一种HBB融合基因修饰的自体造血干细胞、制备方法及其应用与流程
一种hbb融合基因修饰的自体造血干细胞、制备方法及其应用
技术领域
1.本技术涉及一种hbb融合基因修饰的自体造血干细胞、制备方法及其应用,属于遗传工程技术领域。
背景技术:
2.镰状细胞病(scd)和β-地中海贫血(β-tm)是世界上最常见的单基因疾病,每年约有40万孕妇或新生儿受到影响。这些疾病为两类不同的β-珠蛋白基因突变,导致出现异常的血红蛋白结构(scd)或β-珠蛋白链(β-tm)大量减少或缺失。这类遗传性疾病的临床表现通常出现在出生后几个月,此时基因表达从胎儿γ-珠蛋白链转换到形成成人βa-珠蛋白链(hba)。
3.如果有人类白细胞抗原(hla)匹配的同胞供者,同种异体造血干细胞移植(ahsct)目前被推荐作为β-tm的治疗选择。ahsct后无病生存率在儿童受试者中约为88%,成人为65%。然而,只有不到25%的患者有合适的家族内供体。尽管移植结果正在改善,但ahsct仍存在严重不良事件和死亡率的重大风险,两者均随受者年龄和疾病严重程度而增加。严重的不良事件包括移植物衰竭、移植物抗宿主病(gvhd)、清除骨髓和免疫抑制的调节方案的早期或晚期副作用(感染、出血、继发性恶性肿瘤),以及先前存在的器官损害的加重。
4.对于缺乏合适的hla匹配供体的患者,使用自体造血干细胞进行体外基因治疗为潜在的治疗选择带来了希望。基因治疗,通过体外慢病毒将治疗性β-珠蛋白基因衍生物转染到造血干细胞,使红细胞特异性高表达,使我们有望治愈这种遗传性疾病。
5.cn110582305a为美国蓝鸟生物公司的发明专利,主要提供了表达正常血红蛋白的基因治疗载体,其中缺乏hs4 dnase i超敏位点,血红蛋白的表达量较低。
6.cn111447954a专利中主要是对于腺病毒载体的优化,使其具有安全性和有效性。但该专利中hbb基因中的核苷酸点突变或缺失选自264位或265位c的缺失等其他突变,血红蛋白的表达量较低。
技术实现要素:
7.针对现有技术存在的不足,本发明提供一种hbb融合基因修饰的自体造血干细胞、制备方法及其应用,实现以下发明目的:提高人结合珠蛋白的表达量,提高红系集落数量。
8.为解决上述技术问题,本发明采取以下技术方案:一种hbb融合基因修饰的自体造血干细胞,所述hbb融合基因由以下模块依次串联得到:dnase i超敏位点hs2、dnase i超敏位点hs3、dnase i超敏位点hs4、启动子、增强子、hsb t88q;所述dnase i超敏位点hs2的核苷酸序列如序列表中seq id no.2所示;所述dnase i超敏位点hs3的核苷酸序列如序列表中seq id no.3所示;所述dnase i超敏位点hs4的核苷酸序列如序列表中seq id no.4所示;所述启动子的核苷酸序列如序列表中seq id no.5所示;
所述增强子的核苷酸序列如序列表中seq id no.6所示;所述hsb t88q的核苷酸序列如序列表中seq id no.7所示。
9.以下是对上述技术方案的进一步改进:所述自体造血干细胞为cd34
+
细胞。
10.所述制备方法包括重组表达载体pcdh-hbb的构建、慢病毒的包装、重组慢病毒感染cd34
+
细胞。
11.所述重组表达载体pcdh-hbb的构建,将hbb融合基因插入pcdh载体,获得重组表达载体pcdh-hbb。
12.所述慢病毒的包装,采用重组表达载体pcdh-hbb转染复苏后的293t细胞,得到重组慢病毒,所述重组慢病毒的滴度为1.52
×
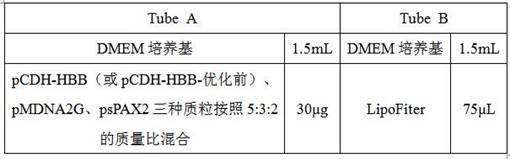
108tu/ml。
13.所述重组慢病毒感染cd34
+
细胞,将重组慢病毒与cd34
+
细胞的比例控制为50:1进行培养、筛选、扩大培养得到hbb融合基因修饰的cd34
+
细胞。
14.所述的hbb融合基因修饰的自体造血干细胞在制备治疗镰状细胞病和β-地中海贫血的药物中的应用。
15.与现有技术相比,本发明取得以下有益效果:本发明利用核苷酸优化后的hbb融合基因修饰的cd34
+
细胞,红系集落数量平均值为540,人结合珠蛋白的含量为985.42mg/l。
附图说明
16.图1为hbb融合基因的示意图;图2为含pcdh-hbb的重组慢病毒对cd34
+
细胞的感染率的流式图;图3为pcdh-hbb修饰的cd34
+
细胞中cd34
+
阳性率的流式图;图4为含pcdh-hbb
ꢀ‑
优化前的重组慢病毒对cd34
+
细胞的感染率的流式图;图5为pcdh-hbb-优化前修饰的cd34
+
细胞中cd34
+
阳性率的流式图;图6为pcr鉴定目标基因的存在的电泳图;图7为hbb融合基因修饰造血干细胞的集落数量形成图。
具体实施方式
