一种苄氨基取代的脂肪酸衍生物及其制备方法与用途与流程
1.本发明属于医药技术领域,具体涉及一种苄氨基取代的脂肪酸衍生物及其制备方法与用途。
技术背景
2.随着恶性肿瘤的发病率和死亡率持续呈现上升的趋势,严重威胁人类的生命健康,目前,对于恶性肿瘤的临床治疗仍然以手术切除、放射治疗和全身化疗为主,其中对于利用化学药物进入机体后,分布至病灶处从而抑制肿瘤细胞的生长和扩散,因为具有见效快、杀伤力强的特点,但是目前抗肿瘤药物对肿瘤细胞的选择低,在杀死肿瘤细胞的同时也对某些正常组织细胞有一定的损伤,从而带来了一系列的副作用和毒性反应。并且,在治疗过程中抗肿瘤的耐药导致药物的抗肿瘤作用疗效降低或失效,甚至肿瘤细胞对此类化疗药物也产生耐药性,从而导致有效的抗肿瘤药物数量、种类减少,而肿瘤细胞类型多样,加上肿瘤细胞发生耐药,因此,寻找到新型的抗肿瘤药物已经成为药物研究者的迫切希望。
3.众所周知,脂肪酸主要存在于动植物的油脂中,包括饱和脂肪酸、单不饱和脂肪酸和多不饱和脂肪酸,其对动植物的正常生理代谢活动起着重要的调节作用,近年来,研究者发现,高级不饱和脂肪酸在抗糖尿病、免疫调节、抗动脉粥样硬化方面有一定的生理调节作用,多数不饱和脂肪酸也具有一定的抗肿瘤的效果(张越华,曾和平.脂肪酸在生命过程中的作用研究进展[j].中国油脂,2006,31(12):6.),而对于单不饱和脂肪酸在抗肿瘤的研究较少,1924年,nakahara\t w指出部分的饱和脂肪酸、单不饱和脂肪酸和反式脂肪酸不仅不能降低抗肿瘤的作用,反而具有增加癌症的风险(https://doi.org/10.1084/jem.40.3.363),j a men
é
ndez等人通过mtt法试验发现,低浓度下的油酸(50μg/ml)能对人乳腺癌细胞株mda-mb-231产生微弱的抑制作用,抑制率约为10%,高浓度下的油酸(125~500μg/ml)则能轻度促进mda-mb-231的增殖,相比之下γ-亚油酸则能浓度依赖性地抑制mda-mb-231的增殖(men
é
ndez ja,del m b m,montero s,et al.effects of gamma-linolenic acid and oleic acid on paclitaxel cytotoxicity in human breast cancer cells[j].european journal of cancer,2001,37(3):402-413.),因此,该研究表明了多不饱和脂肪酸(γ-亚油酸)与单不饱和脂肪酸(油酸)在抗肿瘤效果上的区别,但该研究并不能证明其他单不饱和脂肪酸和多不饱和脂肪酸在抗肿瘤类型及使用剂量,美国专利us201917290963公开了一种含有硫醇后硝基吸电子基团的不饱和脂肪酸在dna修复基因缺陷遗传病因的癌症药物中的应用。综上所述,基于不饱和度与不饱和键的位置存在显著差异,不同不饱和脂肪酸难以从自然界直接获得,且对于抗肿瘤的药理活性也难以预测,因此,对于定向合成一种特定的不饱和脂肪酸对于不饱和脂肪酸在抗癌药物中的研究以及对于癌症患者的治疗具有重要的意义。
技术实现要素:
[0004]
针对上述现有技术中存在的问题,本发明提供了一种苄氨基取代的脂肪酸衍生物
及其制备方法与用途,所述苄氨基取代的脂肪酸衍生物对多种肿瘤细胞株均具有良好的抑制活性。
[0005]
本发明提供一种苄氨基取代的脂肪酸衍生物,命名为5-(苄氧基)-2-己基十六碳-2-烯酸,其分子结构式如下:
[0006][0007]
优选地,所述苄氨基取代的脂肪酸衍生物的结构为(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸,具体分子结构式如下:
[0008][0009]
本发明提供了5-(苄氧基)-2-己基十六碳-2-烯酸的制备方法,具体如下:
[0010]
s1、将3-己基四氢-4-羟基-6-十一烷基-2h-吡喃-2-酮溶于有机溶剂中,在催化剂条件下得到5-己基-2-十一烷基-2,3-二氢吡喃-6-酮;
