓泡5min,密封反应容器并在90℃搅拌17h。将混合物冷却至环境温度并加入(1r)-5-溴-2-[1-(4-异丙基苯基)乙基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉,异构体2(210mg,0.52mmol)、(2-二环己基膦基-2’,4’,6
’‑
三异丙基-1,1’联苯)[2-(2
’‑
氨基-1,1
’‑
联苯)]甲磺酸钯(ii)(23mg,0.026mmol)和磷酸三钾(1m在水中,1.5ml,1.5mmol)。密封反应容器并将混合物在90℃搅拌4h。将反应混合物冷却至环境温度并加入etoac。将有机物用5%柠檬酸水溶液洗涤,经na2so4干燥,过滤并在真空中浓缩。将残余物在scx柱上纯化,用2n的在meoh中的nh3洗脱。使用0-30%的在dcm中的etoac的梯度通过硅胶色谱进一步纯化产物。使用0-40%的在己烷类中的etoac的梯度通过硅胶色谱再次纯化产物。将产物通过sfc(柱:hilic(30
×
150mm,5μm);10%至20%的在co2中的(10mm的在meoh中的nh4hco3,ph 8)梯度)进一步纯化以得到作为白色固体的标题化合物的游离碱(68mg,17%)。es/ms m/z:505[m+h]
+
。
[0111]
将标题化合物的游离碱(68mg,0.14mmol)溶解在acn(3ml)中并加入氯化氢(2m在
乙醚中,0.40ml,0.8mmol)。搅拌2min并在40℃用氮气流除去溶剂。在真空中干燥96h以得到作为深棕色固体的标题化合物(77mg,97%)。es/ms m/z:505[m+h]
+
。
[0112]
制备8
[0113]
2-乙酰氨基-2-[(2-氯-3-甲氧基-苯基)甲基]丙二酸二乙酯
[0114][0115]
在acn(958ml)中混合1-(溴甲基)-2-氯-3-甲氧基-苯(63.9g,271mmol)、乙酰氨基丙二酸二乙酯(60.1g,271mmol)、碳酸钾(75.0g,542mmol)和碘化钾(45.0g,271mmol),并将混合物在氮气下加热至回流保持2h。加入乙酰氨基丙二酸二乙酯(6.6g,29.8mmol)并回流另外36h。浓缩溶剂至1/3体积,并加入水和etoac。分离有机层并将水层用etoac萃取两次。将有机层合并,用水和饱和nacl水溶液洗涤,经na2so4干燥并浓缩以得到作为白色泡沫的标题化合物(105.7g,100%)。es/ms m/z:372[m+h]
+
。
[0116]
制备9
[0117]
5-氯-6-甲氧基-1,2,3,4-四氢异喹啉-3-甲酸;盐酸盐
[0118][0119]
混合2-乙酰氨基-2-[(2-氯-3-甲氧基-苯基)甲基]丙二酸二乙酯(105.6g,284mmol)、乙酸(211ml)和盐酸(37%在h2o中,528ml,6400mmol),并将混合物在氮气下加热至回流保持18h。将混合物冷却至50℃,逐滴加入甲醛(37%在水中,284ml,3790mmol)并在100℃搅拌2h。将混合物冷却至0℃并搅拌30min。将得到的固体过滤,用水洗涤并干燥以得到作为白色固体的标题化合物(68.8g,87%)。es/ms m/z:242[m+h]
+
。
[0120]
制备10
[0121]
5-氯-6-甲氧基-1,2,3,4-四氢异喹啉-3-甲酸甲酯(异构体1和异构体2)
[0122][0123]
将5-氯-6-甲氧基-1,2,3,4-四氢异喹啉-3-甲酸盐酸盐(60.5g,218mmol)悬浮在甲醇(860ml)中并在氮气下在5℃搅拌。逐滴加入亚硫酰氯(77.6g,653mmol)并将混合物在氮气下回流20h。将混合物冷却至环境温度并在真空中除去溶剂。将残余物与etoac一起研磨,然后过滤并将固体用etoac洗涤。将固体在真空下在40℃干燥过夜以得到作为外消旋混合物的标题化合物的盐酸盐(58.8g,92%)。
[0124]
将固体的一部分(15.00g,49.80mmol)悬浮在饱和k2co3水溶液(15ml)中并在环境温度搅拌30min。将固体过滤并用水洗涤。将白色固体在真空中在40℃干燥。将固体溶解在dcm中并使用0-5%的在dcm中的meoh的梯度穿过硅胶塞过滤以产生作为外消旋混合物的标题化合物(12.18g)。es/ms m/z 256[m+h]