17.实施例1 重组表达载体pcdh-hbb的构建hbb融合基因模块示意见图1(完整核酸序列见附录seq id no.1)。
18.hbb融合基因各模块序列(1)dnase i超敏位点hs2 (seq id no.2)(2)dnase i超敏位点hs3(seq id no.3)(3)dnase i超敏位点hs4(seq id no.4)(4)启动子 (seq id no.5)(5)增强子(seq id no.6)(6)hsb t88q (seq id no.7)上述dnase i超敏位点hs2-hs4均是从全长序列中截取的核心序列;本技术对hsb t88q的核苷酸序列进行了密码子优化;
按照上述seq id no.1序列委托上海捷瑞生物工程有限公司合成其整个表达框,插入pcdh载体(由中国疾控中心病毒病预防控制所提供) spei-noti位点,转化,到e.coli(top10),经测序正确后,使用omega公司的质粒提取试剂盒提取质粒,获得重组表达载体pcdh-hbb。本发明中,重组表达载体pcdh-hbb的浓度为1.23
µ
g/
µ
l。
19.将dnase i超敏位点hs2、dnase i超敏位点hs4均采用全长序列,所述dnase i超敏位点hs2的全长核苷酸序列如序列表中seq id no.13所示;所述dnase i超敏位点hs4的全长核苷酸序列如序列表中seq id no.14所示; 将seq id no.13、seq id no.3、seq id no.14、seq id no.5、seq id no.6的核酸序列按顺序连接,再与优化前的hsb t88q的核苷酸序列进行连接,得到优化前的hbb融合基因序列(seq id no.8);载体采用相同的pcdh载体,构建成重组表达载体,命名为pcdh-hbb-优化前,其余操作如上,其质粒浓度为1.05
µ
g/
µ
l。
20.优化前的hsb t88q的核苷酸序列为:将ncbi中序列号为cr541913的第262-264位的密码子调整为cag,得到的序列作为优化前的hsb t88q的核苷酸序列。
21.实施例2 慢病毒的包装及滴度测定1)包装细胞系的复苏本发明使用的包装细胞系为293t细胞。从液氮罐中取出冻存的293t细胞,迅速丢入37℃水浴锅中并快速晃动,尽量在2 min内使细胞溶液完全溶解。将1ml细胞溶液转移到50ml离心管中,加入35ml生理盐水清洗dmso,混匀后离心,离心条件为:1500rpm,5 min。去掉上清,加入5 ml新鲜的高糖dmem(含10 vol% fbs)培养基重悬细胞,转入t75瓶中,每个瓶中继续加入高糖dmem(含10 vol% fbs)培养基定容至10ml。将培养瓶平稳放入37℃、5%co
2 的培养箱中培养。第二天观察细胞存活率,并更换培养基。以后每天观察细胞生长情况,在细胞铺满瓶底的90%时传代,传代2次后细胞用于转染。
22.2)慢病毒的包装及滴度测定当传代2代后,293t细胞铺板达到80%时,用于转染。
23.转染试剂的准备:在5ml离心管中,分别配制a管与b管试剂(tube a andtube b)配好后,放置5min,然后将a管缓慢加入b管,混合均匀。室温放置20min,形成脂质体-dna混合物。将混合物加入培养瓶中,轻微混匀。置于37℃,5%co2培养箱中培养48h,用免疫荧光显微镜观察转染情况,收取全部上清和细胞,转移至50ml离心管,进行离心,离心条件为:4000g离心30min。去除沉淀,浓缩纯化,得到重组慢病毒,分装保存在-80℃冰箱中。利用倍比稀释技术法测定病毒滴度,本发明中含pcdh-hbb的重组慢病毒的滴度为1.52
×
108tu/ml,含pcdh-hbb-优化前的重组慢病毒的滴度为1.46
×
108tu/ml。
24.实施例3 重组慢病毒修饰cd34
+
细胞的制备1. cd34
+
细胞的分选与培养在单细胞采集前,患者服用filgrastim药物,刺激体内细胞的生成,在第5天采集单细胞。采集的细胞用miltenyi 公司提供的cd34 microbead kit分选磁珠分选出cd34
+
细胞,进行细胞计数,将浓度为5
×
105个细胞/ml的细胞培养液接种到六孔板中,每孔中补足到2ml的cd34扩增培养基,放在37℃、5%co2的培养箱中培养2天。六孔板预先用5
µ
g/cm2的fibronectin(购自gibco)置于37℃培养箱中包被6h,cd34扩增培养基为stemspan sfem medium(购自stemcell technologies),含有20ng/ml scf、200ng/ml flt-3l和100ng/ml tpo(均购自mce公司)。
25.2. 重组慢病毒感染cd34
+
细胞cd34
+