[0011]
s2、将5-己基-2-十一烷基-2,3-二氢吡喃-6-酮溶于有机溶剂中,在碱性条件下反应,加入饱和氯化钠溶液洗涤,分液,萃取有机相,浓缩得到化合物;
[0012]
s3、将步骤s2中得到的化合物溶于有机溶剂中,加入溴化苄、叔丁醇钠进行反应,调节溶液呈中性,加入饱和氯化钠,分液、萃取得有机相,浓缩得到的苄氨基取代的脂肪酸衍生物为(r,e)5-(苄氧基)-2-己基十六碳-2-烯酸。
[0013]
进一步地,所述步骤s1中的催化剂选自质子酸或非质子酸;
[0014]
优选地,所述催化剂为质子酸;
[0015]
优选地,所述催化剂可选自硫酸、盐酸、高氯酸、四氟硼酸、冰醋酸、碳酸、nh
4+
、苯磺酸、对甲基苯磺酸中的一种。
[0016]
进一步地,所述步骤s2中的碱性条件为氢氧化钠、氢氧化钾、碳酸氢钠、磷酸氢二钠或氨水的加入形成。
[0017]
进一步地,所述步骤s3中的步骤s2中得到的化合物、溴化苄、叔丁醇钠的摩尔投料比为1:1~5:1~10,优选为1:1.2~2:4~5。
[0018]
本发明还提供了所述步骤s1中5-己基-2-十一烷基-2,3-二氢吡喃-6-酮的另外一种合成途径:将3-己基四氢-4-羟基-6-十一烷基-2h-吡喃-2-酮溶于有机溶剂中,在三苯基膦、偶氮二羧酸酯条件下与亲核试剂反应,将反应得到的化合物进一步在碱性条件中进行反应得到的5-己基-2-十一烷基-2,3-二氢吡喃-6-酮。
[0019]
进一步地,所述步骤中的亲核试剂为含有羧基官能团的化合物,优选为对硝基苯甲酸、n-甲酰基-l-亮氨酸。
[0020]
本发明所述合成步骤中的有机溶剂均选自甲醇、乙醇、异丙醇、正丁醇、石油醚、丙酮、氯仿、二氯甲烷、正庚烷、四氢呋喃、吡啶、二甲基亚砜中的一种。
[0021]
进一步地,本发明所述步骤s1中化合物可为(3s,4s,6r)3-己基-4-羟基-6-十一烷基-2h-吡喃-2-酮或(3s,4s,6s)3-己基-4-羟基-6-十一烷基四氢-2h-吡喃-2-酮。
[0022]
进一步地,本发明所述步骤s1中得到的5-己基-2-十一烷基-2,3-二氢吡喃-6-酮为(2s)-5-己基-2-十一烷基-2,3-二氢吡喃-6-酮或(2r)-5-己基-2-十一烷基-2,3-二氢吡喃-6-酮。
[0023]
基于上述5-(苄氧基)-2-己基十六碳-2-烯酸的制备方法,包括以下3种合成路径:
[0024]
路线一:
[0025][0026]
路线二:
[0027][0028]
路线三:
[0029][0030]
本发明所述的化合物5-(苄氧基)-2-己基十六碳-2-烯酸、(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸在制备抗肿瘤制剂、抗肿瘤药物中的应用;
[0031]
进一步地,所述肿瘤包括肺癌、肝癌、乳腺癌、头颈癌、卵巢癌、慢性粒细胞性白血病、鼻咽癌、肾癌、膀胱癌、胰腺癌,但不仅限于所述肿瘤;
[0032]
进一步地,所述制剂、药物中包含化合物5-(苄氧基)-2-己基十六碳-2-烯酸、5-(苄氧基)-2-己基十六碳-2-烯酸衍生物、其药学上可接受的盐、立体异构体或前药分子、(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸、(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸衍生物、其药学上可接受的盐、立体异构体或前药分子中的一种或多种;
[0033]
进一步地,所述制剂、药物中还可包含一种或多种药学上可接受的载体或药用辅料;
[0034]
进一步地,所述制剂、药物剂型包括片剂、丸剂、散剂、胶囊剂、口服液、注射剂,但不仅限于所述剂型。
[0035]
与现有技术相比,本发明的有益效果如下:
[0036]
(1)本发明采用3-己基四氢-4-羟基-6-十一烷基-2h-吡喃-2-酮为原料,以不同的反应路线合成单不饱和脂肪酸(r,e)5-(苄氧基)-2-己基十六碳-2-烯酸;
[0037]