+
。
[0125]
将外消旋混合物的一部分(10.15g)通过手性sfc(柱:ig(25
×
2cm,5μm);流动相:15%的在co2中的(meoh+0.2%二甲基乙胺);温度:环境温度;流速:80g/min;出口压强:120巴)分离以得到标题化合物的异构体1(第一洗脱,2.69g,26%)和异构体2(第二洗脱,4.10,40%)。分析型手性sfc(柱:ig(10cm
×
4.6mm,5μm);流动相:15-55%的在co2中的(甲醇+0.2%异丙胺);温度:40℃;流速:4ml/min;出口压强:120巴)显示异构体1(保留时间:1.58min)是>98%ee且异构体2(保留时间:1.66min)是95%ee。
[0126]
制备11
[0127]
5-氯-6-甲氧基-3,4-二氢-1h-异喹啉-2,3-二甲酸o2-叔丁基o3-甲基酯
[0128][0129]
将二碳酸二叔丁酯(3.5g,16mmol)加入到5-氯-6-甲氧基-1,2,3,4-四氢异喹啉-3-甲酸甲酯(异构体1,3.6g,14mmol)在dcm(70ml)中的溶液中,并在环境温度搅拌4h。在减压下除去溶剂并使用0-20%的在dcm中的etoac的梯度通过硅胶色谱纯化残余物。将固体用己烷洗涤并过滤以得到作为白色固体的标题化合物(4.55g,90%)。es/ms m/z:256[m+h-boc]
+
。
[0130]
制备12
[0131]
5-氯-3-(羟基甲基)-6-甲氧基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0132][0133]
在环境温度将硼氢化锂(2.0m在thf中,13ml,26mmol)加入到5-氯-6-甲氧基-3,4-二氢-1h-异喹啉-2,3-二甲酸o2-叔丁基o3-甲基酯(4.55g,12.8mmol)在无水thf(40ml)中的溶液中并搅拌6h。加入水并将混合物在环境温度搅拌10min。用水稀释并用etoac萃取两次。将有机层合并,并用水、饱和nahco3水溶液和饱和nacl水溶液洗涤。将有机物经无水na2so4干燥,过滤并在真空中浓缩。将残余物在真空中在环境温度干燥17h以得到作为蜡状固体的标题化合物(4.4g,100%)。es/ms m/z:272[m+h-boc]
+
。
[0134]
制备13
[0135]
5-氯-6-甲氧基-3-(甲氧基甲基)-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0136][0137]
将氢化钠(60%在油中,2.1mg,0.053mmol)加入到5-氯-3-(羟基甲基)-6-甲氧基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯(4.4g,13mmol)和碘甲烷(8.1ml,130mmol)在无水dmf(87ml)中的0℃溶液中,并将反应物搅拌10min。逐滴加入水直到不再观察到气体产生,并将混合物在水和乙酸乙酯之间分配。将有机层用水和饱和nacl水溶液洗涤,经无水na2so4干燥,过滤并在真空中浓缩。将残余物使用0-10%的在dcm中的etoac的梯度通过硅胶色谱纯化以得到作为蜡状固体的标题化合物(4.5g,94%)。es/ms m/z:242[m+h-t-bu]
+
。
[0138]
制备14
[0139]
4-[6-甲氧基-3-(甲氧基甲基)-1,2,3,4-四氢异喹啉-5-基]-2-甲基-苯酚;盐酸盐
[0140][0141]
在可重新密封的试管中将5-氯-6-甲氧基-3-(甲氧基甲基)-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯(2.68g,7.84mmol)和2-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯酚(2.45g,9.42mmol)溶解在2-甲基-2-丁醇(54ml)中,并在搅拌的同时用氮气流净化30min。加入氯(2-二环己基膦基-2
′
,4
′
,6
′‑
三异丙基-1,1
