一种高分子量受阻胺光稳定剂的制备方法与流程
1.本发明属于光稳定剂技术领域,具体涉及一种高分子量受阻胺光稳定剂的制备方法。
背景技术:
2.经过近几十年的快速发展,受阻胺光稳定剂已成为光稳定剂领域里应用最为广泛,市场占有率最大的一个种类。现如今,如何更好地提高受阻胺光稳定剂的光稳效果,更好地使其与高分子聚合材料相互匹配,更好地使其与应用环境相适应,是受阻胺光稳定剂的发展趋势和开发方向。
3.早期开发的传统受阻胺光稳定剂,虽然有着不错的光稳效果,但是其分子量普遍较小,耐抽提性较差,尤其是对厚度较小的高分子材料如农用薄膜,在加工过程中,很容易从高分子材料中逸出,导致其实际添加量不断减少,影响光稳效果。
4.据研究表明,高分子量虽然可以提高受阻胺光稳定剂的耐抽提性、耐迁移性等特点,但如果分子量过高也会有碍其发挥光稳定性能。适当的迁移能力在聚合物材料中,尤其是较厚的制品实际应用中是十分必须的,及时迁移到材料表面补充消耗和散失的光稳定剂,可以有效地防止由外及里的老化过程,而分子量过高将会使得受阻胺光稳定剂的迁移能力下降,影响其使用效果。目前,较为普遍的观点认为2000-3000的相对分子质量是一个较为合适的分布范围。
5.由于传统的受阻胺类光稳定剂是从三丙酮胺的结构基础上发展而来的,而其结构中自带的n-h基团使得其拥有较高的碱性,这种碱性过高的受阻胺光稳定剂,在遇到酸性环境、酸性树脂或者酸性复配剂时就容易导致受阻胺光稳定剂成盐,影响了氮氧自由基的生成数量和生成速率,使其实际含量受损,作用效果降低。导致其无法使用在聚氯乙烯或者涂料等酸性环境中,以及无法和聚合物材料中其他酸性助剂共同使用,限制了传统光稳定剂的使用范围,不能将其功效最大化。
6.所以,在保证光稳定性能的基础上,开发具有更高分子量的受阻胺类光稳定剂,具有更好适配性,更好环境适应性,更高效以及更低生产成本是势在必行的。
7.瑞士的汽巴公司在这一点起步最早,他们在1978年即推出了聚合型受阻胺tinuvin 622,后来又研发了聚合型受阻胺chimassbor944和分子量为2136左右的单体型受阻胺chimassbor 119。同时其他光稳定剂生产公司也都推出了聚合型高相对分子质量产品,如cyasorbuv3346等。这些产品都表现出优秀的应用性能。
8.市场上常见的聚合型受阻胺类光稳定剂虽然性能优异,但其在合成过程中步骤复杂,分子量较难控制。为此,本发明对市场上几种使用广泛的受阻胺类光稳定剂如chimassorb-944、chimassbor 119等合成工艺进行对比研究,通过改进合成工艺,公开了一种高分子量受阻胺光稳定剂的制备方法,使其合成过程更加高效,分子量控制更为容易。本发明公开的受阻胺类光稳定剂分子量为2734.54,同时兼具分子量适宜和合成工艺简便等优点。
技术实现要素:
9.为解决上述问题,本发明公开了一种高分子量受阻胺光稳定剂的制备方法。
10.为达到上述目的,本发明的技术方案如下:
11.本发明的一个目的是提供一种高分子量受阻胺光稳定剂,结构式如式(ⅰ)所示:
[0012][0013]
本发明的另一个目的是提供一种高分子量受阻胺光稳定剂的制备方法,包括以下步骤:
[0014]
s1:通入氢气,叔辛胺和三丙酮胺在催化剂i作用下加氢反应,生成2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺;
[0015][0016]
s2:2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺与三聚氯氰在催化剂ⅱ的作用下进行取代反应,得到6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺反应溶液;
[0017][0018]
s3:将含有6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基
戊烷-2-基)-1,3,5-三嗪-2,4-二胺的有机反应液,与甲醛在催化剂ⅲ作用下进行甲基化反应,得到6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺反应溶液;
[0019][0020]
s4:将含有6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的有机反应溶液,与n,n'-双(3-氨丙基)-乙撑二胺在催化剂ⅳ作用下进行取代反应,得到高分子量受阻胺光稳定剂产物;
[0021][0022]
进一步地,步骤s1中,所述三丙酮胺与叔辛胺与催化剂i与氢气的摩尔比为1:0.5-1.5:0.1-0.9:0.1-1.5。
[0023]
进一步地,步骤s1中,所述催化剂i为骨架铜、骨架镍、骨架钴中的一种或多种,所述加氢反应的温度为85-120℃,加氢反应的时间为8-30h。
[0024]
进一步地,步骤s2中,所述2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺与三聚氯氰与催化剂ⅱ摩尔比为1:0.5-1.2:1-8,所述2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺与溶剂的质量比为1:1-5;2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺的滴加流量为5-50g/min。
