用于病毒感染所致癌症的环形mRNA疫苗开发平台的制作方法
用于病毒感染所致癌症的环形mrna疫苗开发平台
技术领域
1.本发明涉及一种mrna疫苗的开发平台,具体涉及一种病毒感染所致癌症环形mrna疫苗的开发平台。
背景技术:
2.感染被认为全世界肿瘤致病的一类重要因素,预防和采取药物抗感染能有效地进行肿瘤的防治。研究表明,有多达40%的癌症与病毒感染有关。这些包括了许多大家熟知的各种实体肿瘤和血液肿瘤,如鼻咽癌、肝癌、宫颈癌、成人t细胞白血病和卡波西肉瘤等。相关研究表明,病毒只有侵入活体细胞,才能存活并复制更多的病毒。这一过程往往会杀死宿主的细胞,因此这些被感染的细胞是不会癌变的。换句话说,只有那些长期存活在体内的病毒才有致癌的风险。
3.1964年,tony epstein和yvonne barr等人在非洲burkitt淋巴瘤组织中通过电镜,成功分离出了epstern-barr病毒(ebv),这是第一个被发现与人类肿瘤相关的病毒,至今已有半个世纪。随后,其他致瘤病毒也相继发现,如:梅克尔多元癌细胞病毒(mcpyv), 乙肝病毒(hbv),丙肝病毒(hcv),人乳头状瘤病毒(hpv),人t细胞白血病i型病毒(小时tlv-1)和卡波西肉瘤病毒(ks小时v 或hhv-8)等,分别与梅克尔细胞癌、鼻咽癌、肝癌、宫颈癌、成人t细胞白血病和卡波西肉瘤的发生相关。根据解剖部位分析,感染相关的肿瘤平均发生率达到64.4%。其中,ebv相关的鼻咽癌占90%以上, hbv和hcv相关的肝癌和胆管癌占75.9%,hpv相关的宫颈癌为100%。
4.癌症疫苗分为预防性疫苗与治疗性疫苗,接种预防性疫苗可以预防相关病毒的感染,从而大幅度降低肝癌、宫颈癌等的发生风险;而接种治疗性的疫苗,除了治疗癌症,还能将副作用降到最低,甚至无明显的副作用产生。目前市面上的癌症疫苗多为预防性疫苗,如乙肝疫苗,hpv疫苗,靶向疫苗eb病毒糖蛋白gp350等虽然能降低肝癌,宫颈癌和鼻咽癌等的发病率,但这些疫苗并不能用于绝大部分相关病毒感染相关的肿瘤预防和治疗。因此,非常必要建立一种能用于病毒感染所致相关肿瘤治疗疫苗开发的技术平台,从而可以为临床治疗病毒感染相关肿瘤的治疗提供有效疫苗,达到降低病毒感染患者致癌率和改善病毒感染致癌患者治疗效果的目的。
5.目前,主要的肿瘤疫苗有多肽疫苗、mrna疫苗、dna疫苗和树突状细胞疫苗等。其中,mrna 因为其可以灵活的合成,具有很好的应用价值。但是,由于体内rna酶能很快的降解 mrna疫苗,对其效果影响较大。鉴于现有技术存在的缺陷和不足之处,本发明提供了一种病毒感染所致癌症环形mrna疫苗的开发平台,具体涉及一种基于病毒感染相关肿瘤新生抗原和能在人类t细胞表达所述病毒感染相关肿瘤新生抗原的具有特定ires序列的环形mrna分子病毒疫苗的开发。由于环形mrna为封闭环形结构,能够耐受体内rna酶的水解,相较于mrna具有更长的半衰期。这些编码免疫原性多肽的环形mrna是一类新型肿瘤疫苗。本发明所述病毒环形mrna疫苗有望扩大病毒感染患者的受益范围,改善病毒感染相关肿瘤的治疗质量,给病毒感染患者和社会带来更多的病毒感染相关肿瘤治疗收益。
技术实现要素:
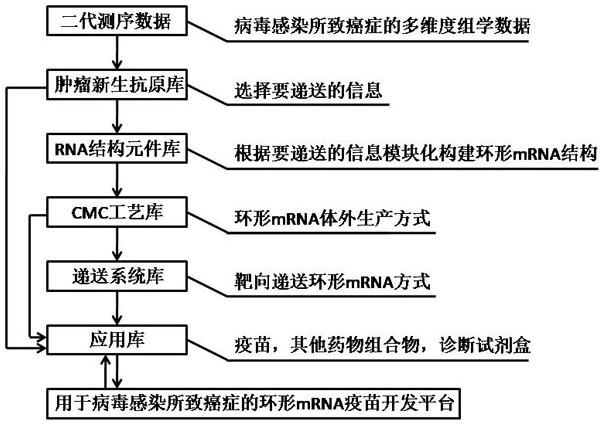
6.发明要解决的问题本发明的目的在于提供一种病毒感染所致癌症环形mrna疫苗的开发平台。通过该开发平台的肿瘤新生抗原库,环形rna元件结构库,cmc工艺库,递送系统库及应用库匹配协调可以展开病毒感染所致癌症肿瘤新生抗原决定簇的辨寻、能编码病毒感染所致肿瘤免疫原性多肽的环形mrna制备及验证,从而来提供能有效治疗病毒感染所致肿瘤的新型环形mrna疫苗。
7.本发明着重解决的问题如下:第一个问题,病毒感染所致癌症的环形mrna疫苗的通用型问题。通过病毒感染所致癌症环形mrna疫苗开发平台的肿瘤新生抗原库提供了的病毒感染所致癌症通用型肿瘤新生抗原。同时通过病毒感染所致癌症环形mrna疫苗开发平台的rna元件库提供了能在人体或小鼠细胞内表达本发明所述通用型肿瘤新生抗原的ires元件。这些通过模块化设计和构建的能表达本发明所述通用型肿瘤新生抗原的环形mrna不仅激活了不同受试者体内病毒感染所致癌症的抗肿瘤免疫反应,而且能引起不同受试者的不同肿瘤的免疫应答反应,从而可以保证环形mrna疫苗有效性的同时具有更广泛的适用性。本发明有望克服现有技术疫苗的治疗需要根据不同个体进行单独的测序及个体化的疫苗制作缺陷,大大降低肿瘤患者的治疗成本和用药等待周期。
8.第二个问题,环形mrna的分离纯化问题。本发明所述的环形mrna是一类具有连续结构的单链rna,其具有增强的稳定性和缺乏与各种细胞蛋白相互作用所必需的末端基序。作为外源性的重组核酸分子,所述环形mrna能够绕过细胞rna感受器,从而避免在rig-1和toll样受体(tlr)感受态细胞和小鼠中引起免疫应答。研究表明,环形mrna制剂的免疫原性和蛋白质表达稳定性取决于纯度。此外,研究证实环形mrna合成过程中残留的杂质可能会引起细胞免疫应答。所以,非常有必要去除环形mrna合成中可能残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质。目前工程级环形mrna的分离纯化采用高效液相色谱尺寸排阻柱法,磷酸酶处理及rna酶r处理的相结合的方法。虽然利用该方法所获得的工程级环形mrna的纯度可以达到90%左右,但这离临床研究和实践的纯度要求存在很大的差距。为了提高分离纯化纯度,本发明病毒感染所致癌症环形mrna疫苗开发平台的cmc工艺库通过在含有所需纯化的环形mrna的液相介质中引入合理强度的声能量或辐射能量,提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度。在本发明中,所合成的环形mrna纯度均大于95%,部分达到99%左右。
9.第三个问题,环形mrna的靶向递送问题。环形mrna带有负电荷,而细胞膜表面也带有负电荷,静电排斥使得环形mrna分子较难穿过细胞膜进入细胞内。但是,环形mrna编码信息中涵盖了核糖体生成蛋白的序列,必须递送至癌细胞内才能编码蛋白和诱发免疫应答。所以,将环形mrna递送到癌细胞内部共有两道屏障:静电排斥致使的膜屏障和递送靶向性。迄今为止环形mrna的递送系统主要基于线性mrna的递送系统。由于环形mrna和线性mrna毕竟是不一样的结构,非常有必要开发能促进环形mrna的癌细胞靶向和癌细胞内的生物分布的递送系统,促进环形mrna疫苗效果的发挥。本发明病毒感染所致癌症环形mrna疫苗开发平台的递送系统库将黑磷作为自我牺牲型辅助剂或添加具有一定结构的有机化合物,在合
理强度的声能量或辐射能量的作用下,结合筛选和使用合理的核酸适配体,提供了具有合理表面荷电的环形mrna脂质体靶向递送系统,使环形mrna对癌细胞的靶向递送率从50%以下提高到85%以上,部分提高到92%左右,极大促进了环形mrna疫苗的效果。
10.第四个问题,环形mrna的应用问题。本发明病毒感染所致癌症环形mrna疫苗开发平台的应用库提供了病毒感染所致癌症检测所用的一种试剂盒,提供了肿瘤新生抗原多肽,环形mrna,线性mrna,重组表达载体及重组宿主细胞的其中一种或多种的合理组合物的应用。
11.为实现前述发明目的,本发明采用的技术方案包括以下几个部分:第一部分,开发平台肿瘤新生抗原库的技术方案。
12.本发明实施例通过开发平台肿瘤新生抗原库提供了一类病毒感染所致癌症的通用肿瘤新生抗原,其包括下列多肽中的任一种或多种的组合:具有seq id 1所示的序列或其保守变异型序列的多肽;具有seq id 2所示的序列或其保守变异型序列的多肽;具有seq id 3所示的序列或其保守变异型序列的多肽;具有seq id 4所示的序列或其保守变异型序列的多肽;具有seq id 5所示的序列或其保守变异型序列的多肽;具有seq id 6所示的序列或其保守变异型序列的多肽;具有seq id 7所示的序列或其保守变异型序列的多肽;具有seq id 8所示的序列或其保守变异型序列的多肽;具有seq id 9所示的序列或其保守变异型序列的多肽;具有seq id 10所示的序列或其保守变异型序列的多肽;具有seq id 11所示的序列或其保守变异型序列的多肽;具有seq id 12所示的序列或其保守变异型序列的多肽;具有seq id 14所示的序列或其保守变异型序列的多肽;具有seq id 15所示的序列或其保守变异型序列的多肽;具有seq id 16所示的序列或其保守变异型序列的多肽;具有seq id 17所示的序列或其保守变异型序列的多肽;具有seq id 18所示的序列或其保守变异型序列的多肽;具有seq id 19所示的序列或其保守变异型序列的多肽;具有seq id 20所示的序列或其保守变异型序列的多肽;具有seq id 21所示的序列或其保守变异型序列的多肽;具有seq id 22所示的序列或其保守变异型序列的多肽;具有seq id 23所示的序列或其保守变异型序列的多肽;具有seq id 24所示的序列或其保守变异型序列的多肽;具有seq id 25所示的序列或其保守变异型序列的多肽;具有seq id 26所示的序列或其保守变异型序列的多肽;具有seq id 27所示的序列或其保守变异型序列的多肽;具有seq id 28所示的序列或其保守变异型序列的多肽;具有seq id 29所示的序列或其保守变异型序列的多肽;
具有seq id 30所示的序列或其保守变异型序列的多肽;具有seq id 31所示的序列或其保守变异型序列的多肽;具有seq id 32所示的序列或其保守变异型序列的多肽;具有seq id 33所示的序列或其保守变异型序列的多肽;具有seq id 34所示的序列或其保守变异型序列的多肽;具有seq id 35所示的序列或其保守变异型序列的多肽;具有seq id 36所示的序列或其保守变异型序列的多肽;具有seq id 37所示的序列或其保守变异型序列的多肽;具有seq id 38所示的序列或其保守变异型序列的多肽;具有seq id 39所示的序列或其保守变异型序列的多肽;具有seq id 40所示的序列或其保守变异型序列的多肽;具有seq id 41所示的序列或其保守变异型序列的多肽;具有seq id 42所示的序列或其保守变异型序列的多肽;具有seq id 43所示的序列或其保守变异型序列的多肽;具有seq id 44所示的序列或其保守变异型序列的多肽;具有seq id 45所示的序列或其保守变异型序列的多肽;具有seq id 46所示的序列或其保守变异型序列的多肽;具有seq id 47所示的序列或其保守变异型序列的多肽;具有seq id 48所示的序列或其保守变异型序列的多肽;具有seq id 49所示的序列或其保守变异型序列的多肽;具有seq id 50所示的序列或其保守变异型序列的多肽;具有seq id 51所示的序列或其保守变异型序列的多肽;具有seq id 52所示的序列或其保守变异型序列的多肽;具有seq id 53所示的序列或其保守变异型序列的多肽;具有seq id 54所示的序列或其保守变异型序列的多肽;具有seq id 55所示的序列或其保守变异型序列的多肽;具有seq id 56所示的序列或其保守变异型序列的多肽;具有seq id57所示的序列或其保守变异型序列的多肽;具有seq id 58所示的序列或其保守变异型序列的多肽;具有seq id 59所示的序列或其保守变异型序列的多肽;具有seq id 60所示的序列或其保守变异型序列的多肽;具有seq id 61所示的序列或其保守变异型序列的多肽;进一步的,所述保守变异型序列为原始序列中的一个或多个氨基酸缺失、错位、插入、缩短或延长而得,其具有至少30%,35%,40%,45%,50%,55%,60%,65%,70%,75%,80%,85%,90%,95%或99%的原始序列同一性的序列。
13.第二部分,开发平台rna元件结构库的技术方案。
14.本发明实施例通过开发平台的rna元件结构库还提供了一类重组核酸分子,所述重组核酸分子能够表达本发明前述实施例所述的seq id no: 1
‑ꢀ
seq id no: 61序列或其保守变异型序列的病毒感染所致癌症的编码一个或两个或多个肿瘤新生抗原多肽。
15.进一步的,所述的重组核酸分子,其所表达的肿瘤新生抗原多肽表达水平为病毒感染所致癌症肿瘤新生抗原多肽在真核细胞中的表达水平。
16.进一步的,所述的重组核酸分子,其含有ires元件;优选地, 所述的重组核酸分子所含的ires元件具有seq id no: 62
‑ꢀ
seq id no: 65所示的任一序列所组成的组中的一种或或两种或多种序列的核苷酸序列;优选地,所述的重组核酸分子所含的ires元件具有seq id no: 62
‑ꢀ
seq id no: 65任一序列所示的序列的反向互补序列的核苷酸序列;优选地,所述的重组核酸分子所含的ires元件具有在高严格性杂交条件或非常高严格性杂交条件下,能够与seq id no: 62
‑ꢀ
seq id no: 65任一序列所示的核苷酸序列杂交的序列的反向互补序列;优选地,所述的重组核酸分子所含的ires元件具有本发明前述实施例所示的核苷酸序列具有至少10%,11%,12%,13%,14%,15%,16%,17%,18%,19%,20%,21%,22%,23%,24%,25%,26%,27%,28%,29%,30%,31%,32%,33%,34%,35%,36%,37%,38%,39%,40%,41%,42%,43%,44%,45%,46%,47%,48%,49%,50%,51%,52%,53%,54%,55%,56%,57%,58%,59%,60%,61%,62%,63%,64%,65%,66%,67%,68%,69%,70%,71%,72%,73%,74%,75%,76%,77%,78%,79%,80%,81%,82%,83%,84%,85%,86%,87%,88%,89%,90%,91%,92%,93%,94%,95%,96%,97%,98%,或99%的序列同一性的序列。