′‑
联苯)[2-(2
′‑
氨基-1,1
′‑
联苯)]钯(ii)(第2代xphos预催化剂,325mg,0.39mmol)和磷酸三钾(1m在水中,16ml,16mmol),密封容器并将混合物在85℃加热2h。将混合物冷却至环境温度,穿过过滤并用etoac冲洗。将有机层用水和饱和nacl水溶液洗涤,经无水na2so4干燥,过滤并在真空中浓缩。使用0-10%的在dcm中的etoac的梯度通过硅胶色谱纯化。将物质溶解在dcm(30ml)中,加入氯化氢(4m在1,4-二氧杂环己烷中,10ml,40mmol)并将混合物在环境温度搅拌1.5h。除去溶剂,然后将固体与etoac一起研磨并过滤。将固体用etoac洗涤以得到作为白色固体的标题化合物(2.6g,91%)。es/ms m/z:314[m+h]
+
。
[0142]
实施例2
[0143]
4-[2-[(2-羟基-4-异丙基-苯基)甲基]-6-甲氧基-3-(甲氧基甲基)-3,4-二氢-1h-异喹啉-5-基]-2-甲基-苯酚;盐酸盐
[0144][0145]
将4-[6-甲氧基-3-(甲氧基甲基)-1,2,3,4-四氢异喹啉-5-基]-2-甲基-苯酚;盐酸盐(157mg,0.45mmol)悬浮在dcm(3ml)中并加入4-异丙基水杨醛(0.11ml,0.71mmol)和三乙胺(0.19ml,1.35mmol)。20min以后,加入三乙酰氧基硼氢化钠(294mg,1.35mmol)并将混合物在环境温度搅拌16h。加入4-异丙基水杨醛(0.060ml,0.39mmol)和三乙酰氧基硼氢化钠(151mg,0.69mmol)并将混合物搅拌6h。将混合物用dcm稀释并用饱和nh4cl水溶液洗涤。将水层用dcm萃取。将有机层合并,经na2so4干燥,过滤并在真空中浓缩。将残余物使用0-25%的在dcm中的etoac的梯度通过硅胶色谱纯化。通过硅胶色谱进一步纯化,用己烷/etoac 3∶1、随后用2∶1洗脱。加载到scx柱上并将化合物用2n的在meoh中的nh3洗脱。将化合物溶解在dcm(1ml)中并加入氯化氢(2m在乙醚中,0.50ml,1.0mmol)。浓缩至干燥并在高真空下在40℃干燥48h以得到作为白色固体的标题化合物(138mg,73%)。es/ms m/z:462[m+h]
+
。
[0146]
制备15
[0147]
2-氟-6-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯酚
[0148][0149]
将4-溴-2-氟-6-甲基-苯酚(24g,94mmol)、双戊酰二硼(28g,110mmol)和乙酸钾(13g,130mmol)溶解在脱气的无水1,4-二氧杂环己烷(200ml)中。加入pd(dppf)cl2·
dcm(5g,6.0mmol)。用氮气在混合物中鼓泡5min并在85℃搅拌2h。将反应混合物用dcm稀释并穿过过滤。将混合物在真空中浓缩并用etoac和水稀释。分离各层并将有机层用水洗涤,经无水mgso4干燥,过滤并在真空中浓缩。将残余物使用0-10%的在己烷类中的mtbe的梯度通过硅胶色谱纯化以得到作为白色固体的标题化合物(14.5g,61%)。es/ms m/z:251[m-h]
+
。
[0150]
实施例3
[0151]
2-氟-4-[(1r)-2-[1-(4-异丙基苯基)乙基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-5-基]-6-甲基-苯酚;盐酸盐
[0152][0153]
用氮气在(1r)-5-溴-2-[1-(4-异丙基苯基)乙基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉(异构体2,31mg,0.077mmol)、2-氟-6-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯酚(30mg,0.12mmol)、磷酸三钾(1m水溶液,0.17ml,0.17mmol)和2-甲基-2-丁醇的混合物中鼓泡5min。加入(2-二环己基膦基-2’,4’,6
’‑
三异丙基-1-1
’‑
联苯)[2-(2
’‑
氨基-1,1
’‑
