一种磁性表面分子印迹电化学传感器及其制备方法和应用
1.本发明属于分子印迹电化学传感器技术领域,特别涉及一种磁性表面分子印迹电化学传感器及其制备方法和应用。
背景技术:
2.四溴双酚a作为一种持久性的有机污染物,对人体及其他水生生物的健康造成重大的影响;传统的检测方法大多采用基于色谱和色谱-质谱联用技术;例如:气相色谱(gc)、气相色谱-质谱联用(gc-ms)和高效液相色谱(hplc);其中,gc和gc-ms作为其最常用的检测技术;但是,色谱法检测存在仪器昂贵,样品前处理繁琐等缺点。
技术实现要素:
3.针对现有技术中存在的技术问题,本发明提供了一种磁性表面分子印迹电化学传感器及其制备方法和应用,以解决现有对四溴双酚a的检测过程,存在检测设备成本高,样品前处理繁琐的技术问题。
4.为达到上述目的,本发明采用的技术方案为:
5.本发明提供了一种磁性表面分子印迹电化学传感器的制备方法,包括以下步骤:
6.步骤1、将磁性fe3o4纳米粒子引入到氧化石墨烯上,制备得到磁性氧化石墨烯纳米材料;
7.步骤2、利用多巴胺对所述磁性氧化石墨烯纳米材料进行修饰,得到聚多巴胺修饰的磁性纳米微球;
8.步骤3、对所述聚多巴胺修饰的磁性纳米微球进行表面叠氮化处理,得到叠氮功能化的磁性载体;
9.步骤4、利用点击化学将raft试剂引入到所述叠氮功能化的磁性载体,聚合,得到磁性表面分子印迹聚合物;
10.步骤5、利用所述磁性表面分子印迹聚合物修饰玻碳电极,得到所述的磁性表面分子印迹电化学传感器。
11.进一步的,步骤1中,通过热溶剂法,将磁性fe3o4纳米粒子引入到氧化石墨烯上,制备得到磁性氧化石墨烯纳米材料;
12.具体过程如下:
13.将氧化石墨烯与乙二醇混合,超声搅拌后,加入fecl3·
6h2o和naac继续超声搅拌;之后,在180-200℃温度下反应1.5-2h,磁分离,洗涤,真空干燥,得到所述磁性氧化石墨烯纳米材料。
14.进一步的,步骤2中,利用多巴胺对所述磁性氧化石墨烯纳米材料进行修饰,得到聚多巴胺修饰的磁性纳米微球的过程,具体如下:
15.将所述磁性氧化石墨烯纳米材料与tris-hcl缓冲液超声混合后,加入盐酸多巴胺,室温下搅拌反应后,磁分离,洗涤,真空干燥,得到所述聚多巴胺修饰的磁性纳米微球;
其中,tris-hcl缓冲液的ph为8.5-9.0。
16.进一步的,步骤3中,对所述聚多巴胺修饰的磁性纳米微球进行表面叠氮化处理,得到叠氮功能化的磁性载体的过程,具体如下:
17.将4-叠氮基丁胺溶解在无水乙醇中,得到4-叠氮基丁胺-乙醇溶液;将所述聚多巴胺修饰的磁性纳米微球与乙醇混合,得到磁性纳米微球的乙醇分散液;将4-叠氮基丁胺-乙醇溶液滴加到所述磁性纳米微球的乙醇分散液中,在氮气保护下,进行叠氮化反应后,磁分离,洗涤,真空干燥,得到叠氮功能化的磁性载体。
18.进一步的,步骤4中,利用点击化学将raft试剂引入到所述叠氮功能化的磁性载体,聚合,得到磁性表面分子印迹聚合物的过程,具体如下:
19.对所述叠氮功能化的磁性载体进行raft试剂功能化处理,得到raft试剂修饰的磁性氧化石墨烯载体;
20.将模板分子和功能单体混合均匀,然后加入所述raft试剂修饰的磁性氧化石墨烯载体、交联剂及引发剂,引发聚合反应,得到磁性分子印迹聚合物;
21.去除模板分子,得到所述磁性表面分子印迹聚合物。
22.进一步的,对所述叠氮功能化的磁性载体进行raft试剂功能化处理,得到raft试剂修饰的磁性氧化石墨烯载体的过程,具体如下:
23.将叠氮功能化的磁性载体分散在二甲基亚砜中,加入raft试剂,超声分散后,依次加入抗坏血酸钠和五水硫酸铜,进行反应,得到反应产物;对反应产物进行磁分离,洗涤,真空干燥,得到所述raft试剂修饰的磁性氧化石墨烯载体。
24.进一步的,将模板分子和功能单体混合均匀,然后加入所述raft试剂修饰的磁性氧化石墨烯载体、交联剂及引发剂,引发聚合反应,得到磁性分子印迹聚合物的过程,具体如下:
25.将模板分子与功能单体分散于甲苯中,搅拌,得到混合分散液;将所述raft试剂修饰的磁性氧化石墨烯载体、交联剂及引发剂分散于所述混合分散液中,在氮气保护下,进行聚合反应,得到磁性分子印迹聚合物;其中,聚合反应温度为60-80℃,反应时间为12-24h;模板分子为四溴双酚a;功能单体为4-乙烯基吡啶,交联剂为乙二醇二甲基丙烯酸酯,引发剂为偶氮二异丁腈。
26.进一步的,步骤5、利用所述磁性表面分子印迹聚合物修饰玻碳电极,得到所述的磁性表面分子印迹电化学传感器的过程,具体如下:
27.将mxene与所述磁性表面分子印迹聚合物混合,配制得到修饰分散液;
28.将玻碳电极浸泡在四氯金酸中,通过循环伏安法,将au纳米颗粒沉积在玻碳电极表面,得到沉积有au纳米颗粒的电极;
29.在沉积有au纳米颗粒的电极上,依次滴涂所述修饰分散液,得到所述磁性表面分子印迹电化学传感器。
30.本发明还提供了一种磁性表面分子印迹电化学传感器,所述磁性表面分子印迹电化学传感器根据所述的一种磁性表面分子印迹电化学传感器的制备方法制备得到。
31.本发明还提供了一种磁性表面分子印迹电化学传感器的应用,利用所述磁性表面分子印迹电化学传感器对环境体系中四溴双酚a的识别与检测。
32.与现有技术相比,本发明的有益效果为:
33.本发明提供了一种磁性表面分子印迹电化学传感器及其制备方法和应用,以氧化石墨烯为载体,引入磁性fe3o4纳米粒子,便于磁性表面分子印迹聚合物在使用过程中从介质中分离出来;通过在磁性氧化石墨烯纳米材料表面包覆聚多巴胺层,既能有效防止fe3o4纳米粒子被氧化,同时也能增加表面官能团使其易于被功能化;通过对所述聚多巴胺修饰的磁性纳米微球进行表面叠氮化处理,在聚多巴胺修饰的磁性纳米微球表面引入叠氮基,通过点击化学连接raft试剂,利用表面引发的raft聚合制备磁性表面分子印迹聚合物;利用所述的磁性表面分子印迹聚合物对玻碳电极进行修饰,获得磁性表面分子印迹电化学传感器;所述磁性表面分子印迹电化学传感器上的磁性表面分子印迹聚合物,对待检测目标具有特异性识别功能,结合电化学分析法,实现对待测样品中待检测目标的分析测定;所述磁性表面分子印迹电化学传感器的识别位点丰富,模板分子传质速率块,能够对待测样品中的四溴双酚a进行高效检测,原理简单,所用原料成本低,检测过程无需任何大型仪器,即可实现对待测样品中四溴双酚a的高灵敏和特异性检测。
34.进一步的,以聚多巴胺修饰的磁性纳米微球为载体,四溴双酚a为模板分子,4-乙烯基吡啶为功能单体,乙二醇二甲基丙烯酸酯为交联剂,偶氮二异丁腈为引发剂进行表面分子印迹的制备,得到的磁性表面分子印迹聚合物对四溴双酚a具有特异性识别功能;利用所述磁性表面分子印迹电化学传感器,并结合电化学分析法,对样品中四溴双酚a进行分析测定,为磁性分子印迹聚合物的制备、复杂样品中目标物质的检测提供了新的思路及方法。
附图说明
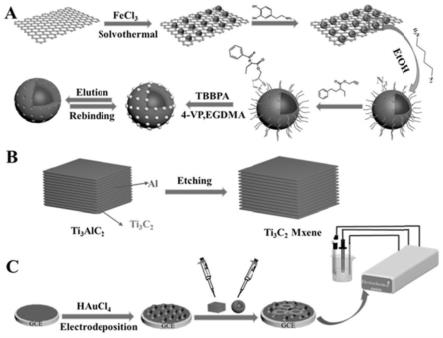
35.图1为实施例1中的磁性表面分子印迹电化学传感器的制备过程示意图;
36.图2为实施例1的氧化石墨烯go(a)、磁性氧化石墨烯纳米材料go@fe3o4(b)、聚多巴胺修饰的磁性纳米微球go@fe3o4@pda(c)以及磁性表面分子印迹聚合物go@fe3o4@mip(d)的透射电子显微镜照片;
37.图3为实施例1中的磁性fe3o4纳米粒子(a)、磁性氧化石墨烯纳米材料go@fe3o4(b)、聚多巴胺修饰的磁性纳米微球go@fe3o4@pda(c)、叠氮功能化的磁性载体go@fe3o4@pda-n3(d),raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb(e)以及磁性表面分子印迹聚合物go@fe3o4@mip(f)的红外光谱图;
38.图4为实施例1中的磁性氧化石墨烯纳米材料go@fe3o4(a)、多巴胺修饰的磁性纳米微球go@fe3o4@pda(b)以及磁性表面分子印迹聚合物go@fe3o4@mip(c)的x射线衍射谱图;
39.图5为实施例1中的1,4-二叠氮丁烷的核磁共振氢谱图;
40.图6为实施例1中的4-叠氮基丁胺的核磁共振氢谱图;
41.图7为实施例1中的炔基化cpdb的核磁共振氢谱图;
42.图8为实施例1中的mxene(a),玻碳电极gce(b),au纳米粒子修饰的玻碳电极au/gce(c)以及磁性表面分子印迹电化学传感器au/mxene/mip/gce(d)的扫描电镜照片;
43.图9为实施例1中玻碳电极gce(a),au纳米粒子修饰的玻碳电极au/gce(b),au及mxene修饰的玻碳电极au/mxene/gce(c),去除模板的au/mxene/mip/gce(d)、包含模板的au/mxene/mip/gce(e)以及磁性表面分子印迹电化学传感器au/mxene/nip/gce(f)的循环伏安曲线;
44.图10为实施例1中的玻碳电极gce(a),au纳米粒子修饰的玻碳电极au/gce(b),au
及mxene修饰的玻碳电极au/mxene/gce(c)以及去除模板的au/mxene/mip/gce(d)的电化学阻抗谱图。