17.本发明实施例通过开发平台rna元件结构库还提供了一种重组表达载体,所述重组表达载体包含有本发明前述实施例所述的重组核酸分子。
18.本发明实施例通过开发平台rna元件结构库还提供了一种线性mrna,所述线性mrna包含本发明前述实施例所述的重组核酸分子;进一步的,所述线性mrna由本发明前述实施例所述的重组表达载体转录形成。
19.本发明实施例通过开发平台rna元件结构库还提供了一种环形mrna,所述环形mrna包含本发明前述实施例所述的重组核酸分子;进一步的,所述环形mrna,由本发明前述实施例所述的重组表达载体转录后环化形成;进一步的,所述环形mrna,由本发明前述实施例所述的线性mrna环化形成。
20.本发明实施例通过开发平台rna元件结构库还提供了一种重组宿主细胞,其包含有本发明前述实施例所述的重组核酸分子;进一步的,所述重组宿主细胞,包含有本发明前述实施例所述的线性mrna;进一步的,所述重组宿主细胞,包含有本发明前述实施例所述的环形mrna。
21.第三部分,开发平台cmc工艺库的技术方案。
22.本发明实施例通过开发平台cmc工艺库匹配了一种重组核酸分子的克级规模合成和分离纯化方法,所述重组核酸分子在克级规模合成分离后获得的纯度大于95%,所述重组核酸分子包括环形mrna及其线性mrna前驱物。
23.进一步地,从病毒感染所致癌症环形mrna疫苗开发平台的cmc工艺库根据成本核算、制备、分离纯化的难易匹配设计环形mrna的克级规模合成工艺路线;优先地,从病毒感染所致癌症的环形mrna疫苗开发平台的rna结构元件库根据环
化效率的大小模块化组装设计所需环形mrna的结构元件及其线性前驱物的结构元件:编码区为筛选出并经验证的病毒感染所致癌症的通用型抗原多肽氨基酸序列,能编码病毒感染所致癌症的通用型抗原多肽的ires元件,利于环化效率提高和增加翻译效率的外显子,间隔区,启动子,同源臂,内含子,以及质粒酶切位点;优先地,合成前面实施例所设计环形mrna线性前驱物的基因片段,并连接到病毒感染所致癌症环形mrna疫苗开发平台cmc工艺库匹配的经济实用的易工程化载体;优先地,将匹配前面实施例所设计环形mrna线性前驱物合成所需的穿刺菌在37摄氏度,适当转速下活化一定时间,随后取活化菌液在37摄氏度,适当转速下培养过夜。然后,抽提质粒;优先地,将前面实施例所设计环形mrna线性前驱物合成所需的一定量质粒,酶(1000units), 10
×
cutsmart buffer和若干体积无酶水混合,并将混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于体外转录;测定质粒酶切线性化后dna浓度;优先地,将将前面实施例达到纯化要求的一定量线性质粒模板,10
×
reaction buffer,浓度为20毫摩尔每升的atp,浓度为20毫摩尔每升的ctp,浓度为20毫摩尔每升的utp,浓度为20毫摩尔每升的gtp,t7 rna polymerase mix,及若干体积无核酸酶水构成的混合液在37摄氏度孵育一定时间,然后加入dnase i在37摄氏度消化一定时间。将所获得的混合物进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度;优先地,将前面实施例达到纯化要求的一定量线性mrna溶液,20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的混合溶液在55摄氏度加热一定时间。将所获得的混合物采用进行纯化。
24.进一步地,将前面实施例合成的环形mrna进行质检,确保所合成的环形mrna达到疫苗原材料的质控要求;优先地,对将前面实施例合成的环形mrna进行od值测定;优先地,对将前面实施例合成的环形mrna通过1%变性琼脂糖凝胶电泳进行大小进行鉴定;优先地,对将前面实施例合成的环形mrna采用高效液相色谱和质谱联用对环形mrna的纯度进行鉴定和杂质分析;进一步确定环形mrna的环化效率;优先地,对将前面实施例合成的环形mrna和其线性mrna前驱物进行pcr扩增,对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序,与前面实施例所设计的环形mrna和线性mrna序列进行比较,形成闭环质控过程评价。
25.进一步地,所述重组核酸分子分离纯化的方法为硅膜离心柱法;优先地,所述重组核酸分子硅膜离心柱法的分离纯化步骤为:第一步, 吸取重组核酸分子待分离纯化的溶液转移至硅膜柱中,离心;第二步,加入乙醇至硅膜柱中,离心,重复数次,直至保证硅膜柱内无液体;第三步,把双蒸水或甲酰胺加至硅膜柱,离心,用一干净离心管收集洗脱液;第四步,在洗脱液中加入一定体积的乙酸钠和异丙醇,在-20摄氏度沉淀过夜,
12000rpm 离心数分钟, 用75%乙醇洗沉淀,真空干燥得到较纯的环形mrna。
26.进一步地,所述重组核酸分子分离纯化的方法为磁分离方法;优先地,所述重组核酸分子磁分离方法的分离纯化步骤为:第一步,在depc处理过的eppendof管中,加入所合成的rna粗产物和无rnase的水至一定体积,在65摄氏度加热一定时间;第二步,加入适量生物素标记的探针和适量20
×
ssc于rna粗产物中轻轻混匀,室温放置一段时间;第三步,将sa-pmps轻晃开后,放入磁性分离架中,使sa-pmps集中于试管一侧, 小心去除上清,然后用一定体积0.5
×
ssc漂洗sa-pmps,用磁性分离架集中磁珠,去除上清,漂洗数次;第四步,将漂洗过的sa-pmps重新悬浮于一定体积0.5
×
ssc中,加入第二步中褪火的生物素标记的探针,轻轻混匀,室温放置一段时间,每隔数分钟轻轻混匀一次;第五步,用磁性分离架捕获磁珠,吸弃上清,用适量0.1
×
ssc漂洗sa-pmps,用磁性分离架集中磁珠,吸弃上清,漂洗数次;第六步,将漂洗过的sa-pmps重新悬浮于一定体积depc水中,用磁性分离架捕获磁珠,将洗脱的水相吸至一新管中。重复洗涤一次,吸取水相,两次水相合并; 第七步,在洗脱液中加入一定体积的乙酸钠和异丙醇,在-20摄氏度沉淀过夜,12000rpm 离心数分钟, 用75%乙醇洗沉淀,真空干燥得到较纯的环形mrna。
27.进一步地,所述重组核酸分子分离纯化的方法为亲和柱层析法;优先地,所述重组核酸分子亲和柱层析法的分离纯化步骤为:第一步,分别用depc处理层析柱, 0.1摩尔每升氢氧化钠处理纤维素,用1
×
上样液洗柱至ph《8.0,无菌水溶解重组核酸分子,65摄氏度温浴5分钟后,迅速冷却至室温,加等体积2
×
上样液,上样,分步收集;第二步,1倍体积1
×
上样液洗柱,分步收集。od260沉淀收集液,确定收集液的该值是否接近0,如该值仍很大,则上样液继续洗柱,直至该值接近0;第三步,2-3倍柱床体积的洗脱液,洗柱,分步收集。测定各收集管的od260,将有吸光值的各管合并;第四步,在洗脱液中加入适量体积的乙酸钠和异丙醇,在-20摄氏度沉淀过夜,12000rpm 离心数分钟, 用75%乙醇洗沉淀,真空干燥得到较纯的环形mrna。
28.进一步地,所述重组核酸分子分离纯化的方法为尺寸排阻柱方法;优先地,所述重组核酸分子尺寸排阻柱法的分离纯化步骤为:第一步,用洗脱缓冲液冲洗柱子;第二步,装入需分离纯化的重组核酸分子样品;第三步,向分离柱中添加pbs或缓冲液进行洗脱;第四步,收集空隙体积后的液体为较高纯度的重组核酸分子。
29.优选地,所述洗脱缓冲液,其为tris-edta缓冲液。
30.优选地,所述洗脱缓冲液,其为柠檬酸盐缓冲液。
31.优选地,所述洗脱缓冲液,其为乙酸盐缓冲液。
32.优选地,所述洗脱缓冲液,其为组氨酸盐缓冲液。
33.优选地,所述洗脱缓冲液,其为硼酸盐缓冲液。
34.优选地,所述洗脱缓冲液,其为甲酸盐缓冲液。
35.优选地,所述洗脱缓冲液,其为琥珀酸盐缓冲液。
36.优选地,所述洗脱缓冲液,其为碳酸盐缓冲液。
37.优选地,所述洗脱缓冲液,其为苹果酸盐缓冲液。
38.优选地,所述洗脱缓冲液,其为天冬氨酸盐缓冲液。
39.优选地,所述洗脱缓冲液,其为heppso缓冲液。
40.优选地,所述洗脱缓冲液,其为hepes缓冲液。
41.优选地,所述洗脱缓冲液,其为中性缓冲盐水。
42.进一步的,所述的重组核酸分子,通过磁分离方法和尺寸排阻柱相结合的方法进行分离纯化。
43.进一步的,所述的重组核酸分子,通过尺寸排阻柱和磷酸酶处理相结合的方法进行分离纯化。
44.进一步的,所述的重组核酸分子,通过磁分离方法和尺寸排阻柱相结合的方法进行分离纯化。
45.进一步的,所述的重组核酸分子,通过尺寸排阻柱和特殊场技术相结合的方法进行分离纯化。
46.进一步的,所述的重组核酸分子,通过尺寸排阻柱法,磷酸酶处理及rna酶r处理 相结合的方法进行分离纯化。
47.进一步的,所述的重组核酸分子,通过高效液相色谱及电导检测相结合的方法进行分离纯化。
48.进一步的,所述的重组核酸分子,通过磁分离方法,尺寸排阻柱,特殊场技术及rna酶r处理 相结合的方法进行分离纯化。
49.进一步的,所述的重组核酸分子,通过磁分离方法,特殊场技术及尺寸排阻柱相结合的方法进行分离纯化。
50.进一步的,所述的重组核酸分子,通过尺寸排阻柱法,特殊场技术,磷酸酶处理及rna酶r处理 相结合的方法进行分离纯化。
51.进一步的,所述的重组核酸分子,通过磁分离方法,特殊场技术,磷酸酶处理及rna酶r处理 相结合的方法进行分离纯化。
52.进一步的,所述的重组核酸分子,通过亲和柱层析法,特殊场技术,磷酸酶处理及rna酶r处理 相结合的方法进行分离纯化。
53.进一步的,所述的重组核酸分子,通过硅膜离心柱法,特殊场技术,磷酸酶处理及rna酶r处理 相结合的方法进行分离纯化。
54.优选地,其中所述特殊场技术的范围为:超声波技术或等离子体技术。
55.更优选地,其中所述超声波技术的超声波声强度小于100毫瓦每平方厘米。在一个实施方式中,其中所述超声波技术的声强度为98毫瓦每平方厘米。在某些实施方式中,其中所述超声波技术的声强度分别为95毫瓦每平方厘米,92毫瓦每平方厘米,90毫瓦每平方厘米,87毫瓦每平方厘米,84毫瓦每平方厘米,80毫瓦每平方厘米,76毫瓦每平方厘米,73毫瓦
每平方厘米,70毫瓦每平方厘米, 68毫瓦每平方厘米,65毫瓦每平方厘米,63毫瓦每平方厘米,60毫瓦每平方厘米,57毫瓦每平方厘米,55毫瓦每平方厘米,52毫瓦每平方厘米,48毫瓦每平方厘米,44毫瓦每平方厘米,40毫瓦每平方厘米,36毫瓦每平方厘米,32毫瓦每平方厘米,28毫瓦每平方厘米,24毫瓦每平方厘米,20毫瓦每平方厘米,16毫瓦每平方厘米。在某些实施方式中,其中所述超声波技术的声强度分别为:12毫瓦每平方厘米,10毫瓦每平方厘米,8毫瓦每平方厘米,6毫瓦每平方厘米,4毫瓦每平方厘米,2毫瓦每平方厘米,1.8毫瓦每平方厘米,1.5毫瓦每平方厘米,1.2毫瓦每平方厘米,1.1毫瓦每平方厘米,1毫瓦每平方厘米,0.9毫瓦每平方厘米,0.8毫瓦每平方厘米,0.7毫瓦每平方厘米,0.6毫瓦每平方厘米,0.5毫瓦每平方厘米,0.4毫瓦每平方厘米,0.3毫瓦每平方厘米,0.2毫瓦每平方厘米,0.1毫瓦每平方厘米,0.095毫瓦每平方厘米,0.09毫瓦每平方厘米,0.085毫瓦每平方厘米,0.08毫瓦每平方厘米,0.075毫瓦每平方厘米,0.07毫瓦每平方厘米,0.065毫瓦每平方厘米, 0.06毫瓦每平方厘米,0.055毫瓦每平方厘米。在某些实施方式中,其中所述超声波技术的声强度分别为:0.052毫瓦每平方厘米,0.05毫瓦每平方厘米,0.048毫瓦每平方厘米,0.045毫瓦每平方厘米,0.042毫瓦每平方厘米,0.04毫瓦每平方厘米,0.038毫瓦每平方厘米,0.035毫瓦每平方厘米,0.033毫瓦每平方厘米,0.031毫瓦每平方厘米,0.028毫瓦每平方厘米,0.026毫瓦每平方厘米,0.024毫瓦每平方厘米,0.022毫瓦每平方厘米,0.02毫瓦每平方厘米,0.018毫瓦每平方厘米,0.016毫瓦每平方厘米,0.014毫瓦每平方厘米,0.012毫瓦每平方厘米,10微瓦每平方厘米,9微瓦每平方厘米,8微瓦每平方厘米,7微瓦每平方厘米,6微瓦每平方厘米,5微瓦每平方厘米,4微瓦每平方厘米,3微瓦每平方厘米,2微瓦每平方厘米,1微瓦每平方厘米。
56.更优选地,其中所述等离子体技术的等离子体产生方式为: 气相方式或液相方式。
57.更优选地,其中气相等离子体的辐射能量小于1.2焦每平方厘米。在一个实施方式中,其中所述气相等离子体产生的辐射能量为1.1焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为1.08焦每平方厘米,1.05焦每平方厘米,1.02焦每平方厘米,1焦每平方厘米,0.98焦每平方厘米,0.95焦每平方厘米,0.9焦每平方厘米,0.88焦每平方厘米,0.85焦每平方厘米,0.83焦每平方厘米,0.8焦每平方厘米,0.78焦每平方厘米,0.75焦每平方厘米,0.72焦每平方厘米,0.7焦每平方厘米,0.68焦每平方厘米,0.65焦每平方厘米,0.62焦每平方厘米,0.6焦每平方厘米,0.57焦每平方厘米,0.55焦每平方厘米,0.52焦每平方厘米,0.5焦每平方厘米,0.47焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:0.45焦每平方厘米,0.43焦每平方厘米,0.4焦每平方厘米,0.38焦每平方厘米,0.35焦每平方厘米,0.33焦每平方厘米,0.3焦每平方厘米,0.28焦每平方厘米,0.25焦每平方厘米,0.22焦每平方厘米,0.2焦每平方厘米,0.18焦每平方厘米,0.15焦每平方厘米,0.12焦每平方厘米,0.1焦每平方厘米,0.08焦每平方厘米,0.06焦每平方厘米,0.04焦每平方厘米,0.03焦每平方厘米,0.027焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:0.025焦每平方厘米,0.023焦每平方厘米,0.02焦每平方厘米,0.018焦每平方厘米,0.015焦每平方厘米,0.013焦每平方厘米,0.01焦每平方厘米,0.008焦每平方厘米,0.007焦每平方厘米,0.006焦每平方厘米,0.005焦每平方厘米,0.004焦每平方厘米,0.003焦每平方厘米,0.002焦每平方厘米,