联苯)]甲磺酸钯(ii)(xphos pd g3,4mg,0.5μmol)并将混合物加热至100℃保持3h。将混合物冷却至室温并倒在scx柱上。将柱用meoh洗脱并然后用在meoh中的nh3(2m)洗脱。在真空中从nh3/meoh洗脱液中除去溶剂。将残余物使用7-30%的在己烷类中的etoac的梯度通过硅胶色谱纯化以得到标题化合物的游离碱(28mg),然后加入acn并加入氯化氢(2m在乙醚中,0.20ml,0.31mmol)。用氮气流除去溶剂并将物质在40℃在高真空下干燥过夜以得到作为白色固体的标题化合物(26mg,85%)。es/ms m/z:448[m+h]
+
。
[0154][0155]
制备17
[0156]
2-苄氧基-1-溴-3-氟-5-碘-苯
[0157][0158]
将2-溴-6-氟-4-碘-苯酚(3.66g,11.5mmol)和碳酸钾(3.2g,23mmol)溶解在dmf(23ml)中并在室温搅拌5min。通过注射器加入苄基溴(2.1ml,18mmol)并在室温搅拌2小时。加入acoet和水,分离各层并将有机层用水洗涤,经mgso4干燥,过滤并在真空中浓缩以得到棕色油。通过快速色谱纯化,用0-20%的在己烷中的dcm的梯度洗脱以得到作为白色固体的标题化合物(4.53g,96%收率)。
[0159]
制备18
[0160]
(1r)-6-甲氧基-1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0161]
手性的
[0162][0163]
在微波管中在脱气的1,4-二氧杂环己烷(20ml)中合并制备4(1000mg,2.81mmol)、4,4,5,5-四甲基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1,3,2-二氧杂硼杂环戊烷(1000mg,3.94mmol)和乙酸钾(520mg,5.30mmol)。加入三(亚苄基丙酮)二钯(0)(160mg,0.17mmol)和三环己基膦(120mg,0.42mmol)。将混合物脱气5分钟,加盖并在微波中在150℃加热1小时。通过快速色谱纯化,用0-20%的在己烷中的甲基叔丁基醚的梯度洗脱以得到作为黄色油的标题化合物(942mg,75%收率)。ms(esi)m/z:304[m+h-(t-bu)]
+
。
[0164]
制备19
[0165]
(1r)-5-(4-苄氧基-3-溴-5-氟-苯基)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0166]
手性的
[0167][0168]
在可重新密封的试管中将(1r)-6-甲氧基-1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯(3380mg,8.38mmol)、2-苄氧基-1-溴-3-氟-5-碘-苯(4100mg,10.07mmol)、碳酸钠(2.7g,25.0mmol)溶解在1,4-二氧杂环己烷(67ml)和水(17ml)中。用氮气在溶液中鼓泡5分钟并加入四(三苯基膦)钯(0)(500mg,0.43mmol)。给试管加盖并在90℃搅拌5h。浓缩,吸附到上并通过快速色谱纯化,用0-30%的在己烷中的甲基叔丁基醚的梯度洗脱以得到作为白色固体的标题化合物(3.07g,90%收率)。ms(esi)m/z:502[m+h-(t-bu)]
+
。
[0169]
制备20
[0170]
(1r)-5-(4-苄氧基-3-氟-5-异丙烯基-苯基)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0171]
手性的
[0172][0173]
在可重新密封的试管中将(1r)-5-(4-苄氧基-3-溴-5-氟-苯基)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯(150mg,0.27mmol)、2-异丙烯基-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(100mg,0.59mmol)、碳酸钠(85mg,0.80mmol)溶解在1,4-二氧杂环己烷(4ml)和水(0.27ml)中。用氮气鼓泡5分钟并加入四(三苯基膦)钯(0))(17mg,0.015mmol),加盖并在90℃搅拌18h。浓缩并吸附在上,并通过快速色谱纯化,用20-60%的在己烷中的甲基叔丁基醚的梯度洗脱以得到作为无色油的标题化合物(200mg,49%收率)。ms(esi)m/z:418[m+h-boc]+。