具体实施方式
45.为了使本发明所解决的技术问题,技术方案及有益效果更加清楚明白,以下具体实施例,对本发明进行进一步的详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
46.本发明提供了一种磁性表面分子印迹电化学传感器,包括玻碳电极及磁性表面分子印迹聚合物;所述磁性表面分子印迹聚合物修饰在所述玻碳电极上;所述磁性表面分析印迹聚合物,以聚多巴胺修饰的磁性纳米微球为载体,通过叠氮基丁胺对聚多巴胺修饰的磁性纳米微球进行表面修饰,然后经点击化学将炔基化的raft试剂修饰到聚多巴胺修饰的磁性纳米微球表面,最后经raft聚合反应制备得到。
47.本发明还提供了一种磁性表面分子印迹电化学传感器的制备方法,包括以下步骤:
48.步骤1、利用hummers法,制备得到氧化石墨烯go。
49.步骤2、利用溶剂热法,将fe3o4磁性纳米粒子引入到氧化石墨烯go上,制备得到磁性氧化石墨烯纳米材料go@fe3o4。
50.其中,制备磁性氧化石墨烯纳米材料go@fe3o4的过程,具体如下:
51.将氧化石墨烯go与乙二醇混合,超声搅拌30min后,加入fecl3·
6h2o和naac继续超声搅拌15min;之后,在180-200℃温度下反应1.5-2h,经磁分离,洗涤,真空干燥,得到所述磁性氧化石墨烯纳米材料go@fe3o4。
52.步骤3、利用多巴胺对所述磁性氧化石墨烯纳米材料进行修饰,得到聚多巴胺修饰的磁性纳米微球go@fe3o4@pda;具体的,将所述磁性氧化石墨烯纳米材料go@fe3o4与tris-hcl缓冲液超声混合30min后,加入盐酸多巴胺,在室温条件下,进行搅拌反应12h后,经磁分离,洗涤,真空干燥,得到所述聚多巴胺修饰的磁性纳米微球go@fe3o4@pda;其中,tris-hcl缓冲液的ph为8.5-9.0。
53.步骤4、对所述聚多巴胺修饰的磁性纳米微球进行表面叠氮化处理,得到叠氮功能化的磁性载体;具体过程如下:
54.将4-叠氮基丁胺溶解在无水乙醇中,得到4-叠氮基丁胺-乙醇溶液;将所述聚多巴胺修饰的磁性纳米微球与乙醇混合,得到磁性纳米微球的乙醇分散液;将4-叠氮基丁胺-乙醇溶液滴加到所述磁性纳米微球的乙醇分散液中,在氮气保护下,进行叠氮化反应后,经磁分离,洗涤,真空干燥,得到叠氮功能化的磁性载体。
55.步骤5、利用点击化学将raft试剂引入到所述叠氮功能化的磁性载体,聚合,得到磁性表面分子印迹聚合物;具体包括以下步骤:
56.步骤51、对所述叠氮功能化的磁性载体进行raft试剂功能化处理,得到raft试剂修饰的磁性氧化石墨烯载体;具体的,将叠氮功能化的磁性载体分散在二甲基亚砜中,加入raft试剂,超声分散30min后,依次加入抗坏血酸钠和五水硫酸铜,在温度50℃的条件下反应24h,得到反应产物;对反应产物进行磁分离,洗涤,真空干燥,得到所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb。
57.步骤52、将模板分子和功能单体混合均匀,然后加入所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb、交联剂及引发剂,引发聚合反应,得到磁性分子印迹聚合物;具体过程如下:
58.将模板分子与功能单体分散于甲苯中,搅拌,得到混合分散液;将所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb、交联剂及引发剂分散于所述混合分散液中,在氮气保护下,进行聚合反应,得到磁性分子印迹聚合物;其中,聚合反应温度为60-80℃,反应时间为12-24h;模板分子为四溴双酚a;功能单体为4-乙烯基吡啶,交联剂为乙二醇二甲基丙烯酸酯,引发剂为偶氮二异丁腈。
59.步骤53、去除所述磁性分子印迹聚合物的模板分子,得到所述磁性表面分子印迹聚合物go@fe3o4@mip。
60.步骤6、利用所述磁性表面分子印迹聚合物go@fe3o4@mip对玻碳电极进行修饰,得到所述的磁性表面分子印迹电化学传感器;具体过程如下:
61.将玻碳电极作为工作电极,铂丝电极作为对电极,ag/agcl电极作为参比电极,置于铁氰化钾溶液中在-0.3-0.8v的电位范围内测cv,直至氧化还原峰δe<76mv,得到氧化还原后的玻碳电极;之后,将氧化还原后的玻碳电极置于0.05μm的al2o3的粉末上,进行抛光打磨,并用氮气吹干后,以haucl4为前驱体电沉积au,并用mxene修饰所述氧化还原后的玻碳电极,得到沉积有au纳米颗粒的电极;在所述沉积有au纳米颗粒的电极表面滴涂体积为5μl,浓度为5mg/ml的磁性表面分子印迹聚合物go@fe3o4@mip分散液,干燥后,获得所述的磁性表面分子印迹电化学传感器au/mxene/mip/gce。