0.001焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:950微焦每平方厘米,925微焦每平方厘米,900微焦每平方厘米,875微焦每平方厘米,850微焦每平方厘米,825微焦每平方厘米,800微焦每平方厘米,775微焦每平方厘米,750微焦每平方厘米,725微焦每平方厘米,700焦每平方厘米,675微焦每平方厘米,650微焦每平方厘米,625微焦每平方厘米,600微焦每平方厘米,575微焦每平方厘米,550微焦每平方厘米,525微焦每平方厘米,500微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:475微焦每平方厘米,450微焦每平方厘米,425焦每平方厘米,400微焦每平方厘米, 375微焦每平方厘米,350微焦每平方厘米,325微焦每平方厘米,300微焦每平方厘米,275微焦每平方厘米,250微焦每平方厘米,225微焦每平方厘米,200微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:190微焦每平方厘米,180微焦每平方厘米,170微焦每平方厘米,160微焦每平方厘米,150微焦每平方厘米,140微焦每平方厘米,130微焦每平方厘米,120微焦每平方厘米,110微焦每平方厘米,100微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:98微焦每平方厘米,95微焦每平方厘米,93微焦每平方厘米,92微焦每平方厘米,90微焦每平方厘米,87微焦每平方厘米,85微焦每平方厘米,82微焦每平方厘米,80微焦每平方厘米,78微焦每平方厘米,75微焦每平方厘米,72微焦每平方厘米,70微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:68微焦每平方厘米,65微焦每平方厘米,63微焦每平方厘米,60微焦每平方厘米,57微焦每平方厘米,54微焦每平方厘米,50微焦每平方厘米,47微焦每平方厘米,43微焦每平方厘米,40微焦每平方厘米,36微焦每平方厘米,32微焦每平方厘米,28微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:25微焦每平方厘米,23微焦每平方厘米,20微焦每平方厘米,17微焦每平方厘米,14微焦每平方厘米,10微焦每平方厘米,9微焦每平方厘米,7微焦每平方厘米,6微焦每平方厘米,5微焦每平方厘米,4微焦每平方厘米,3微焦每平方厘米,2微焦每平方厘米,1微焦每平方厘米。
58.更优选地,其中液相相等离子体产生的辐射能量小于1焦每平方厘米。在一个实施方式中,其中所述气相等离子体产生的辐射能量为0.99焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为0.98焦每平方厘米,0.95焦每平方厘米,0.9焦每平方厘米,0.88焦每平方厘米,0.85焦每平方厘米,0.83焦每平方厘米,0.8焦每平方厘米,0.78焦每平方厘米,0.75焦每平方厘米,0.72焦每平方厘米,0.7焦每平方厘米,0.68焦每平方厘米,0.65焦每平方厘米,0.62焦每平方厘米,0.6焦每平方厘米,0.57焦每平方厘米,0.55焦每平方厘米,0.52焦每平方厘米,0.5焦每平方厘米,0.47焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:0.45焦每平方厘米,0.43焦每平方厘米,0.4焦每平方厘米,0.38焦每平方厘米,0.35焦每平方厘米,0.33焦每平方厘米,0.3焦每平方厘米,0.28焦每平方厘米,0.25焦每平方厘米,0.22焦每平方厘米,0.2焦每平方厘米,0.18焦每平方厘米,0.15焦每平方厘米,0.12焦每平方厘米,0.1焦每平方厘米,0.08焦每平方厘米,0.06焦每平方厘米,0.04焦每平方厘米,0.03焦每平方厘米,0.027焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:0.025焦每平方厘米,0.023焦每平方厘米,0.02焦每平方厘米,0.018焦每平方厘米,0.015焦每平方厘米,0.013焦每平方厘米,0.01焦每平方厘米,0.008焦每平方厘米,0.007焦每平方厘米,
0.006焦每平方厘米,0.005焦每平方厘米,0.004焦每平方厘米,0.003焦每平方厘米,0.002焦每平方厘米,0.001焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:950微焦每平方厘米,925微焦每平方厘米,900微焦每平方厘米,875微焦每平方厘米,850微焦每平方厘米,825微焦每平方厘米,800微焦每平方厘米,775微焦每平方厘米,750微焦每平方厘米,725微焦每平方厘米,700焦每平方厘米,675微焦每平方厘米,650微焦每平方厘米,625微焦每平方厘米,600微焦每平方厘米,575微焦每平方厘米,550微焦每平方厘米,525微焦每平方厘米,500微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:475微焦每平方厘米,450微焦每平方厘米,425焦每平方厘米,400微焦每平方厘米, 375微焦每平方厘米,350微焦每平方厘米,325微焦每平方厘米,300微焦每平方厘米,275微焦每平方厘米,250微焦每平方厘米,225微焦每平方厘米,200微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:190微焦每平方厘米,180微焦每平方厘米,170微焦每平方厘米,160微焦每平方厘米,150微焦每平方厘米,140微焦每平方厘米,130微焦每平方厘米,120微焦每平方厘米,110微焦每平方厘米,100微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:98微焦每平方厘米,95微焦每平方厘米,93微焦每平方厘米,92微焦每平方厘米,90微焦每平方厘米,87微焦每平方厘米,85微焦每平方厘米,82微焦每平方厘米,80微焦每平方厘米,78微焦每平方厘米,75微焦每平方厘米,72微焦每平方厘米,70微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:68微焦每平方厘米,65微焦每平方厘米,63微焦每平方厘米,60微焦每平方厘米,57微焦每平方厘米,54微焦每平方厘米,50微焦每平方厘米,47微焦每平方厘米,43微焦每平方厘米,40微焦每平方厘米,36微焦每平方厘米,32微焦每平方厘米,28微焦每平方厘米。在某些实施方式中,其中所述气相等离子体产生的辐射能量分别为:25微焦每平方厘米,23微焦每平方厘米,20微焦每平方厘米,17微焦每平方厘米,14微焦每平方厘米,10微焦每平方厘米,9微焦每平方厘米,7微焦每平方厘米,6微焦每平方厘米,5微焦每平方厘米,4微焦每平方厘米,3微焦每平方厘米,2微焦每平方厘米,1微焦每平方厘米。
59.进一步地,其中所述特殊场技术中包含有气体通入过程;优选地,所述气体为氢气;优选地,所述气体为氮气;优选地,所述气体为空气;优选地,所述气体为氧气;优选地,所述气体为氩气;优选地,所述气体为氦气;优选地,所述气体为氨气;优选地,所述气体为上述实施例所述一种气体跟另一种气体的合理混合物。
60.进一步地,其中所述特殊场技术中包含有搅拌过程;优选地,所述的搅拌其方式为:机械搅拌和磁力搅拌,优选为机械搅拌。
61.更优选地,其中所述搅拌速率为:采用超声波技术搅拌的速率为10-500rpm;采用气相等离子体技术搅拌的速率为1-40rpm;采用液相等离子体技术搅拌的速率为1-300rpm。在一个实施方式中,其中所述超声波技术搅拌的速率为12rpm。在某些实施方式中,其中所
述超声波技术搅拌的速率分别为15rpm,25rpm,35rpm,50rpm,65rpm,80rpm,100rpm,125rpm,140rpm,160rpm,180rpm。在某些实施方式中,其中所述超声波技术搅拌的速率为分别为:200rpm,230rpm,250rpm,300rpm,350rpm,450rpm。在一个实施方式中,其中所述气相等离子体技术搅拌的速率为2rpm。在某些实施方式中,其中所述气相等离子体技术搅拌的速率分别为3rpm,4rpm,5rpm,6rpm,7rpm,8rpm,10rpm,12rpm,16rpm,20rpm。在某些实施方式中,其中所述气相等离子体技术搅拌的速率分别为23rpm,25rpm,30rpm,32rpm,35rpm,38rpm。在一个实施方式中,其中所述液相等离子体技术搅拌的速率为2rpm。在某些实施方式中,其中所述液相等离子体技术搅拌的速率分别为5rpm,7rpm,10rpm,12rpm,15rpm,18rpm,20rpm,23rpm,25pm,28rpm,30rpm。在某些实施方式中,其中所述液相等离子体技术搅拌的速率分别为32rpm,35rpm,40rpm,42rpm,45rpm,48rpm,50rpm,53rpm,55pm,58rpm,60rpm。在某些实施方式中,其中所述液相等离子体技术搅拌的速率为分别为:63rpm,65rpm,68rpm,70rpm,75rpm,80rpm,90rpm,100rpm 。在某些实施方式中,其中所述液相等离子体技术搅拌的速率为分别为:110rpm,120rpm,130rpm,150rpm,175rpm,200rpm,225rpm,250rpm,275rpm。
62.更优选地,其中所述处理的一段时间为:采用超声波技术处理的时间为1-120分钟;采用气相等离子体技术处理的时间5秒-60分钟;采用液相等离子体技术处理的时间1秒-60分钟。在一个实施方式中,其中所述超声波技术处理的时间为2分钟。在某些实施方式中,其中所述超声波技术处理的时间分别为3分钟,4分钟,5分钟,6分钟,7分钟,8分钟,9分钟,10分钟,12分钟,15分钟,18分钟。在某些实施方式中,其中所述超声波技术处理的时间分别为20分钟,22分钟,25分钟,27分钟,30分钟,33分钟,35分钟,40分钟,45分钟,47分钟。在某些实施方式中,其中所述超声波技术处理的时间分别为50分钟,55分钟,57分钟,60分钟,63分钟,67分钟,70分钟,75分钟,80分钟,85分钟。在某些实施方式中,其中所述超声波技术处理的时间分别为90分钟,95分钟,100分钟,105分钟,110分钟,115分钟,118分钟。在一个实施方式中,其中所述采用气相等离子体技术处理的时间为7秒钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为9秒钟,11秒钟,13秒钟,15秒钟,20秒钟,25秒钟,30秒钟,35秒钟,40秒钟,45秒钟,50秒钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为55秒钟,60秒钟,1.5分钟,2分钟,3分钟,4分钟,5分钟,6分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为7分钟,8分钟,9分钟,10分钟,12分钟,15分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为17分钟,18分钟,20分钟,21分钟,22分钟,25分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为27分钟,30分钟,32分钟,35分钟,37分钟,40分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为43分钟,45分钟,47分钟,50分钟,52分钟,55分钟,58分钟。在一个实施方式中,其中所述采用液相等离子体技术处理的时间为2秒钟。在某些实施方式中,其中所述液相等离子体技术处理的时间分别为3秒钟,4秒钟,5秒钟,6秒钟,7秒钟,8秒钟,9秒钟,10秒。在某些实施方式中,其中所述液相等离子体技术处理的时间分别为11秒钟,13秒钟,15秒钟,20秒钟,25秒钟,30秒钟,35秒钟,40秒钟,45秒钟,50秒钟。在某些实施方式中,其中所述液相等离子体技术处理的时间分别为55秒钟,60秒钟,1.5分钟,2分钟,3分钟,4分钟,5分钟,6分钟。在某些实施方式中,其中所述液相等离子体技术处理的时间分别为7分钟,8分钟,9分钟,10分钟,12分钟,15分钟。在某些实施方式中,其
中所述液相等离子体技术处理的时间分别为17分钟,18分钟,20分钟,21分钟,22分钟,25分钟。在某些实施方式中,其中所述液相等离子体技术处理的时间分别为27分钟,30分钟,32分钟,35分钟,37分钟,40分钟。在某些实施方式中,其中所述液相等离子体技术处理的时间分别为43分钟,45分钟,47分钟,50分钟,52分钟,55分钟,58分钟。
63.进一步地,所述特殊场技术的液相介质采用pbs缓冲液。
64.进一步地,所述特殊场技术的液相介质采用约4.5-6.5范围内的含盐水性缓冲液。
65.更优选地,所述含盐水性缓冲液,其为tris-edta缓冲液。