[0174]
制备21
[0175]
(1r)-5-(3-氟-4-羟基-5-异丙基-苯基)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯
[0176]
手性的
[0177][0178]
将(1r)-5-(4-苄氧基-3-氟-5-异丙烯基-苯基)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸叔丁酯(270mg,0.25mmol)溶解在甲醇(5ml)中,加入钯(10%在c上,400mg,0.36mmol)。将混合物在氢气气氛(1大气压)下在室温搅拌18小时。经过滤并用乙醇洗涤。在真空中除去溶剂以得到作为棕色油的标题化合物(134mg,60%收率)。ms(esi)m/z:374[m+h-(t-bu)]
+
[0179]
制备22
[0180]
2-氟-6-异丙基-4-[(1r)-6-甲氧基-1-甲基-1,2,3,4-四氢异喹啉-5-基]苯酚
[0181]
手性的
[0182][0183]
将(1r)-5-(3-氟-4-羟基-5-异丙基-苯基)-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-2-甲酸酯(134mg,0.25mmol)溶解在dcm(1.2ml)中,加入盐酸(4m在二氧杂环己烷中,0.3ml,1mmo))。将混合物在室温搅拌2小时。除去溶剂并通过scx纯化,用甲醇洗脱,随后用2n的在甲醇中的nh3洗脱。合并碱性级分并除去溶剂以得到标题化合物(120mg,68质量%,99%收率)。ms(esi)m/z:330[m+h]
+
[0184]
制备23
[0185]
4-[(1r)-2-[(4,4-二甲基环己基)甲基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-5-基]-2-氟-6-异丙基-苯酚
[0186][0187]
将4,4-二甲基环己烷-1-甲醛(77mg,0.49mmol)加入到2-氟-6-异丙基-4-[(1r)-6-甲氧基-1-甲基-1,2,3,4-四氢异喹啉-5-基]苯酚(120mg,0.25mmol)在dcm(3ml)中的溶液中。将混合物在室温搅拌30min并加入三乙酰氧基硼氢化钠(100mg,0.47mmol)。将反应物在室温搅拌4小时。加入水和二氯甲烷。将水层用二氯甲烷萃取。将有机层合并,用饱和nacl水溶液洗涤,经无水硫酸镁干燥,过滤并在真空中浓缩。通过快速色谱纯化,用0%至20%的在己烷中的丙酮的梯度洗脱以得到作为无色油的标题化合物(90mg,78%收率)。ms(esi)m/z:454[m+h]
+
[0188]
实施例4
[0189]
合成4-[(1r)-2-[(4,4-二甲基环己基)甲基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-5-基]-2-氟-6-异丙基-苯酚;盐酸盐
[0190]
手性的
[0191]
[0192]
将4-[(1r)-2-[(4,4-二甲基环己基)甲基]-6-甲氧基-1-甲基-3,4-二氢-1h-异喹啉-5-基]-2-氟-6-异丙基-苯酚(90mg,0.19mmol)溶解在acn(1ml)中,加入盐酸(2m在乙醚中,0.18ml,0.36mmo))。将混合物在室温声处理5分钟。除去溶剂并将物质在45℃在真空中干燥18小时以得到作为黄褐色固体的标题化合物(90mg,94%收率)。ms(esi)m/z:454[m+h]
+
。
[0193]
生物学测定
[0194]
人gip受体增强剂测定
[0195]
在表达人gipr的hek293克隆细胞系(ncbi登录号np_000155)中使用camp形成确定gip受体(gipr)功能活性。所述测定测量在ec