62.本发明所述的磁性表面分子印迹电化学传感器的制备方法,首先通过溶剂热法把fe3o4磁性纳米颗粒引入到hummers法制备的go上制备磁性石墨烯纳米材料,磁性颗粒的存在能够方便将磁性表面分子印迹聚合物从介质中分离出来,通过包覆聚多巴胺方便在go@fe3o4@pda的表面引入叠氮基;通过点击化学将炔基化raft试剂修饰到在叠氮功能化磁性石墨烯载体上,然后通过表面引发的raft聚合制备分子印迹聚合物层;以haucl4为前驱体通过电化学沉积法将金纳米颗粒修饰到玻碳电极上,然后滴涂mxene到电极表面,au及mxene的修饰能够显著增加电极的响应电流,滴涂分子印迹聚合物之后,获得所述磁性表面分子印迹电化学传感器;本发明所述的磁性表面分子印迹电化学传感器的应用中,以[fe(cn)6]
3-/
4-为探针,通过循环伏安法和差示脉冲伏安法对环境体系中四溴双酚a(tbbpa)进行分析测定。
[0063]
实施例1
[0064]
如附图1所示,本实施例1提供了一种磁性表面分子印迹电化学传感器的制备方法,具体包括以下步骤:
[0065]
步骤1、采用hummers法,制备得到氧化石墨烯go;
[0066]
氧化石墨烯go的制备过程,具体如下:
[0067]
将2g的nano3和96ml的浓h2so4以及2g的石墨磷片混合,在0℃条件下搅拌30min,得到混合体系a;
[0068]
将12g的kmno4分3次加入到所述混合体系a中,搅拌1.5h后,再在35℃恒温条件下反应2h;之后,加入80ml纯净水,待反应体系到达90℃后继续反应1h,得到体系一;
[0069]
将20ml的h2o2加入到200ml的去离子水中,将20ml的浓盐酸加入92ml的去离子水
中,随后依次加入到上述体系一中,继续搅拌至室温,停止搅拌,静置分层,对下层沉淀物质离心、洗涤至中性,冷冻干燥后得到所述氧化石墨烯go。
[0070]
步骤2、采用溶剂热法,将fe3o4磁性纳米粒子引入到氧化石墨烯go上,制备得到磁性氧化石墨烯纳米材料go@fe3o4;具体过程如下:
[0071]
将200mg的氧化石墨烯go加入到60ml的乙二醇中,超声搅拌30min,得到混合体系b;
[0072]
将0.973g的fecl3·
6h2o和1.947g的naac加入到混合体系b中,超声搅拌15min至全部溶解,得到墨绿色的溶液;
[0073]
将所述墨绿色的溶液倒入反应釜中,在180-200℃下反应1.5-2h;冷却后磁分离、洗涤、真空干燥;得到磁性氧化石墨烯纳米材料go@fe3o4;其中,洗涤过程,采用去离子水和乙醇各清洗三次;真空干燥的温度为60℃,真空干燥的时间为12h。
[0074]
步骤3、利用多巴胺对所述磁性氧化石墨烯纳米材料进行修饰,得到聚多巴胺修饰的磁性纳米微球go@fe3o4@pda;制备过程,具体如下:
[0075]
将100mg的磁性氧化石墨烯纳米材料go@fe3o4加入到90ml的tris-hcl缓冲溶液中;其中,tris-hcl缓冲溶液的ph为8.5-9.0;超声搅拌30min后,加入500mg的盐酸多巴胺,在室温条件下,进行搅拌反应12h后,得到反应产物;对反应产物进行磁分离,洗涤,真空干燥;得到所述聚多巴胺修饰的磁性纳米微球go@fe3o4@pda,其中洗涤依次用乙醇清洗两次,去离子水清洗一次,真空干燥的温度为60℃,真空干燥的时间为8h。
[0076]
步骤4、对所述聚多巴胺修饰的磁性纳米微球进行表面叠氮化处理,得到叠氮功能化的磁性载体;具体过程如下:
[0077]
将100mg的所述聚多巴胺修饰的磁性纳米微球go@fe3o4@pda加入到25ml的乙醇中并超声搅拌30min,得到磁性纳米微球的乙醇分散液;
[0078]
将250mg的4-叠氮基丁胺溶解到5ml的无水乙醇中,得到4-叠氮基丁胺-乙醇溶液;
[0079]
将4-叠氮基丁胺-乙醇溶液滴加到所述磁性纳米微球的乙醇分散液中,在氮气保护下,进行叠氮化反应18h后,经磁分离,洗涤,真空干燥,得到叠氮功能化的磁性载体go@fe3o4@pda-n3;其中,洗涤过程,采用乙醇清洗三次;真空干燥的温度为60℃,真空干燥的时间为8h。
[0080]
本实施例1中,所述4-叠氮基丁胺的制备过程,具体包括以下步骤:
[0081]