[0025]
进一步地,步骤s2中,所述催化剂ⅱ为32wt%液碱,液碱的滴加流量为5-50g/min;
所述取代反应的溶剂为二甲苯、甲苯中的一种或多种;所述取代反应的温度为80-110℃,取代反应的时间为24-60h。
[0026]
所述步骤s2中,取代反应完成后,进行分水、水洗,水洗的量为2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺重量的0.5-1.6倍,水洗温度为60-90℃,水洗时间为0.1-5h。
[0027]
进一步地,步骤s3中,所述6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺反应溶液与甲醛与催化剂ⅲ的质量比为1:0.01-1.1:0.02-0.09。
[0028]
进一步地,步骤s3中,所述催化剂ⅲ为甲酸,硫酸二甲酯,碘甲烷,溴甲烷中的一种或多种,催化剂ⅲ的滴加流量为5-20g/min,所述甲基化反应的温度为65-120℃,甲基化反应的时间为12-26h。
[0029]
所述步骤s3中,甲基化反应完成后,进行分水、水洗,水洗的量为6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺反应溶液重量的0.1-2倍,水洗温度为60-85℃,水洗时间为0.1-5h。
[0030]
进一步地,步骤s4中,所述含有6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺反应溶液与n,n'-双(3-氨丙基)-乙撑二胺与催化剂ⅳ的质量比为1:0.01-1.2:0.02-0.8。
[0031]
进一步地,步骤s4中,所述催化剂ⅳ为32wt%液碱,所述取代反应的温度为155-255℃,取代反应的时间为8-30h。
[0032]
所述步骤s4中,取代反应完成后,进行分水、水洗,水洗的量为含有6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺反应溶液重量的0.2-3倍,水洗温度为65-85℃,水洗时间为1-10h。
[0033]
本发明的有益效果为:
[0034]
1、本发明采用的原料三丙酮胺为本公司基础原料,资源丰富且质量优异;
[0035]
2、本发明制备的高分子量受阻胺光稳定剂产品具有优异的抗氧化效果;
[0036]
3、制备方法简单,原子利用率高,方法绿色环保。
附图说明
[0037]
图1为本发明实施例1步骤s1中制备得到的目标产物的红外图谱;
[0038]
图2为本发明实施例1步骤s2中制备得到的目标产物的红外图谱;
[0039]
图3为本发明实施例1步骤s3中制备得到的目标产物的红外图谱;
[0040]
图4为本发明实施例1步骤s4中制备得到的目标产物的红外图谱。
具体实施方式
[0041]
下面结合附图和具体实施方式,进一步阐明本发明,应理解下述具体实施方式仅用于说明本发明而不用于限制本发明的范围。
[0042]
实施例1
[0043]
s1:向3l加氢釜中投入450g叔辛胺,450g的三丙酮胺,80g骨架铜,氮气置换,开启搅拌并升温至95℃,通氢气5.5g,然后保温反应10h,蒸馏脱溶,制得739.34g的2,2,6,6-四
甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺。
[0044]
结构式如下所示:
[0045][0046]
2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺的红外图谱如图1所示,在2955cm-1
、1455cm-1
、1398cm-1
吸收峰,表明存在-ch3、-ch
2-的伸缩、弯曲振动vc-h。在3260cm-1
处有一个vn-h的吸收峰,在1237cm-1
处呈现vc-n的特征吸收峰,表明分子中有二级胺存在。
[0047]
s2:向5l烧瓶中投入600g二甲苯,115g的三聚氯氰,开启搅拌滴加264g 2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺,滴加时控制反应温度为85℃,控制滴加流量为10g/min,滴加132g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺后,开始同时滴加584g液碱(质量分数32%),液碱滴加流量控制为15g/min。然后升温至90℃反应30h,反应结束后,分水,分水结束后,向有机层加入200g的75℃热水进行搅拌水洗0.5h,水洗结束后,进行分水,制得955g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液。
[0048]
结构式如下所示:
[0049][0050]