66.更优选地,所述含盐水性缓冲液,其为柠檬酸盐缓冲液。
67.更优选地,所述含盐水性缓冲液,其为乙酸盐缓冲液。
68.更优选地,所述含盐水性缓冲液,其为组氨酸盐缓冲液。
69.更优选地,所述含盐水性缓冲液,其为硼酸盐缓冲液。
70.更优选地,所述含盐水性缓冲液,其为甲酸盐缓冲液。
71.更优选地,所述含盐水性缓冲液,其为琥珀酸盐缓冲液。
72.更优选地,所述含盐水性缓冲液,其为碳酸盐缓冲液。
73.更优选地,所述含盐水性缓冲液,其为苹果酸盐缓冲液。
74.更优选地,所述含盐水性缓冲液,其为天冬氨酸盐缓冲液。
75.更优选地,所述含盐水性缓冲液,其为heppso缓冲液。
76.更优选地,所述含盐水性缓冲液,其为hepes缓冲液。
77.更优选地,所述含盐水性缓冲液,其为中性缓冲盐水。
78.更优选地,所述含盐水性缓冲液ph,其为4-7范围。在一些实施例中,ph为3.5。在一些实施例中,ph分别为4.5,5,5.5,6, 6.5,7.5。
79.更优选地,所述含盐水性缓冲液,为上述实施例所述含盐水性缓冲液中的一种跟另一种或多种之间的合理含盐水性缓冲液混合物。
80.第四部分,开发平台递送系统库的技术方案。
81.本发明实施例通过开发平台递送系统库匹配了一种环形mrna分子疫苗的递送系统。
82.其中,所述的环形mrna分子疫苗,其为用于癌症治疗的疫苗。
83.进一步地,所述的环形mrna分子疫苗,其包括作为活性成分的疫苗佐剂。
84.更进一步地,所述的环形mrna分子疫苗,其包括病毒感染所致癌症的肿瘤新生抗原多肽。
85.更进一步地,所述的环形mrna分子疫苗,其包括编码病毒感染所致癌症的肿瘤新生抗原多肽的重组核酸分子。
86.进一步地,所述递送系统具有病毒感染所致癌症细胞的递送靶向性。
87.进一步的,所述的递送系统,其包括时释性、缓释性和持续释放的递送系统,使得重组核酸分子药物的递送发生在待治疗的部位敏化之前,并且有足够的时间引起敏化。
88.更进一步地,所述的释放的递送系统,其包括基于聚合物的系统。
89.优选地,所述聚合物为聚丙交酯-乙交酯、共聚草酸酯、聚酯酰胺、聚原酸酯、聚己内酯、聚羟基丁酸和聚酸酐中的一种或二种或多种混合物。
90.更进一步地,所述的释放的递送系统,其包括基于非聚合物的系统。
91.优选地,所述的非聚合物,其是脂质。
92.更优选地,所述脂质,其是固醇,如胆固醇、胆固醇酯。
93.更优选地,所述脂质,其是脂肪酸或中性脂肪,如单甘油二酯和甘油三酯。
94.更进一步地,所述的释放的递送系统,其包括进行了表面合理改性的递送系统。
95.优选地,所述递送系统的表面合理改性,其包括递送系统的表面荷电调控;优选地,所述递送系统的表面合理改性,其包括递送系统的粒径调控;优选地,所述递送系统的表面合理改性,其包括采用含声能量的技术手段;更优选地,所述含声能量的技术手段为超声波技术手段。
96.优选地,所述递送系统的表面合理改性,其包括采用含辐射能量的技术手段;更优选地,所述含辐射能量的技术手段为等离子体技术手段,所述等离子体技术手段为气相等离子体技术手段或液相相等离子体技术手段。
97.优选地,所述递送系统的表面合理改性,其包括采用一些能辅助在声能量或辐射能量存在情况下协助递送系统合理改性的自我牺牲型物质,其包括采用黑磷为自我牺牲型辅助物质。
98.更优选地,所述递送系统的表面合理改性,其特征在于,包括如下步骤:第一步,将黑磷和非聚合物按照一定比例进行混合,得到混合物a;第二步,将富氧水溶液加入或将含氧的气体通入第一步中的混合物a中,然后在一定温度下结合特殊场技术进行搅拌处理一段时间,得到混合液a;第三步,将异双官能化聚乙二醇加入第二步获得的混合液a,然后在一定温度下进行超声搅拌处理,得到混合液b;第四步,将核酸适配体加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna进行混合。
99.更优选地,其中所述黑磷的产品形式为黑磷量子点、黑磷粉末、黑磷晶体、黑磷纳米片、石墨烯状黑磷中的一种或多种之间的合理组合物;优选石墨烯状黑磷。
100.更优选地,其中所述脂质产品的形式选自包含以下的列表:peg-dmg、peg-c-dmg、peg-c-dma、peg-dspe、peg-pe、peg 修饰的神经酰胺、peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、peg-s-dmg、daa、 peg-c-domg,galnac-peg-dsg,dope、dspc、dppc、dopc、dppg、popc、pope、dppe、dmpe、dspe、dlin-mc3-dma、dlindma、dodma、dlin-mc2-mpz、dlin-kc2-dma、dotap、dc-chol, otma、dotap、反式dope,popg,磷脂酰丝氨酸、二油酰基磷脂酰丝氨酸、磷脂酰肌醇、磷脂酸、磷脂酰甘油、dopg、二豆蔻酰磷脂酰甘油的一种及它们之间的混合物。
101.更优选地,在一个实施方式中,所述黑磷和脂质体的摩尔比例为:1-20:40-120。在某些实施方式中,所述黑磷和脂质体的摩尔比例为:4-8.6:61-81。在另一实施方式中,所述黑磷和脂质体的摩尔比例分别为:5:63, 5:65, 5:67, 5:69, 5:71, 5:73, 5:75, 5:77, 5:79, 5:80中的一种。在某些实施方式中,所述黑磷和脂质体的摩尔比例分别为6:62、6:64、6:66、6:68、6:70、6:72、6:74、6:76、6:78、6:80中的一种。在某些实施方式中,所述黑磷和脂质体的摩尔比例为7:63, 7:65, 7:67, 7:69, 7:71, 7:73, 7:75, 7:77, 7:79, 7:80中的一种。在某些实施方式中,所述黑磷和脂质体的摩尔比例为8.2:63, 8.2:65, 8.2:
67, 8.2:69, 8.2:71, 8.2:73, 8.2:75, 8.2:77, 8.2:79, 8.2:80中的一种。
102.更优选地,在一个实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dotap和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dppc,dotap和dopc的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dppg,dotap和popc的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为pope,dotap和dmpe的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspe,dotap和sope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为为反式dope,dotap和popg的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dli-mc3-dma和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dppc,dlindma和dopc的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dppg,dodma和popc的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为pope,dlin-mc2-mpz和dmpe的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspe,dlin-kc2-dma和sope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为为反式dope,dc-chol和popg的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为为反式dope,otma和popg的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,dotap和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,dotap和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,dotap和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,dotap和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,dotap和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,dotap和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,dotap和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,dotap和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,dotap和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popg,dotap和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,dotap,otma和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,dotap,otma和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,dotap,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,dotap,otma和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,dotap,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,dotap,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,dotap,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,dotap,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,dotap,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popg,dotap,otma和dope的混合。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,dodma,otma和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,dodma,otma和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,dodma,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,dodma,otma和dope的混合物。在某些实施方式中,与黑磷混
合的脂质体产品为dspc,pope,dodma,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,dodma,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,dodma,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,dodma,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,dodma,otma和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popg,dodma,otma和dope的混合。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popg,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope的混合。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,磷脂酰肌醇、磷脂酸和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,磷脂酰肌醇、磷脂酸和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popg,磷脂酰肌醇、磷脂酸和dope的混合。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,磷脂酰甘油、dopg和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,磷脂酰甘油、dopg和dope的混合物。在某些实施方