将100ml的n,n-二甲基酰胺,50ml的水、14ml的1,4-二溴丁烷以及16g的nan3混合,油浴加热至80℃,并在该温度下磁力搅拌20h;之后,冷却至室温后,用饱和nacl溶液洗涤4次,得到混合体系c;用正己烷萃取混合体系c,得到萃取液a;采用用mgso4干燥所述萃取液a的有机层,过滤后蒸发溶剂得到1,4-二叠加氮基丁烷;将4.2g的1,4-二叠氮基丁烷、20ml的乙酸乙酯以及20ml的乙醚,在0℃条件下下溶于60ml盐酸中,然后,再缓慢添加7.8g的三苯基膦,在室温下反应20h,得到反应混合物;采用乙醚对所述反应混合液洗涤两次,之后,采用用naoh溶液将洗涤后的反应混合液的ph值调节到13,然后再用二氯甲烷萃取,得到萃取液b;将所述萃取液b的有机相合并,经无水硫酸镁干燥后过滤,除去溶剂得到4-叠氮基丁胺。
[0082]
步骤5、利用点击化学将raft试剂引入到所述叠氮功能化的磁性载体,得到raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb;具体过程如下:
[0083]
将180mg的叠氮功能化的磁性载体go@fe3o4@pda-n3和50mg的炔丙基-4-乙基戊酸二硫代苯甲酸酯加入到20ml的二甲基亚砜中,超声分散30min,得到混合体系d;
[0084]
将17.8mg的抗坏血酸钠和4.5mg的cuso4·
5h2o都分别加入到1ml的水中溶解,然后依次注加入到混合体系d中,氮气保护下机械搅拌进行反应,得到反应产物;其中,反应温度为50℃,反应时间为24h;
[0085]
对所述反应产物进行磁分离,洗涤,真空干燥得到所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb;其中,洗涤过程,采用乙醇清洗三次;真空干燥温度为60℃,真空干燥的时间为8h。
[0086]
步骤6、将模板分子和功能单体混合均匀,然后加入所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb、交联剂及引发剂,引发聚合反应,得到磁性分子印迹聚合物;具体过程如下:
[0087]
将所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb加入到40ml的无水甲苯中,超声分散15min;然后加入0.138g的4-乙烯基吡啶和0.225g的tbbpa,氮气保护下室温搅拌7h,得到混合溶液;然后将10mmol的乙二醇二甲基丙烯酸酯和0.08mmol的偶氮二异丁腈加入到所述混合溶液中,氮气保护下60-80℃,进行聚合反应12-24h;反应结束后用磁铁分离产物,并用无水乙醇清洗3次所得产品,得到磁性分子印迹聚合物。
[0088]
步骤7、去除所述磁性分子印迹聚合物的模板分子,得到所述磁性表面分子印迹聚合物go@fe3o4@mip;具体的,采用醋酸-甲醇的混合溶剂洗涤所述磁性分子印迹聚合物,然后再用无水乙醇清洗至ph=7,并用真空干燥箱干燥,得到所述磁性表面分子印迹聚合物go@fe3o4@mip;其中,醋酸-甲醇的混合溶剂采用冰醋酸和甲醇按照体积比为1:9的比例混合得到。
[0089]
步骤8、利用所述磁性表面分子印迹聚合物go@fe3o4@mip对玻碳电极进行修饰,得到所述的磁性表面分子印迹电化学传感器;具体过程如下:
[0090]
将裸的玻碳电极gce,采用0.05μm的氧化铝浆抛光成镜面,并用乙醇、超纯水超声清洗;
[0091]
在含有8.5mg ml-1
的haucl4的溶液中通过cv在-0.3-0.8v范围内,以100mvs-1
的扫描速度扫描5个循环,将au纳米颗粒电沉积到裸gce表面,得到改性gce;之后,将改性gce用去离子水冲洗,红外灯干燥后获得au纳米粒子修饰的玻碳电极au/gce;
[0092]
将体积为5μl的1mg/mlmxene悬浮液滴涂在au纳米粒子修饰的玻碳电极au/gce表面,在红外烤灯下干燥,得到au及mxene修饰的玻碳电极au/mxene/gce。
[0093]
将体积为5μl的0.8mg/ml所述磁性表面分子印迹聚合物go@fe3o4@mip的悬浮液滴涂到au及mxene修饰的玻碳电极au/mxene/gce表面,在红外灯下干燥,得到磁性表面分子印迹电化学传感器au/mxene/mip/gce。
[0094]