6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的红外图谱如图2所示,在2965cm-1
、1483cm-1
、1369cm-1
吸收峰,表明存在-ch3、-ch
2-的伸缩、弯曲振动vc-h。在1400~1600cm-1
处的峰为苯环的特征峰,在1242cm-1
处呈现vc-n的特征吸收峰,在700~750cm-1
为vc-cl的特征吸收峰。
[0051]
s3:向装有955g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液的烧瓶中加入80g甲醛,控制反应温度为70℃,然后滴加60g甲酸(质量分数为94%),甲酸滴加流量控制为5g/min,滴加完毕后,升温至95℃,反应15h。反应结束后,分水。然后向有机层加入220g的72℃热水,进行搅拌水洗1h,水洗结束后,进行分水,制得960g的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液。
[0052]
结构式如下所示:
[0053][0054]
6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的红外图谱如图3所示,在2969cm-1
、1485cm-1
、1358cm-1
吸收峰,表明存在-ch3、-ch
2-的伸缩、弯曲振动vc-h,是受阻胺基团的特征吸收峰。在1400~1600cm-1
处的峰为苯环的特征峰,在1291cm-1
处呈现vc-n的特征吸收峰,在700~750cm-1
为vc-cl的特征吸收峰。
[0055]
s4:将960g的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液转移至5l高压釜中,并加入110g液碱(质量分数为32%),加入30g的n,n'-双(3-氨丙基)-乙撑二胺,氮气置换,开启搅拌并升温至185℃并保温反应8h;反应结束后,分水。然后向有机层加入250g的73℃热水进行搅拌水洗1h,水洗结束后,进行分水,有机层脱溶后即得349.2g的成品,收率为96%。
[0056]
结构式如下所示:
[0057][0058]
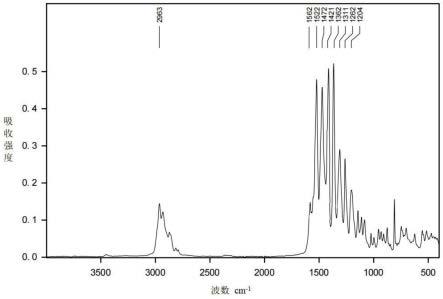
目标产物的的红外图谱如图4所示,在2963cm-1
、1472cm-1
、1362cm-1
吸收峰,表明存在-ch3、-ch
2-的伸缩、弯曲振动vc-h,是受阻胺基团的特征吸收峰,在1400~1600cm-1
处的峰为苯环的特征峰,在1262cm-1
处呈现vc-n的特征吸收峰。
[0059]
实施例2
[0060]
s1:向5l加氢釜中投入1380g叔辛胺,1400g的三丙酮胺,120g骨架铜,氮气置换,开启搅拌并升温至90℃,通氢气22.5g,然后保温反应15h,蒸馏脱溶,制得1938.12g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺。
[0061]
s2:向10l玻璃反应釜中投入3200g二甲苯,1430g的三聚氯氰,开启搅拌滴加1938.12g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺,滴加时控制反应温度为95℃,控制滴加流量为15g/min,滴加750g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺后,开始同时滴加1875g液碱(质量分数32%),液碱滴加流量控制为15g/min。然后升温至90℃反应36h,反应结束后,分水,分水结束后,向有机层加入1800g的85℃热水进行搅拌水洗2h,水洗结束后,进行分水,制得6945.8g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液。
[0062]
s3:向装有6945.8g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液的烧瓶中加入230g甲醛,控制反应温度为85℃,然后滴加305g硫酸二甲酯,硫酸二甲酯滴加流量控制为15g/min,滴加完毕后,升温至95℃,反应12h。反应结束后,分水。然后向有机层加入2600g的75℃热水,进行搅拌水洗1.5h,水洗结束后,进行分水,制得6782.7g的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液。
[0063]
s4:将6782.7g的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液转移至20l反应釜中,并加入580g液碱(质量分数为32%),加入490g的n,n'-双(3-氨丙基)-乙撑二胺,氮气置换,开启搅拌并升温至185℃并保温反应8h;反应结束后,分水。然后向有机层加入2050g的80℃热水进行搅拌水洗1h,水洗结束后,进行分水,有机层脱溶后即得3576.9g的成品,收率为98.8%。
[0064]
实施例3
[0065]