(2)本发明的单不饱和脂肪酸(r,e)5-(苄氧基)-2-己基十六碳-2-烯酸相比亚油酸、油酸以及其钠盐的抗肿瘤活性更高,其40μm的剂量对胰腺癌panc-1细胞株的抑制率高于90%以上,对头颈癌tca8113细胞、乳腺癌mcf-7细胞的抑制率高于80%以上,对肝癌hepg2细胞、鼻咽癌cne-2细胞株、肾癌a498细胞株、膀胱癌t24细胞株的抑制率在70~80%之间,对卵巢癌skov3细胞的抑制活性也可达65.8%,对肺癌a549细胞、cml k562细胞株的抑制活性分别为38.11%、44.51%。
[0038]
(3)本发明制备的(r,e)5-(苄氧基)-2-己基十六碳-2-烯酸可与盐酸特拉唑嗪、烟酸占替诺等药物产生协同增效抗胰腺癌效果。
附图说明
[0039]
图1为本发明实施例1制备得到的化合物ⅳ的1h-nmr(全δ范围);
[0040]
图2为本发明实施例1制备得到的化合物ⅳ的1h-nmr(低δ区域);
[0041]
图3为本发明实施例1化合物ⅳ的
13
c-nmr。
[0042]
图4为本发明实施例1化合物ⅳ的质谱图
具体实施方式
[0043]
本发明下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例中所用到的各种常用化学试剂,均为市售产品。
[0044]
除非另有定义,本发明所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不用于限制本发明。
[0045]
为使本发明的目的、技术方案和优点更加清楚明了,下面结合具体实施方式,对本发明进一步详细说明。应该理解,这些描述只是示例性的,而并非要限制本发明的范围。此外,在以下说明中,省略了对公知结构和技术的描述,以避免不必要地混淆本发明的概念。
[0046]
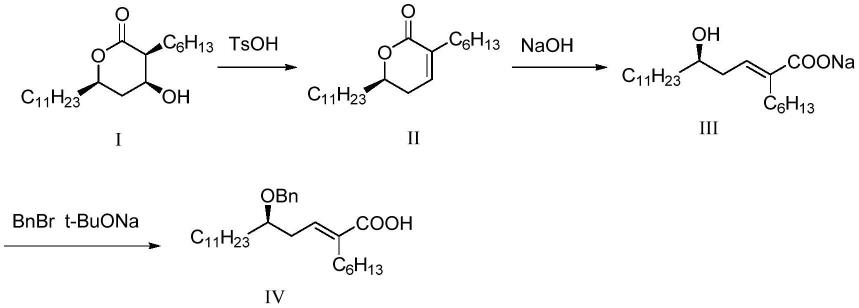
实施例1(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸的制备
[0047]
反应流程如下:
[0048][0049]
具体制备步骤:
[0050]
s1、将10g化合物i溶于200ml氯仿中,加入对甲苯磺酸(tsoh)4.0g于64
±
2℃反应20h,浓缩至无馏份,加入100ml二氯甲烷复溶,水洗至中性,浓缩得10g浓缩物,用15ml乙酸乙酯复溶,于16℃加入75ml正庚烷,继续降温至0℃搅拌1h,过滤,滤饼于石油醚/乙酸乙酯(2:1,v/v)洗脱液体系下进行硅胶柱柱层析分离,浓缩得化合物ii(2.92g)。
[0051]
s2、将1.0g化合物ii溶于7ml四氢呋喃中,加入2.05ml 2mol/l naoh于42.5
±
2.5℃反应87h,反应完成,用饱和氯化钠水溶液洗至有机相层为中性,分液,浓缩有机相得化合物iii。
[0052]
s3、将1.14g化合物iii溶于10ml四氢呋喃中,加入0.8g溴化苄(bnbr)和1.25g叔丁醇钠(t-buona),27.5
±
2.5℃反应20h,反应完成,加入稀盐酸溶液(0.1mol/l)调至中性,加入饱和氯化钠水溶液,分液,有机相浓缩后用石油醚/乙酸乙酯体系(2:1,v/v)进行硅胶柱柱层析分离,浓缩得0.5g(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸。
[0053]
实施例2(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸的制备
[0054]
具体反应流程如下:
[0055][0056]
s1、将20g化合物i、19.2g三苯基膦(ph3p)、9.36g对硝基苯甲酸混悬于150ml四氢呋喃中,于0℃加入19.9g偶氮二甲酸二异丙酯(diad),加毕,反应1.0h,反应完成,加入150ml乙酸乙酯,依次用饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤,浓缩有机相得浓缩物53.2g,进行石油醚/乙酸乙酯体系(2:1,v/v)柱层析分离得化合物v(10.5g)。