如附图2所示,附图2中给出了实施例1的氧化石墨烯go(a)、磁性氧化石墨烯纳米材料go@fe3o4(b)、聚多巴胺修饰的磁性纳米微球go@fe3o4@pda(c)以及磁性表面分子印迹聚合物go@fe3o4@mip(d)的透射电子显微镜照片;从附图2(a)中可以看出,所述氧化石墨烯go为片层结构;从附图2(b)中可以看出,fe3o4纳米微球分布于氧化石墨烯go表面,粒径约在25nm左右,且具有良好的分散性;从附图2(c)中可以看出,在磁性纳米粒子表面成功的被聚多巴胺包覆,形成非常明显的核-壳结构;从附图2(b)中可以看出,通过raft聚合制备go@
fe3o4@mip后,聚多巴胺修饰的磁性氧化石墨烯表面变得粗糙,形成约10nm左右的壳层。
[0095]
如附图3所示,附图3中给出了实施例1中的磁性fe3o4纳米粒子(a)、磁性氧化石墨烯纳米材料go@fe3o4(b)、聚多巴胺修饰的磁性纳米微球go@fe3o4@pda(c)、叠氮功能化的磁性载体go@fe3o4@pda-n3(d),raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb(e)以及磁性表面分子印迹聚合物go@fe3o4@mip(f)的红外光谱图;从附图3(a)中可以看出,560cm-1
处的吸收峰是fe3o4中fe-o键的特征吸收峰,表明fe3o4成功制备;从附图3(b)中可以看出,3420cm-1
处的特征峰可归属于受氢键影响的-oh的伸缩振动峰,位于1740cm-1
的是go表面羧基中c=o的伸缩振动特征吸收峰,1630cm-1
处的吸收峰归属于go中c-c伸缩振动峰,1035cm-1
处的特征峰归属于c-o-c环氧基的伸缩振动峰;560cm-1
处的吸收峰是fe3o4中fe-o键的特征吸收峰。表明fe3o4磁性纳米颗粒成功引入到氧化石墨烯;从附图3(c)中可以看出,go@fe3o4@pda的红外谱图在3231cm-1
、1510cm-1
、1284cm-1
出现新的吸收峰,3231cm-1
处的特征峰归属于n-h的伸缩振动峰,1510cm-1
为n-h的弯曲振动吸收峰,1284cm-1
处的特征峰归属于聚多巴胺中酚羟基伸缩振动吸收峰。表明聚多巴胺的成功包覆;从附图3(d)中可以看出,2100cm-1
处的特征峰归属于叠氮基的伸缩振动峰。表明成功叠氮基被成功引入;从附图3(e)中可以看出,1166cm-1
和1218cm-1
处的伸缩振动峰分别对应于raft试剂中c=s和c-s-c的特征吸收峰,表明炔基化的raft试剂通过点击化学被成功地引入到了磁性纳米粒子的表面;从附图3(f)中可以看出,1415cm-1
处的特征峰是吡啶环的特征吸收峰,这表明在go@fe3o4载体表面上成功的制备了分子印迹聚合物。
[0096]
如附图4所示,附图4中给出了实施例1中的磁性氧化石墨烯纳米材料go@fe3o4(a)、多巴胺修饰的磁性纳米微球go@fe3o4@pda(b)以及磁性表面分子印迹聚合物go@fe3o4@mip(c)的x射线衍射谱图;从附图4(a)中可以看出,位于2θ=30.1
°
、35.5
°
、43.2
°
、53.7
°
、57
°
及62.8
°
的特征衍射峰分别对应于(220)、(331)、(400)、(511)、(440)和(220)晶面,这很好地与fe3o4纳米颗粒的晶型相匹配。2θ=25.2
°
处的衍射峰为go特征峰,这表明fe3o4纳米粒子成功地修饰到go片层上;从附图4(b)及附图4(c)中可以看出包覆了pda层和mip之后,衍射谱图并无明显变化,说明包覆层并没有破坏fe3o4的晶体结构。
[0097]
如附图5-7所示,附图5中给出了实施例1中的1,4-二叠氮丁烷的谱图,附图6中给出了实施例中的4-叠氮基丁胺的谱图,附图7中给出了实施例中的炔基化cpdb的1h-nmr的谱图;从附图5中可以看出,1,4-二叠氮基丁烷的核磁共振氢谱图可以看出,各个氢都得到了归属,表明目标产物被成功合成;1h nmr(400mhz;cdcl3):1.69(m,4h,c-ch
2-c),3.35(t,4h,2
×‑
ch
2-n3);从附图6中可以看出,4-叠氮基丁胺的各个氢都得到了归属;1h nmr(400mhz;cdcl3):1.35(s,2h,nh2),1.5(s,2h,ch
2-ch
2-ch2),1.7(m,2h,n
3-ch
2-ch2),2.74(t,2h,ch
2-ch
2-nh2),3.3(t,2h,n
3-ch2);从附图7中可以看出,炔基化cpdb的各个氢都得到了归属,证明炔基化cpdb被成功合成;1h nmr(400mhz;cdcl3):1.11(td,3h,-c-ch3),2.11(m,2h,-c-ch