s1:向3l加氢釜中投入680g叔辛胺,590g的三丙酮胺,40g骨架镍,氮气置换,开启搅拌并升温至90℃,通氢气8.8g,然后保温反应12h,蒸馏脱溶,制得939.6g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺。
[0066]
s2:向10l玻璃反应釜中投入800g二甲苯,635g的三聚氯氰,开启搅拌滴加939.6g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺,滴加时控制反应温度为95℃,控制滴加流量为15g/min,滴加450g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺后,开始同时滴加600g液碱(质量分数32%),液碱滴加流量控制为15g/min。然后升温至90℃反应30h,反应结束后,分水,分水结束后,向有机层加入690g的85℃热水进行搅拌水洗2h,水洗结束后,进行分水,制得1805.8g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液。
[0067]
s3:向装有1805.8g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液的烧瓶中加入90g甲醛,控制反应温度为82℃,然后滴加56g溴甲烷,溴甲烷滴加流量控制为12g/min,滴加完毕后,升温至90℃,反应16h。反应结束后,分水。然后向有机层加入1000g的75℃热水,进行搅拌水洗0.5h,水洗结束后,进行分水,制得1690.1g的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液。
[0068]
s4:将1690.1的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的二甲苯溶液转移至5l反应釜中,并加入320g液碱(质量分数为32%),加入60g的n,n'-双(3-氨丙基)-乙撑二胺,氮气置换,开启搅拌并升温至185℃并保温反应12h;反应结束后,分水。然后向有机层加入600g的80℃热水进行搅拌水洗1h,水洗结束后,进行分水,有机层脱溶后即得852.2g的成品,收率为94.76%。
[0069]
实施例4
[0070]
s1:向5l加氢釜中投入450g叔辛胺,450g的三丙酮胺,80g骨架镍,氮气置换,开启搅拌并升温至95℃,通氢气5.5g,然后保温反应10h,蒸馏脱溶,制得696.5g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺。
[0071]
s2:向5l烧瓶中投入600g甲苯,115g的三聚氯氰,开启搅拌滴加264g 2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺,滴加时控制反应温度为85℃,控制滴加流量为10g/min,滴加132g的2,2,6,6-四甲基-n-(2,4,4-三甲基戊烷-2-基)哌啶-4-胺后,开始同时滴加187g液碱(质量分数32%),液碱滴加流量控制为15g/min。然后升温至90℃反应30h,反应结束后,分水,分水结束后,向有机层加入200g的75℃热水进行搅拌水洗0.5h,水洗结束后,进行分水,制得925.8g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的甲苯溶液。
[0072]
s3:向装有925.8g的6-氯-n2,n4-双(2,2,6,6-四甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的甲苯溶液的烧瓶中加入80g甲醛,控制反应温度为70℃,然后滴加60g甲酸(质量分数为94%),甲酸滴加流量控制为5g/min,滴加完毕后,升温至95℃,反应15h。反应结束后,分水。然后向有机层加入220g的72℃热水,进行搅拌水洗1h,水洗结束后,进行分水,制得926.4g的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的甲苯溶液。
[0073]
s4:将926.4g的6-氯-n2,n4-双(1,2,2,6,6-五甲基哌啶-4-基)-n2,n4-双(2,4,4-三甲基戊烷-2-基)-1,3,5-三嗪-2,4-二胺的甲苯溶液转移至5l高压釜中,并加入110g液碱(质量分数为32%),加入30g的n,n'-双(3-氨丙基)-乙撑二胺,氮气置换,开启搅拌并升温至185℃并保温反应8h;反应结束后,分水。然后向有机层加入250g的73℃热水进行搅拌水洗1h,水洗结束后,进行分水,有机层脱溶后即得318.9g的成品,收率96.7%。
[0074]
需要说明的是,以上内容仅仅说明了本发明的技术思想,不能以此限定本发明的保护范围,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰均落入本发明权利要求书的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1