[0057]
s2、将10.5g化合物v溶于60ml甲醇中,加入4.35g碳酸钾于37.5
±
2.5℃反应3.5h,反应完成,用稀盐酸溶液(0.1mol/l)调至中性,浓缩后用110ml乙酸乙酯复溶,加入用饱和氯化钠,分液取有机相浓缩得化合物ii(6.2g)。
[0058]
s3、将6.2g化合物ii溶于62ml四氢呋喃中,加入13.5ml 2mol/l氢氧化钠于35℃反应74h,反应完成,用饱和氯化钠水溶液洗至中性,分液、浓缩有机相得化合物iii(7.1g)。
[0059]
s4、将6.8g化合物iii溶于68ml四氢呋喃中,加入4.8g溴化苄(bnbr)和7.5g叔丁醇钠(t-buona),27.5
±
2.5℃反应20h,反应完成,加入稀盐酸溶液(0.1mol/l)调至中性,加入饱和氯化钠水溶液,分液,浓缩有机相得8.9g浓缩物,用石油醚/乙酸乙酯体系(2:1,v/v)进行硅胶柱柱层析分离,浓缩得2.27g(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸。
[0060]
实施例3(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸的制备
[0061]
具体反应流程如下:
[0062][0063]
s1、将20g化合物i、19.2g三苯基膦(ph3p)、9.5g n-甲酰基-l-亮氨酸混悬于100ml四氢呋喃,于0℃加入19.9g偶氮二甲酸二异丙酯(diad),加毕,反应1.5h,反应完成,浓缩反应液,加入120ml正庚烷复溶,并用甲醇水溶液洗后,分液、浓缩正庚烷相,将浓缩物于石油醚/乙酸乙酯体系(2:1,v/v)硅胶柱柱层析分离,馏分浓缩得化合物vi(12.5g)。
[0064]
s2、将12.3g化合物vi溶于55ml四氢呋喃中,加入38ml 1mol/l氢氧化钠于27.5
±
2.5℃反应3.5h,反应完成,用稀盐酸溶液(0.1mol/l)调至中性,加入饱和氯化钠,分液、浓缩有机相后于石油醚/乙酸乙酯体系(2:1,v/v)硅胶柱柱层析分离得化合物ii(5.4g)。
[0065]
s3、将5.4g化合物ii溶于50ml四氢呋喃中,加入11.2ml 2mol/l氢氧化钠于35℃反应78h,反应完成,用饱和氯化钠水溶液洗至中性,分液、浓缩有机相得化合物iii(6.7g)。
[0066]
s4、将6.0g化合物iii溶于67ml四氢呋喃中,加入4.2g溴化苄(bnbr)和6.98g叔丁醇钠(t-buona),27.5
±
2.5℃反应20h,反应完成,加入稀盐酸溶液(0.1mol/l)调至中性,加入氯化钠水溶液,分液、浓缩有机相后用石油醚/乙酸乙酯体系(2:1,v/v)进行硅胶柱柱层析分离,浓缩馏分得2.5g(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸。
[0067]
试验例一、(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸的分子结构鉴定
[0068]
本发明实施例1~3制备得到的(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸,经核磁共振、液质检测对其进行化学结构确证:
[0069][0070]1h-nmr、
13
c-nmr均采用bruker advance500型核磁共振波谱仪,样品溶解溶剂为cdcl3,测定结果见表1~2以及附图1~3;
[0071]
表1:核磁共振氢谱1h-nmr数据
[0072][0073]
注:*多重性:s,单峰;d,二重峰;t,三重峰;m,多重峰;br,宽峰。
[0074]
表2:核磁共振碳谱
13
c-nmr数据
[0075]
[0076][0077]
由表1和表2检测结果可知:
[0078]1h-nmr给出9组峰,其积分面积比(由低场至高场)为4:1:1:2:1:2:2:28:6,共47个质子,结合-cooh上的质子,共48个质子,与结构中质子数相同;
[0079]
13
c-nmr给出24组峰,共有48个碳,与结构中碳原子数相同。
[0080]
lc-ms检测采用流动相为乙腈-磷酸-水(860/0.05/140)的流动相体系,检测结果见图4。
[0081]
试验例二、(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸的细胞毒性试验
[0082]
(1)细胞吸光值测定及结果
[0083]
受试样品:亚油酸、油酸、化合物iii((r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸钠盐)、化合物iv((r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸);
[0084]
实验步骤:用细胞培养液将对数生长期的各肿瘤细胞配成浓度为4
×
104个
·
ml-1
细胞悬浮液,每孔100μl接种于96孔培养板,置37℃、5%co2培养箱中培养24h;吸出每孔中原培养液,每孔加入200μl受试样品溶液(以dmso为溶剂),继续置37℃、体积分数5%co2培养箱中培养1d,每组设3复孔。1d后1块培养板加入mtt 20μl(5mg
·
ml-1)/孔,在37℃体积分数5%co2孵育箱中继续培养4h后,吸除原液,加入二甲基亚砜200μl/孔,振荡5min后,在酶标读数仪上在450nm处测吸光值a,三复孔的a值依次编号为a1、a2与a3,试验均在平行条件下进行,且设置1空白对照(空白对照加入50μldmso),细胞毒性由吸光值大小判定,结果见表3所示:
[0085]
表3:吸光度试验结果
[0086]
[0087][0088]
(2)肿瘤细胞抑制率测定及结果
[0089]
采用公式:抑制率=1-受试物吸光度/空白吸光度,计算亚油酸、油酸、化合物ⅲ与化合物ⅳ在对应的受试浓度下对各肿瘤细胞株的抑制率,结果如表4所示:
[0090]
表4:受试物对肿瘤细胞的抑制率
[0091][0092]
由表4结果可知,化合物iv对胰腺癌panc-1细胞株的抑制率高于90%以上,对头颈癌tca8113细胞、乳腺癌mcf-7细胞的抑制率高达80%以上,对肝癌hepg2细胞、鼻咽癌cne-2细胞株、肾癌a498细胞株、膀胱癌t24细胞株的抑制率在70~80%之间,对卵巢癌skov3细胞的抑制活性也可达65.8%,相对而言,化合物iv对肺癌a549细胞、cml k562细胞株的抑制活性较低,而相比于亚油酸、油酸以及化合物iii,本发明制备的化合物ⅳ(r,e)-5-(苄氧基)-2-己基十六碳-2-烯酸具有显著的抗肿瘤效果。
[0093]
(3)肿瘤细胞毒性比较结果
[0094]
就化合物ⅳ与其他受试物的细胞毒性比较优劣结果而言,若化合物ⅳ抑制率≥或≈其他化合物,则认为化合物ⅳ的细胞毒性优于后者,标记为“+”;其他情况则认为无法判断优劣,标记为
“‑”
,结果如表5所示;
[0095]
表5:化合物ⅳ对肿瘤细胞毒性比较结果
[0096]
细胞株亚油酸油酸化合物ⅲ肺癌a549细胞
‑‑‑
肝癌hepg2细胞++-乳腺癌mcf-7细胞+++头颈癌tca8113细胞+++卵巢癌skov3细胞+
‑‑
cml k562细胞株+
‑‑
鼻咽癌cne-2细胞株++-肾癌a498细胞株+-+膀胱癌t24细胞株+-+胰腺癌panc-1细胞株+++
[0097]
从表5所示,化合物ⅳ对除肺癌a549细胞以外的其他受试细胞株的细胞毒性优于亚油酸,对5种细胞株(hepg2、mcf-7、tca8113、cne-2、panc-1)的细胞毒性优于油酸,对5种细胞株(mcf-7、tca8113、a498、t24、panc-1)的细胞毒性优于化合物ⅲ。
[0098]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1