2-c),2.51(td,1h,-c≡ch),4.75(m,3h,-ch-coo-ch
2-c),7.39(tt,2h,ar-h),7.55(ddq,1h,ar-h),8.00(dq,2h,ar-h)。
[0098]
如附图8所示,附图8中给出了实施例1中的mxene(a),玻碳电极gce(b),au纳米粒子修饰的玻碳电极au/gce(c)以及磁性表面分子印迹电化学传感器au/mxene/mip/gce(d)的扫描电镜照片;从附图8(a)中可以看出,制备的mxene呈现出类似于手风琴独特的层状结构,表明mxene的成功制备;从附图8(b)、8(c)中可以看出,与裸玻碳电极相比,电沉积au后,
gce表面出现了大量颗粒,这表明au纳米颗粒成功沉积在了电极表面;从附图8(d)中可以看出,滴涂mxene和mip悬浮液后,电极表面变得更加粗糙,出现了大量的球状颗粒,这证实了au/mxene/mip/gce已被成功构筑。
[0099]
如附图9所示,附图9中给出了实施例1中玻碳电极gce(a),au纳米粒子修饰的玻碳电极au/gce(b),au及mxene修饰的玻碳电极au/mxene/gce(c),去除模板的au/mxene/mip/gce(d)、包含模板的au/mxene/mip/gce(e)以及磁性表面分子印迹电化学传感器au/mxene/nip/gce(f)的循环伏安曲线;从附图9(a)、9(b)中可以看出,金修饰电极后,响应电流远大于裸电极的响应电流,是由于金沉积后[fe(cn)6]
3-/4-的电子转移率提高;从附图9(c)中可以看出,au/mxene复合材料修饰电极,电流进一步增加;从附图9(d)、9(e)中可以看出,当mip薄膜滴涂在修饰电极上时,去除模板的au/mxene/mip/gce的峰值电流值发生降低,峰值电流的降低是因为mip膜的不导电性,导致电子转移速率降低;当与tbbpa结合后,由于印迹腔的阻塞,减少了质量和电子传输,电流响应值发生了明显降低;从附图7(f)中可以看出,在tbbpa结合后的峰值电流与au/mxene/nip/gce基本相同,这是由于au/mxene/nip/gce表面没有印迹腔存在。
[0100]
如附图10所示,附图10中给出了实施例1中的玻碳电极gce(a),au纳米粒子修饰的玻碳电极au/gce(b),au及mxene修饰的玻碳电极au/mxene/gce(c)以及去除模板的au/mxene/mip/gce(d)的电化学阻抗谱图;从附图10(a)、10(b)中可以看出,经au沉积的修饰电极的nyquist环明显小于裸电极,表明au的沉积致使电导率显著增加;从附图10(c)可以看出,au/mxene/gce的nyquist环是最小的,这表明au/mxene作为修饰材料修饰电极有效地提高了电荷转移能力;从附图10(d)中可以看出,对于au/mxene/mip/gce的半圆半径(rct)出现增加,这是因为mip膜的导电性较差,进而导致电子转移能力降低。
[0101]
实施例2
[0102]
本实施例2与实施例1步骤大体相同,不同之处在于步骤8中pbs的酸碱度。
[0103]
具体为,配置ph分别为6.0、6.5、7.0、7.5及8.0的含有5.0mmol l-1
[fe(cn)6]
3-/
4-缓冲溶液,在玻碳电极表面修饰au/mxene以及所述磁性表面分子印迹聚合物go@fe3o4@mip,分别将ph为6.0、6.5、7.0、7.5及8.0的磷酸盐缓冲液作为本次试验电解液,以修饰后的玻碳电极为工作电极,铂丝电极为对电极,ag/agcl电极为参比电极,电化学法测定tbbpa的最佳ph值,在其中ph=7时的电流响应最好,被选做最佳酸碱度。
[0104]
实施例3
[0105]
本实施例3与实施例1步骤大体相同,不同之处在于步骤8中电极置于待测溶液中的时间。
[0106]
具体的,将电极置于ph=7.5的磷酸缓冲溶液(5mmol l-1
[fe(cn)6]
3-/4-)的时间分别为0min,2min,3min,5min,7min,9min以及10min;通过差示脉冲伏安法检测,结果表明,随着孵化时间的增加,响应电流减小,在5min后峰值电流不再变化,这是因为mip电极表面存在印迹孔促进了tbbpa结合,导致电子转移率下降。因此,选择5min为最佳孵化时间。
[0107]
在实际应用中,本发明所述的一种磁性表面分子印迹电化学传感器,高选择性能和高灵敏度表明有希望在复杂环境中对tbbpa进行识别和检测。
[0108]
对比例1
[0109]