细胞感染之前,更换培养基,每孔加入2ml含有100ng/ml的il-3、50ng/ml的il-1α、100ng/ml的il-6、20ng/ml的scf、200ng/ml的flt-3l、100ng/ml的tpo的stemspan sfem medium对其进行预刺激培养24h,进行慢病毒感染实验。
26.从-80℃拿出实施例2制备的重组慢病毒解冻后加入cd34扩增培养基,将病毒滴度稀释到5
×
107tu/ml,用稀释后的重组慢病毒液重悬1
×
106个cd34
+
细胞,得到细胞悬液。将细胞悬液加入到6孔板中,每孔1ml,使病毒颗粒数与cd34
+
细胞数比例为50:1,37℃,5%的co2培养箱中培养6h后,用cd34扩增培养基稀释一倍(每孔加入1ml培养基),继续培养1天后更换含有300ng/ml嘌呤霉素的cd34扩增培养基筛选4天,更换cd34扩增培养基进行扩大培养3天。
27.分别得到pcdh-hbb修饰的cd34
+
细胞、pcdh-hbb-优化前修饰的cd34
+
细胞,通过流式细胞术检测cd34
+
阳性率和gfp的表达率。以未进行慢病毒感染的cd34
+
细胞作为阴性对照。本发明中pcdh-hbb对cd34
+
细胞的感染率为55.1%,cd34
+
阳性率为91.6%;pcdh-hbb-优化前对cd34
+
细胞的感染率为54.1%,cd34
+
阳性率为89.8%(见图2-5)。
28.实施例4 pcr鉴定基因修饰的cd34
+
细胞中hsb的存在分别收集扩增培养的pcdh-hbb修饰的cd34
+
细胞和pcdh-hbb-优化前修饰的cd34
+
细胞,提取细胞基因组,利用各自的特异引物进行pcr鉴定,其中pcdh-hbb的引物序列为hbb-f:tacagaggtgtggcaagcag(seq id no.9)和hbb-r:accactttctgataggcagc(seq id no.10),pcdh-hbb-优化前的引物序列为b-f:agatggctctgccctgactt(seq id no.11)和b-r:cgttcaccttgccccacagg(seq id no.12)。结果如图6 所示,pcr能够扩增到与目的基因大小相同的片段,说明pcdh-hbb基因和pcdh-hbb-优化前基因均整合进入了cd34
+
细胞中。
29.实施例5 cd34
+
细胞集落形成实验(1)分别收集慢病毒感染后的cd34
+
细胞和未感染的cd34
+
细胞,计数。
30.(2)使用200
µ
l imdm+2%fbs重悬3000个cd34
+
细胞。
31.(3)在细胞悬液中加入1ml甲基纤维素,混匀,加入2.5mm培养盘。
32.(4)把培养盘放入37℃、5%co2细胞培养箱中培养14天,培养过程中禁止移动。
33.(5)14天过后观察集落形态,计数红系集落数量(如图7)。
34.结果如图7所示,hbb融合基因修饰的cd34
+
细胞中红系集落数量平均值为540,而hbb-优化前基因修饰的cd34
+
细胞中红系集落数量平均值为370。
35.实施例6 基因修饰的cd34
+
细胞中人结合珠蛋白表达量检测将基因修饰的cd34
+
细胞培养一周后,检测其人结合珠蛋白的表达量。利用人结合珠蛋白elisa试剂盒(购自上海沪震生物科技有限公司)检测人结合珠蛋白的表达量。各收集一孔细胞及上清,将细胞裂解后离心,取上清进行检测。具体步骤如下:1. 标准品的加样:设置标准品孔和样本孔,标准品孔各加不同浓度的标准品50μl。
36.2. 加样:分别设空白孔(空白对照孔不加样品及酶标试剂,其余各步操作相同)、待测样品孔。在酶标包被板上待测样品孔中先加样品稀释液40μl,然后再加待测样品10μl(样品终稀释度为5倍)。加样将样品加于酶标板孔底部,尽量不触及孔壁,轻轻晃动混匀。
37.3. 加酶:每孔加入酶标试剂100μl,空白孔除外。
38.4. 温育:用封板膜封板后置37℃温育60分钟。
39.5. 配液:将20倍浓缩洗涤液用蒸馏水20倍稀释后备用。
40.6. 洗涤:小心揭掉封板膜,弃去液体,甩干,每孔加满洗涤液,静置30秒后弃去,如此重复5次,拍干。
41.7. 显色:每孔先加入显色剂a50μl,再加入显色剂b50μl,轻轻震荡混匀,37℃避光显色15分钟。
42.8. 终止:每孔加终止液50μl,终止反应。
43.9. 测定:以空白孔调零,450nm波长依序测量各孔的吸光度(od值)。
44.经计算,pcdh-hbb修饰的cd34
+
细胞中人结合珠蛋白含量为985.42mg/l,pcdh-hbb-优化前修饰的cd34
+
细胞中人结合珠蛋白含量为658.23mg/l。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1