式中,所述与黑磷混合的脂质体产品为dspc,sope,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popg,磷脂酰甘油、dopg和dope的混合。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,二豆蔻酰磷脂酰甘油,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg-dmg,dppc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg-dmg,dopc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,dppg,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,popc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为peg-dmg,pope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,dmpe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,ospe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,sope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,反式dope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,二豆蔻酰磷脂酰甘油,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与
domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope的混合物。在另一实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg、二豆蔻酰磷脂酰甘油和dope的混合。
103.更优选地,在一个实施方式中,其中所述与黑磷混合的脂质体产品dspc,dotap和dope之间的摩尔比例为1:27-37:33-43。在一个实施方式中,其中所述与黑磷混合的脂质体产品dspc,dotap和dope之间的摩尔比例为1:30:37。在一个实施方式中,其中所述与黑磷混合的脂质体产品dspc,dotap和dope之间的摩尔比例为1:33:37。在一个实施方式中,其中所述与黑磷混合的脂质体产品dspc,dotap和dope之间的摩尔比例为1:35:37。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dotap和dope之间的摩尔比例为1:35:35。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dotap和dope之间的摩尔比例为1:35:39。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dotap和dope之间的摩尔比例为1:35:41。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dotap和dopc之间的摩尔比例为1:20-30:32-40。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dotap和dopc之间的摩尔比例为1:25:36。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dotap和dopc之间的摩尔比例为1:22:36。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dotap和dopc之间的摩尔比例为1:28:36。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dotap和dopc之间的摩尔比例为1:25:34。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dotap和dopc之间的摩尔比例为1:25:36。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dotap和dopc之间的摩尔比例为1:25:38。在另一实施方式中,所述与黑磷混合的脂质体产品dppg,dotap和popc之间的摩尔比例为1:25-35:35-50。在某些实施方式中,所述与黑磷混合的脂质体产品pope,dotap和dmpe之间的摩尔比例为1:22-33:30-38。在某些实施方式中,与黑磷混合的脂质体产品dspe,dotap和sope之间的摩尔比例为1:21-25:35-50。在某些实施方式中,所述与黑磷混合的脂质体产品为反式dope,dotap和popg之间的摩尔比例为1:18-27:30-40。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dli-mc3-dma和dope之间的摩尔比例为1:19-26:32-42。在某些实施方式中,其中所述与黑磷混合的脂质体产品dppc,dlindma和dopc之间的摩尔比例为1:21-28:35-45。在另一实施方式中,所述与黑磷混合的脂质体产品为dppg,dodma和popc之间的摩尔比例为1:23-32:30-38。在某些实施方式中,所述与黑磷混合的脂质体产品pope,dlin-mc2-mpz和dmpe之间的摩尔比例为1:20-27:33-43。在某些实
施方式中,与黑磷混合的脂质体产品dspe,dlin-kc2-dma和sope之间的摩尔比例为1:16-21:30-40。在某些实施方式中,所述与黑磷混合的脂质体产品为为反式dope,dc-chol和popg之间的摩尔比例为1:18-27:32-39。在某些实施方式中,所述与黑磷混合的脂质体产品为为反式dope,otma和popg之间的摩尔比例为1:15-22:28-35。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dppc,dotap和dope之间的摩尔比例为1:5-12:27-37:33-43。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,dotap和dope之间的摩尔比例为1:2-8:27-37:33-43。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,dotap和dope之间的摩尔比例为1:1-10:27-37:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,dotap和dope之间的摩尔比例为1:5-15:27-37:33-43。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,dotap和dope之间的摩尔比例为1:4-16:27-37:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,dotap和dope之间的摩尔比例为1:3-13:27-37:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,dotap和dope之间的摩尔比例为1:2-8:27-37:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,dotap和dope之间的摩尔比例为1:10-18:27-37:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,dotap和dope之间的摩尔比例为1:3-9:27-37:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popg,dotap和dope之间的摩尔比例为1:2-10:27-37:33-43。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,dotap,otma和dope之间的摩尔比例为1:3-9:17-24:10-25:33-43。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,dotap,otma和dope之间的摩尔比例为1:5-15:17-24:10-25:33-43。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,dotap,otma和dope之间的摩尔比例为1:1-7:17-24:10-25:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,dotap,otma和dope之间的摩尔比例为1:3-13:17-24:10-25:33-43。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,dotap,otma和dope之间的摩尔比例为1:2-11:17-24:10-25:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,dotap,otma和dope之间的摩尔比例为1:6-14:17-24:10-25:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,dotap,otma和dope之间的摩尔比例为1:1-5:17-24:10-25:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,dotap,otma和dope之间的摩尔比例为1:2-9:17-24:10-25:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,dotap,otma和dope之间的摩尔比例为1:2-9:17-24:10-25:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,popg,dotap,otma和dope之间的摩尔比例为1:3-11:12-21:9-20:24-35。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dppc,dodma,otma和dope之间的摩尔比例为1:4-9:8-18:7-17:27-37。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dopc,dodma,otma和dope之间的摩尔比例为1:7-13:7-21:11-23:24-39。在另一实施方式中,所述与黑磷混合的脂质体产品dspc,dppg,dodma,otma和dope之间的摩尔比例为1:5-9:11-18:14-29:27-40。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,popc,dodma,otma和dope之间的摩尔比例为1:2-7:5-15:10-20:25-45。在某些实施方式中,与黑磷混合的脂质体产品dspc,pope,dodma,otma和dope之间的摩尔比例为1:5-15:12-22:12-32:30-40。在某些实施
方式中,所述与黑磷混合的脂质体产品dspc,dmpe,dodma,otma和dope之间的摩尔比例为1:2-12:6-15:10-22:26-39。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,ospe,dodma,otma和dope之间的摩尔比例为1:5-15:9-18:15-27:30-50。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,sope,dodma,otma和dope之间的摩尔比例为1:5-15:9-18:15-27:30-50。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,反式dope,dodma,otma和dope之间的摩尔比例为1:2-6:5-20:13-25:28-43。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,popg,dodma,otma和dope之间的摩尔比例为1:3-10:4-14:10-20:25-35。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dppc,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:2-9:6-10:13-23:30-40。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dopc,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:1-5:3-15:18-28:28-43。在另一实施方式中,所述与黑磷混合的脂质体产品dspc,dppg,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:2-9:12-25:20-33:34-47。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,popc,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:1-7:10-20:22-32:33-45。