对比例1与实施例1步骤大体相同,不同之处在于步骤6中功能单体的选择;
[0110]
具体如下:
[0111]
将所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb加入到40ml甲苯中超声分散处理15min,然后加入0.113g的甲基丙烯酸和0.225g的tbbpa,氮气保护下室温搅拌7h,得到混合溶液;然后将10mmol的乙二醇二甲基丙烯酸酯和0.08mmol的偶氮二异丁腈加入到上述溶液,氮气保护下60-80℃,进行聚合反应12-24h反应聚合;反应结束后用磁铁分离产物,并用无水乙醇清洗3次所得产品,得到分子印迹聚合物。
[0112]
对对比例1制备得到的磁性表面分子印迹聚合物go@fe3o4@mip通过差示脉冲伏安法进行性能评估,发现所得到的传感器的电流响应为0.38ma。
[0113]
对比例2
[0114]
对比例2与实施例1步骤大体相同,不同之处在于步骤6中功能单体投加量;
[0115]
具体如下:
[0116]
将所述raft试剂修饰的磁性氧化石墨烯载体go@fe3o4@pda-cpdb加入到40ml的甲苯中超声分散处理15min;然后加入0.138g的4-乙烯基吡啶和0.225g的tbbpa,氮气保护下室温搅拌7h,得到混合溶液;然后将10mmol的乙二醇二甲基丙烯酸酯和0.08mmol的偶氮二异丁腈加入到上述混合溶液,氮气保护下60-80℃,进行聚合反应12-24h反应聚合;反应结束后用磁铁分离产物,并用无水乙醇清洗3次所得产品,得到分子印迹聚合物。
[0117]
对上述对比例制备得到的磁性表面分子印迹聚合物go@fe3o4@mip通过差示脉冲伏安法进行性能评估,发现所得到的传感器的电流响应为0.25ma;与甲基丙烯酸作为功能单体相比,4-乙烯基吡啶具有更高的亲和力。
[0118]
电化学灵敏度检测试验
[0119]
在电化学灵敏度检测试验中,配置七组浓度分别为0nm,0.05nm,0.1nm,0.5nm,1nm,5nm以及10nm的tbbpa溶液(保持电解液的ph为7.5);用制备好的电化学传感器在溶液中孵化5min,然后使用差示脉冲伏安法分别在七组溶液中进行检测,得到了不同的电流响应。
[0120]
由此得到tbbpa的响应电流与tbbpa浓度的对数的线性关系,该线性回归方程的表达式为:
[0121]
△
i(μa)=8.963logc
tbbpa
(nm)+22.567(r2=0.991)
[0122]
其中,检测限为0.0144nm。
[0123]
由此可知,该电化学传感器有着较低的检测限,也表明了本发明的磁性表面分子印迹电化学传感器能够在复杂的环境中实现检测痕量检测。
[0124]
电化学特异性检测试验
[0125]
采用选择tbbpa的结构类似物,例如:双酚a、4,4-联苯二酚及对叔丁基苯酚来研究印迹聚合物的选择性;向浓度为10.0nm的tbbpa溶液中加入十倍量的双酚a、4,4-联苯二酚以及对叔丁基苯酚中,在上述三种电解液中,用同样结构的磁性表面分子印迹电化学传感器进行差示脉冲伏安法来检测;此外,用只含有10.0nm tbbpa的电解液作为对比;通过dpv检测响应电流,结果发现其类似干扰物的存在对tbbpa的检测结果不会造成干扰,原因是合成的mip表面产生特定的识别腔完全匹配于tbbpa的形状,大小和空间排布,而双酚a、4,4-联苯二酚以及对叔丁基苯酚不与识别位点互补;因此,不会发生特异性结合,这表明本发明所述的磁性表面分子印迹电化学传感器能够在复杂环境中特异性识别tbbpa。
[0126]
本发明所述的一种磁性表面分子印迹电化学传感器及其制备方法,通过在氧化石墨烯上采用聚多巴胺包裹磁性纳米微球fe3o4为载体制备磁性表面分子印迹聚合物,制备的印迹材料能够应用在电化学传感器上特异性识别tbbpa。磁性纳米粒子的存在使得印迹材料可以通过外加磁场快速分离,表面印迹方法增加了印迹材料的结合位点;印迹层的表层分布也使得印迹位点容易接近,达到迅速识别模板分子的效果;au以及mxene的修饰增强了模板分子的电化学响应,此传感器效识别位点丰富,吸附容量较大,模板分子传质速率快,吸附效率高,可高效检测环境样品中的tbbpa。
[0127]
上述实施例仅仅是能够实现本发明技术方案的实施方式之一,本发明所要求保护的范围并不仅仅受本实施例的限制,还包括在本发明所公开的技术范围内,任何熟悉本技术领域的技术人员所容易想到的变化、替换及其他实施方式。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1