20
剂量的gipr激动剂gip(1-42)存在下化合物诱导的camp产生。将表达hgipr受体的细胞用25pm的gip(1-42)(labcyte echo直接稀释物)处理,并确定20μl测定体积(最终的dmso浓度是1.0%)的在dmem(gibco目录号31053)中的试验化合物(在dmso中的10点浓度-应答曲线,3倍labcyte echo直接稀释物,384孔板corning目录号3570)的剂量应答,所述dmem补充了1x glutamax
tm
(gibco目录号35050)、0.1%牛酪蛋白(sigma c4765-10ml)、250μm ibmx(3-异丁基-1-甲基黄嘌呤,acros目录号228420010)和20mm hepes(gibco目录号15630)。在37℃温育30min以后,使用cisbio camp dynamic 2htrf测定试剂盒(62am4pej)定量地确定引起的细胞内camp增加。简而言之,通过加入在细胞裂解缓冲液(10μl)中的camp-d2缀合物,随后加入也在细胞裂解缓冲液(10μl)中的抗体抗-camp-eu
3+-穴状化合物,检测细胞内的camp水平。将得到的竞争性测定在室温温育至少60min,然后使用perkinelmer仪器用在320nm的激发以及在665nm和620nm的发射进行检测。envision单位(在665nm/620nm的发射*10,000)与存在的camp量成反比,并使用camp标准曲线转换为nm camp/孔。将在每个孔中产生的camp的量(nm)转换为观察到的最大应答的百分比。通过使用最大应答百分比相对于添加的化合物的浓度进行非线性回归分析而导出相对ec
50
值和顶端百分比(percent top)(e
max
),拟合至四参数逻辑方程。当在上述camp测定中试验实施例1和3的化合物时的ec
50
和e
max
数据显示在表1中。这些数据指示,实施例1和3的化合物是人gip受体的正变构调节剂。
[0196]
表1.hek293 gipr细胞系细胞内camp应答
[0197]
实施例ec
50
(nm)和sem(n)e
max
(%)
±
sem(n)实施例111.9(5.6,n=4)101
±
7(n=4)实施例337.5(22.1,n=4)91.0
±
8(n=4)
[0198]
人glp-1受体增强剂测定
[0199]
在表达人glp-1r的hek293克隆细胞系(ncbi登录号np_002053)中使用camp形成确定glp-1受体(glp-1r)功能活性。所述测定测量在ec
20
剂量的glp-1r激动剂胃泌酸调节素存在下化合物诱导的camp产生。将表达glp-1r的细胞用5nm的glp-1r激动剂胃泌酸调节素(bachem目录号h-6058)处理,并确定20μl测定体积(最终的dmso浓度是0.56%)的在dmem(gibco目录号31053)中的试验化合物(在dmso中的10点浓度-应答曲线,3倍labcyte echo直接稀释物,384孔板corning目录号3570)的剂量应答,所述dmem补充了2mm l-谷氨酰胺(gibco目录号25030)、0.25%胎牛血清(gibco目录号16000-044)、0.05%级分v牛血清白蛋白(gibco目录号15260)、1x青霉素/链霉素(hyclone目录号sv30010)、250μm ibmx(3-异丁基-1-甲基黄嘌呤,acros目录号228420010)和20mm hepes(hyclone目录号sh30237.01)。在
室温温育60min以后,使用定制的cisbio camp hybrid htrf测定试剂盒(60miszzz)定量地确定引起的细胞内camp增加。简而言之,通过加入在细胞裂解缓冲液(10μl)中的dynamic 2 camp-d2缀合物,随后加入也在细胞裂解缓冲液(10μl)中的hirange抗体抗-camp-eu
3+-穴状化合物,检测细胞内的camp水平。将得到的竞争性测定在室温温育至少60min,然后使用perkinelmer仪器用在320nm的激发以及在665nm和620nm的发射进行检测。envision单位(在665nm/620nm的发射*10,000)与存在的camp量成反比,并使用camp标准曲线转换为nmcamp/孔。将在每个孔中产生的camp的量(nm)转换为观察到的最大应答的百分比。通过使用最大应答百分比相对于添加的化合物的浓度进行非线性回归分析导出相对ec