在某些实施方式中,与黑磷混合的脂质体产品dspc,pope,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:5-12:8-14:18-26:30-38。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,dmpe,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:2-8:6-12:15-20:26-33。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:4-14:8-18:19-29:30-39。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:3-13:13-23:14-24:28-36。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,反式dope,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:5-10:11-20:24-33:30-40。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,popg,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope之间的摩尔比例为1:8-12:13-25:20-30:35-43。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dppc,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:5-15:10-20:17-28:30-38。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:2-11:9-17:14-24:28-36。在另一实施方式中,所述与黑磷混合的脂质体产品dspc,dppg,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:4-14:5-15:16-23:32-41。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,popc,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:2-10:7-23:12-20:27-35。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:5-15:17-30:16-32:30-43。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,dmpe,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:8-12:13-23:21-30:32-40。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,ospe,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:10-21:8-18:15-28:30-39。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:5-15:10-23:18-32:35-45。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:2-13:8-18:12-21:28-38。在某些实施方式中,所述
与黑磷混合的脂质体产品dspc,popg,磷脂酰肌醇、磷脂酸和dope之间的摩尔比例为1:4-12:10-21:17-27:30-44。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dppc,磷脂酰甘油、dopg和dope之间的摩尔比例为1:8-16:13-20:15-35:28-38。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,磷脂酰甘油、dopg和dope之间的摩尔比例为1:4-12:10-18:13-24:20-35。在另一实施方式中,所述与黑磷混合的脂质体产品dspc,dppg,磷脂酰甘油、dopg和dope之间的摩尔比例为1:6-16:13-23:18-29:30-46。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,磷脂酰甘油、dopg和dope之间的摩尔比例为1:4-12:13-20:15-25:26-39。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,磷脂酰甘油、dopg和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,磷脂酰甘油、dopg和dope之间的摩尔比例为1:2-13:10-22:21-34:32-44。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,磷脂酰甘油、dopg和dope之间的摩尔比例为1:4-10:8-18:11-21:22-35。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,sope,磷脂酰甘油、dopg和dope之间的摩尔比例为1:5-15:6-22:13-19:27-39。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,反式dope,磷脂酰甘油、dopg和dope之间的摩尔比例为1:3-9:5-15:20-30:25-45。在某些实施方式中,所述与黑磷混合的脂质体产品dspc,popg,磷脂酰甘油、dopg和dope之间的摩尔比例为1:5-17:5-18:17-33:30-41。在某些实施方式中,其中所述与黑磷混合的脂质体产品dspc,dppc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-13:8-23:23-37:20-35。在某些实施方式中,其中所述与黑磷混合的脂质体产品为dspc,dopc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:9-20:25-30:30-40。在另一实施方式中,所述与黑磷混合的脂质体产品为dspc,dppg,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-14:10-27:17-28:32-42。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,popc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-17:12-25:20-33:30-42。在某些实施方式中,与黑磷混合的脂质体产品为dspc,pope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-10:15-30:23-30:28-38。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,dmpe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope的混合物。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,ospe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-18:14-24:21-29:29-46。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,sope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-14:15-25:20-27:30-36。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,反式dope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:6-18:12-28:24-34:33-46。在某些实施方式中,所述与黑磷混合的脂质体产品为dspc,二豆蔻酰磷脂酰甘油,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:7-21:10-19:20-30:30-36。在某些实施方式中,其中所述与黑磷混合的脂质体产品peg-dmg,dppc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:6-16:8-17:22-34:33-47。在某些实施方式中,其中所述与黑磷混合的脂质体产品peg-dmg,dopc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:7-12:11-21:20-30:30-40。在另一实施方式中,所述与黑磷混合的脂质体产品peg-dmg,dppg,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-14:10-17:16-26:22-34。在某些实施方式中,所述与黑磷混合的脂质体产品
peg-dmg,popc,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:13-23:14-24:29-39。在某些实施方式中,与黑磷混合的脂质体产品为peg-dmg,pope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-10:14-21:12-22:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,dmpe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:16-26:17-27:30-45。在某些实施方式中,所述与黑磷混合的脂质体产品peg-dmg,ospe,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:2-12:10-20:13-23:27-37。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,sope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-9:13-23:18-29:30-39。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,反式dope,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:6-12:10-20:16-26:32-42。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dmg,二豆蔻酰磷脂酰甘油,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-14:8-18:10-27:25-35。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:2-10:5-15:13-26:29-38。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-13:10-24:21-29:26-36。在另一实施方式中,所述与黑磷混合的脂质体产品peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:8-18:12-25:25-30:32-42。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-14:11-19:20-28:30-38。在某些实施方式中,与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-13:10-15:24-33:35-45。在某些实施方式中,所述与黑磷混合的脂质体产品peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:2-12:11-19:21-35:30-42。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-10:15-22:25-37:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-14:12-19:20-30:31-41。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-13:10-22:25-35:35-44。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-c-dmg、peg-c-dma,磷脂酰甘油、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:16-25:24-34:30-39。在某些实施方式中,其中所述与黑磷混合的脂质体产品peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:2-12:18-28:28-38:30-40。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:16-26:29-39:32-45。在另一实施方式中,所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-14:10-20:26-36:37-47。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:8-18:14-24:28-38:28-38。在某些实施方式中,与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆
蔻酰磷脂酰甘油和dope之间的摩尔比例为1:6-12:13-18:20-30:33-43。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:1-6:7-15:21-31:32-42。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-9:13-25:25-35:36-40。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-8:10-16:20-30:30-38。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-10:12-24:26-36:33-42。在某些实施方式中,所述与黑磷混合的脂质体产品为peg-dspe、peg-pe、peg 修饰的神经酰胺、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-13:8-18:20-30:31-41。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:2-12:6-16:25-35:35-40。在某些实施方式中,其中所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-9:10-15:20-30:32-42。在另一实施方式中,所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:12-22:23-28:27-37。在某些实施方式中,所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-12:15-28:27-33:35-42。在某些实施方式中,与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-13:11-24:25-35:30-40。在某些实施方式中,所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:13-23:23-33:31-41。在某些实施方式中,所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-10:11-17:26-36:29-39。在某些实施方式中,所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:6-16:12-17:26-36:29-39。在某些实施方式中,所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-13:10-20:20-30:27-37。在某些实施方式中,所述与黑磷混合的脂质体产品为peg修饰的二烷基胺、peg修饰的二酰基甘油、peg-dpg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:4-10:12-18:22-28:26-36。 在某些实施方式中,其中所述与黑磷混合的脂质体产品daa、 peg-c-domg,galnac-peg-dsg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:8-18:16-24:25-35:33-43。在某些实施方式中,其中所述与黑磷混合的脂质体产品daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-13:10-20:21-31:28-37。在另一实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:2-12:8-18:15-25:32-35。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:3-13:6-16:12-24:30-38。在某些实施方式中,与黑磷混合的脂质体产品为daa、 peg-c-domg,
galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-10:6-16:12-24:30-38。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:6-13:8-19:16-30:32-42。在某些实施方式中,所述与黑磷混合的脂质体产品daa、 peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-15:10-22:18-28:39-45。在某些实施方式中,所述与黑磷混合的脂质体产品daa、 peg-c-domg,galnac-peg-dsg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:6-16:12-18:13-23:29-39。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:8-12:18-28:23-33:34-44。在某些实施方式中,所述与黑磷混合的脂质体产品为daa、 peg-c-domg,galnac-peg-dsg、二豆蔻酰磷脂酰甘油和dope之间的摩尔比例为1:5-10:15-25:20-32:30-40。
104.更优选地,在一个实施方式中,其中所述富氧水溶液的氧浓度大于80 毫克每升。在某些实施方式中,其中所述富氧水溶液的氧浓度分别为80 毫克每升,82 毫克每升,84 毫克每升,86 毫克每升,88 毫克每升,90 毫克每升,92毫克每升,88 毫克每升,90 毫克每升,92毫克每升,96 毫克每升,100毫克每升,88 毫克每升,90 毫克每升,92毫克每升,88 毫克每升,90 毫克每升,92毫克每升,96 毫克每升,100毫克每升,105毫克每升,110毫克每升,115毫克每升,120毫克每升。
105.更优选地,其中所述富氧水溶液的溶质选自包含以下的列表:氯化钠、磷酸氢二钠、磷酸氢二钾、氯化钾、氯化镁、氯化钙、硫酸镁和它们之间的混合物,及上述溶质中加入蔗糖,葡萄糖及蔗糖和葡萄糖之间的混合物。在一个实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钠的质量浓度为:0.3%-1.2%。在某些实施方式中,其中所述富氧水溶液的溶质为磷酸氢二钠,磷酸氢二钠的质量浓度为:1.0%-3.39%。在某些实施方式中,其中所述富氧水溶液的溶质为磷酸氢二钾,磷酸氢二钾的质量浓度为:0.8%-5.02%。在某些实施方式中,其中所述富氧水溶液的溶质为磷酸氢二钾,磷酸氢二钾的质量浓度为:0.5%-6.77%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钾,氯化钾的质量浓度为:0.5%-1.41%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化镁,氯化镁的质量浓度为:2.9%-7.52%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钙,氯化钙的质量浓度为:3.9%-8.97%。在某些实施方式中,其中所述富氧水溶液的溶质为硫酸镁,硫酸镁的质量浓度为:1.5%-12.13%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠和蔗糖,氯化钠和蔗糖的质量浓度分别为:0.2%-1.5%和0.1%-3.2%。在某些实施方式中,其中所述富氧水溶液的溶质为磷酸氢二钠和蔗糖,磷酸氢二钠和蔗糖的质量浓度分别为:1.3%-3.0%和0.4%-4.5%。在某些实施方式中,其中所述富氧水溶液的溶质为磷酸氢二钾和蔗糖,磷酸氢二钾和蔗糖的质量浓度分别为:0.5%-3.9%和1.1%-6.9%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钾和蔗糖,氯化钾和蔗糖的质量浓度分别为:0.9%-2.53%和0.5%-5.1%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化镁和蔗糖,氯化镁和蔗糖的质量浓度分别为:3.3%-6.4%和1.2%-8.7%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钙和蔗糖,氯化钙和蔗糖的质量浓度分别为:1.2%-10.2%和3.3%-10.5%。在某些实施方式中,其中所述富氧水溶液的溶质为硫酸镁和蔗糖,硫酸镁和蔗糖的质量浓度分别为:4.0%-8.0%和2.5%-15.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠和葡萄
糖,氯化钠和葡萄糖的质量浓度分别为:0.5%-1.0%和0.5%-5.0%。在某些实施方式中,其中所述富氧水溶液的溶质为磷酸氢二钠和葡萄糖,磷酸氢二钠和葡萄糖的质量浓度分别为:1.0%-4.5%和0.5%-9.0%。在某些实施方式中,其中所述富氧水溶液的溶质为磷酸氢二钾和葡萄糖,磷酸氢二钾和葡萄糖的质量浓度分别为:1.3%-6.6%和0.1%-9.9%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钾和葡萄糖,氯化钾和葡萄糖的质量浓度分别为:0.5%-4.5%和1.9%-7.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化镁和葡萄糖,氯化镁和葡萄糖的质量浓度分别为:4.5%-8.5%和2.0%-10.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钙和葡萄糖,氯化钙和葡萄糖的质量浓度分别为:2.5%-8.7%和2.1%-8.4%。在某些实施方式中,其中所述富氧水溶液的溶质为硫酸镁和葡萄糖,硫酸镁和葡萄糖的质量浓度分别为:2.0%-12.0%和3.0%-13.2%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钾和葡萄糖,氯化钠,氯化钾和葡萄糖的质量浓度分别为:0.2%-0.6%,0.3%-1.2%和1.5%-8.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,磷酸氢二钠和葡萄糖,氯化钠,磷酸氢二钠和葡萄糖的质量浓度分别为:0.4%-1.0%,1.5%-6.0%和2.0%-8.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,磷酸氢二钾和葡萄糖,氯化钠,磷酸氢二钾和葡萄糖的质量浓度分别为:0.1%-1.0%,2.1%-7.7%和2.1%-10.2%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钾和葡萄糖,氯化钠,氯化钾和葡萄糖的质量浓度分别为:0.6%-1.2%,0.8%-3.5%和4.0%-10.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化镁和葡萄糖,氯化钠,氯化镁和葡萄糖的质量浓度分别为:0.2%-1.0%,7.5%-12.0%和4.0%-16.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钙和葡萄糖,氯化钠,氯化钙和葡萄糖的质量浓度分别为:0.5%-1.5%,4.0%-10.2%和3.5%-9.1%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,硫酸镁和葡萄糖,氯化钠,硫酸镁和葡萄糖的质量浓度分别为:0.6%-1.2%,4.5%-10.1%和5.2%-10.1%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,蔗糖和葡萄糖,氯化钠,蔗糖和葡萄糖的质量浓度分别为:0.4%-1.2%,1.3%-3.6%和2.1%-7.2%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,蔗糖和葡萄糖,氯化钠,蔗糖和葡萄糖的质量浓度分别为:0.2%-0.9%,0.8%-7.2%和1.2%-6.4%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,蔗糖和葡萄糖,氯化钠,蔗糖和葡萄糖的质量浓度分别为:0.25%-1.25%,1.0%-6.1%和1.0%-6.7%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,蔗糖和葡萄糖,氯化钠,蔗糖和葡萄糖的质量浓度分别为:0.5%-1.0%,1.1%-2.8%和3.2%-11.1%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,蔗糖和葡萄糖,氯化钠,蔗糖和葡萄糖的质量浓度分别为:0.7%-1.5%,1.2%-8.5%和5.5%-12.8%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,蔗糖和葡萄糖,氯化钠,蔗糖和葡萄糖的质量浓度分别为:1.0%-1.6%,3.2%-8.8%和5.1%-10.4%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,蔗糖和葡萄糖,氯化钠,蔗糖和葡萄糖的质量浓度分别为:0.3%-0.9%,2.1%-7.7%和6.1%-11.7%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,磷酸氢二钠,蔗糖和葡萄糖,氯化钠,磷酸氢二钠,蔗糖和葡萄糖的质量浓度分别为:0.5%-1.0%,0.8%-2.9%,1.1%-3.8%和4.0%-6.8%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,磷酸氢二钠,蔗糖和葡萄糖,氯化钠,磷酸氢二钠,蔗糖和葡萄糖的质量浓度分别为:0.4%-1.3%,0.4%-5.5%,1.5%-6.5%和2.8%-8.1%。在某些实施方式中,其中所述富氧水溶