50
值和顶端百分比(e
max
),拟合至四参数逻辑方程。当在上述camp测定中试验实施例2的化合物时的ec
50
和e
max
数据显示在表2中。该数据指示,实施例2的化合物是人glp-1受体的正变构调节剂。
[0200]
表2.hek293 glp-1r细胞系细胞内camp应答
[0201]
实施例ec
50
(nm)和sem(n)e
max
(%)
±
sem(n)实施例215.7(8.1,n=10)181
±
35(n=10)
[0202]
本发明考虑的某些实施方案如下:
[0203]
1.下式的化合物或其药学上可接受的盐:
[0204][0205]
其中
[0206]
r1选自下组:f和h;
[0207]
r2选自下组:ch3、ch3chch3和-nhch2ch(ch3)2;
[0208]
r3选自下组:h和-ch2och3;
[0209]
r4选自下组:h和ch3;
[0210]
r5选自下组:h和ch3;
[0211]
r6选自下组:h和oh;
[0212]
r7选自下组:
[0213]
2.下式的化合物或其药学上可接受的盐:
[0214][0215]
其中
[0216]
r1选自下组:f和h;
[0217]
r2选自下组:ch3和-nhch2ch(ch3)2;
[0218]
r3选自下组:h和-ch2och3;
[0219]
r4选自下组:h和ch3;
[0220]
r5选自下组:h和ch3;
[0221]
r6选自下组:h和oh。
[0222]
3.实施方案1至2中的任一个的化合物或其药学上可接受的盐,其中r4是ch3。
[0223]
4.实施方案1至3中的任一个的化合物或其药学上可接受的盐,其中r5是ch3。
[0224]
5.实施方案1至4中的任一个的化合物或其药学上可接受的盐,其中r6是h。
[0225]
6.实施方案1至5中的任一个的化合物或其药学上可接受的盐,其中r1是f。
[0226]
7.实施方案1至6中的任一个的化合物或其药学上可接受的盐,其中r3是h。
[0227]
8.实施方案1至7中的任一个的化合物或其药学上可接受的盐,其中r2是nhch2ch(ch3)2。
[0228]
9.实施方案1至7中的任一个的化合物或其药学上可接受的盐,其中r2是ch3。
[0229]
10.实施方案1的化合物或其药学上可接受的盐,其中所述化合物选自下组:
[0230]
[0231]
11.实施方案10的化合物,其中所述化合物是
[0232]
或其药学上可接受的盐。
[0233]
12.实施方案1的化合物或其药学上可接受的盐,其中所述化合物是
[0234][0235]
13.实施方案12的化合物,其中所述化合物是:
[0236][0237]
或其药学上可接受的盐。
[0238]
14.实施方案1至13中的任一个的化合物,其中所述化合物是盐酸盐。
[0239]
15.药物组合物,其包含实施方案1至14中的任一个的化合物或其药学上可接受的盐和至少一种药学上可接受的载体、稀释剂或赋形剂。
[0240]
16.用于治疗哺乳动物中的ii型糖尿病的方法,所述方法包括给所述哺乳动物施用有效量的实施方案1至14中的任一个的化合物或其药学上可接受的盐。
[0241]
17.用于疗法中的实施方案1至14中的任一个的化合物或其药学上可接受的盐。
[0242]
18.用于治疗ii型糖尿病的实施方案1至14中的任一个的化合物或其药学上可接受的盐。
[0243]
19.实施方案1至14中的任一个的化合物或其药学上可接受的盐在制备药物中的用途,所述药物用于治疗ii型糖尿病。
[0244]
20.用于受体的正变构调节的实施方案1至14中的任一个的化合物或其药学上可接受的盐,所述受体选自下组:glp1、gip和胰高血糖素。
[0245]
21.用于glp1受体的正变构调节的实施方案2至14中的任一个的化合物或其药学上可接受的盐。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1