液的溶质为氯化钠,磷酸氢二钾,蔗糖和葡萄糖,氯化钠,磷酸氢二钾,蔗糖和葡萄糖的质量浓度分别为:0.5%-1.4%,1.3%-3.6%,1.5%-8.0%和1.3%-7.2%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钾,蔗糖和葡萄糖,氯化钠,氯化钾,蔗糖和葡萄糖的质量浓度分别为:0.3%-0.9%,1.5%-1.8%,2.7%-4.9%和5.8%-10.3%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化镁,蔗糖和葡萄糖,氯化钠,氯化镁,蔗糖和葡萄糖的质量浓度分别为:0.3%-1.2%,1.1%-2.1%,3.1%-8.1%和4.8%-10.2%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钙,蔗糖和葡萄糖,氯化钠,氯化钙,蔗糖和葡萄糖的质量浓度分别为:0.6%-1.3%,0.9%-3.5%,1.1%-6.5%和4.7%-8.1%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,硫酸镁,蔗糖和葡萄糖,氯化钠,硫酸镁,蔗糖和葡萄糖的质量浓度分别为:0.5%-1.5%,0.9%-1.6%,2.5%-7.0%和4.2%-10.7%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,磷酸氢二钠,磷酸氢二钾,蔗糖和葡萄糖,氯化钠,磷酸氢二钠,磷酸氢二钾,蔗糖和葡萄糖的质量浓度分别为:0.1%-0.9%,0.4%-1.4%,0.6%-1.9%,3.2%-5.9%和4.8%-9.0%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,磷酸氢二钠,氯化钾,蔗糖和葡萄糖,氯化钠,磷酸氢二钠,氯化钾,蔗糖和葡萄糖的质量浓度分别为:0.1%-1.0%,0.6%-4.8%,1.0%-2.5%,3.0%-8.9%和1.5%-7.5%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,磷酸氢二钠,磷酸氢二钾,蔗糖和葡萄糖,氯化钠,磷酸氢二钠,磷酸氢二钾,蔗糖和葡萄糖的质量浓度分别为:0.8%-1.1%,0.7%-4.8%,2.1%-6.7%和2.8%-8.8%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钾,磷酸氢二钾,蔗糖和葡萄糖,氯化钠,氯化钾,磷酸氢二钾,蔗糖和葡萄糖的质量浓度分别为:0.6%-1.4%,1.1%-1.6%,2.7%-5.8%,4.9%-9.2%和3.3%-8.7%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化镁,磷酸氢二钠,蔗糖和葡萄糖,氯化钠,氯化镁,磷酸氢二钠,蔗糖和葡萄糖的质量浓度分别为:0.8%-1.5%,2.3%-4.2%,1.7%-7.9%,2.9%-10.2%和6.7%-12.3%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,氯化钙,磷酸氢二钠,蔗糖和葡萄糖,氯化钠,氯化钙,蔗糖和葡萄糖的质量浓度分别为:0.1%-1.1%,1.9%-4.2%,3.3%-7.8%,4.5%-8.9%和7.9%-13.9%。在某些实施方式中,其中所述富氧水溶液的溶质为氯化钠,硫酸镁,磷酸氢二钠,蔗糖和葡萄糖,氯化钠,硫酸镁,磷酸氢二钠,蔗糖和葡萄糖的质量浓度分别为:0.3%-1.0%,1.2%-3.6%,0.6%-1.9%,4.6%-9.2%和3.3%-9.1%。
106.更优选地,其中所述含氧气体的产品范围为:纯氧,空气或氧气跟惰性气体的混合物。
107.更优选地,其中所述特殊场技术的范围为:超声波技术或等离子体技术。
108.更优选地,其中所述超声波技术的超声波功率密度大于0.35瓦每平方厘米。在一个实施方式中,其中所述超声波技术的超声波功率密度为0.35瓦每平方厘米。在某些实施方式中,其中所述超声波技术的超声波功率密度分别为0.35瓦每平方厘米,0.37瓦每平方厘米,0.40瓦每平方厘米,0.44瓦每平方厘米,0.48瓦每平方厘米,0.52瓦每平方厘米,0.56瓦每平方厘米,0.60瓦每平方厘米,0.64瓦每平方厘米,0.68瓦每平方厘米,0.72瓦每平方厘米,0.76瓦每平方厘米,0.80瓦每平方厘米,0.84瓦每平方厘米,0.88瓦每平方厘米,0.92瓦每平方厘米,0.96瓦每平方厘米,1.00瓦每平方厘米,1.05瓦每平方厘米,1.10瓦每平方厘米,1.16瓦每平方厘米,1.20瓦每平方厘米,1.26瓦每平方厘米,1.32瓦每平方厘米,1.40瓦每平方厘米。在某些实施方式中,其中所述超声波技术的超声波功率密度分别为:1.5瓦
每平方厘米,2.0瓦每平方厘米,3.0瓦每平方厘米,4.0瓦每平方厘米,5.0瓦每平方厘米,6.0瓦每平方厘米,7.5瓦每平方厘米,9.0瓦每平方厘米,10.5瓦每平方厘米,12.0瓦每平方厘米。
109.更优选地,其中所述等离子体技术的等离子体产生方式为:气相方式或液相方式。
110.更优选地,其中气相等离子体产生的电子和离子的能量大于10电子伏特。在一个实施方式中,其中所述气相等离子体产生的电子和离子的能量为10电子伏特。在某些实施方式中,其中所述气相等离子体产生的电子和离子的能量分别为12电子伏特,14电子伏特,16电子伏特,18电子伏特,20电子伏特,22电子伏特,24电子伏特,26电子伏特,28电子伏特,30电子伏特,32电子伏特,34电子伏特,36电子伏特,38电子伏特,40电子伏特。在某些实施方式中,其中所述气相等离子体产生的电子和离子的能量分别为:42电子伏特,47电子伏特,52电子伏特,57电子伏特,60电子伏特,63电子伏特,68电子伏特,72电子伏特,76电子伏特,80电子伏特,84电子伏特,88电子伏特,92电子伏特,96电子伏特,100电子伏特。
111.更优选地,其中液相相等离子体产生的电子和离子的功率密度大于0.1瓦每平方厘米。在一个实施方式中,其中所述液相等离子体产生的电子和离子的功率密度为0.1瓦每平方厘米。在某些实施方式中,其中所述液相等离子体产生的电子和离子的功率密度分别为0.15瓦每平方厘米,0.17瓦每平方厘米,0.19瓦每平方厘米,0.21瓦每平方厘米,0.23瓦每平方厘米,0.25瓦每平方厘米,0.27瓦每平方厘米,0.30瓦每平方厘米,0.32瓦每平方厘米,0.34瓦每平方厘米,0.36瓦每平方厘米,0.38瓦每平方厘米,0.40瓦每平方厘米,0.44瓦每平方厘米,0.46瓦每平方厘米,0.49瓦每平方厘米,0.52瓦每平方厘米,0.55瓦每平方厘米,0.58瓦每平方厘米,0.63瓦每平方厘米,0.68瓦每平方厘米,0.70瓦每平方厘米,0.74瓦每平方厘米,0.78瓦每平方厘米,0.80瓦每平方厘米。在某些实施方式中,其中所述液相等离子体产生的电子和离子的功率密度分别为:0.82瓦每平方厘米,0.87瓦每平方厘米,0. 92瓦每平方厘米,0.97瓦每平方厘米,1.00瓦每平方厘米,1.05瓦每平方厘米,1.1瓦每平方厘米,1.15瓦每平方厘米,1.2瓦每平方厘米,1.25瓦每平方厘米。在某些实施方式中,其中所述液相等离子体产生的电子和离子的功率密度分别为:1.27瓦每平方厘米,1.37瓦每平方厘米,1.47瓦每平方厘米,1.57瓦每平方厘米,1.60瓦每平方厘米,1.75瓦每平方厘米,1.90瓦每平方厘米,2.00瓦每平方厘米,2.2瓦每平方厘米,2.5瓦每平方厘米。
112.更优选地,其中所述搅拌方式为:机械搅拌和磁力搅拌,优选为机械搅拌。
113.更优选地,其中所述搅拌速率为:采用超声波技术搅拌的速率为50-700rpm;采用气相等离子体技术搅拌的速率为10-40rpm;采用液相等离子体技术搅拌的速率为50-400rpm。在一个实施方式中,其中所述超声波技术搅拌的速率为52rpm。在某些实施方式中,其中所述超声波技术搅拌的速率分别为55rpm,65rpm,75rpm,80rpm,90rpm,100rpm,110rpm,125rpm,140rpm,160rpm,180rpm。在某些实施方式中,其中所述超声波技术搅拌的速率为分别为:185rpm,200rpm,230rpm,250rpm,300rpm,350rpm,450rpm,550rpm,600rpm,650rpm,700rpm。在一个实施方式中,其中所述气相等离子体技术搅拌的速率为10rpm。在某些实施方式中,其中所述气相等离子体技术搅拌的速率分别为12rpm,15rpm,18rpm,20rpm,23rpm,27rpm,30rpm,34rpm,36rpm,38rpm,40rpm。在一个实施方式中,其中所述液相等离子体技术搅拌的速率为52rpm。在某些实施方式中,其中所述液相等离子体技术搅拌的速率分别为52rpm,55rpm,65rpm,70rpm,80rpm,90rpm,100rpm,120rpm,140rpm,160rpm,200rpm。在
某些实施方式中,其中所述液相等离子体技术搅拌的速率为分别为:205rpm,215rpm,240rpm,270rpm,300rpm,330rpm,360rpm,380rpm,400rpm。
114.更优选地,其中所述处理的一段时间为:采用超声波技术处理的时间大于45分钟;采用气相等离子体技术处理的时间大于15分钟;采用液相等离子体技术处理的时间大于5分钟。在一个实施方式中,其中所述超声波技术处理的时间为45分钟。在某些实施方式中,其中所述超声波技术处理的时间分别为46分钟,48分钟,50分钟,53分钟,55分钟,58分钟,60分钟,65分钟,70分钟,75分钟,80分钟。在某些实施方式中,其中所述超声波技术处理的时间分别为82分钟,92分钟,100分钟,105分钟,110分钟,115分钟,120分钟,130分钟,150分钟,180分钟。在一个实施方式中,其中所述采用气相等离子体技术处理的时间为15分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为16分钟,18分钟,20分钟,23分钟,25分钟,28分钟,30分钟,35分钟,40分钟,45分钟,50分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为52分钟,58分钟,70分钟,90分钟,105分钟,120分钟,135分钟,150分钟。在一个实施方式中,其中所述采用液相等离子体技术处理的时间为5分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为6分钟, 8分钟,10分钟,13分钟,15分钟,18分钟,20分钟,25分钟,30分钟。在某些实施方式中,其中所述气相等离子体技术处理的时间分别为32分钟,38分钟,40分钟,50分钟,60分钟。
115.更优选地,其中所述异双官能化聚乙二醇选自包含以下的列表:amino-peg4-alcohol、nh2-peg2-c2-boc、amino-peg4-ch2cooh、nh2-(peg)n-cooh (n=1-12)及其之间的混合物。
116.更优选地,其中所述的一定温度下进行超声搅拌处理的温度为:-20摄氏度以上。在一个实施方式中,其中所述超声搅拌处理的温度为-20摄氏度。在某些实施方式中,其中所述超声搅拌处理的温度分别为-20摄氏度,-18摄氏度,-15摄氏度,-13摄氏度,-10摄氏度,-8摄氏度,-6摄氏度,-4摄氏度,-2摄氏度。在某些实施方式中,所述超声搅拌处理的温度分别为-1摄氏度,0摄氏度,2摄氏度,4摄氏度,6摄氏度,8摄氏度,10摄氏度,12摄氏度,14摄氏度。在某些实施方式中,所述超声搅拌处理的温度分别为16摄氏度,19摄氏度,22摄氏度,25摄氏度,28摄氏度,30摄氏度。
117.更优选地,其中所述的一定温度下进行超声搅拌处理的超声波功率为:超声波功率密度大于0.1瓦每平方厘米。在一个实施方式中,其中所述超声波技术的超声波功率密度为0.1瓦每平方厘米。在某些实施方式中,其中所述超声波技术的超声波功率密度分别为0.12瓦每平方厘米,0.15瓦每平方厘米,0.18瓦每平方厘米,0.2瓦每平方厘米,0.25瓦每平方厘米,0.3瓦每平方厘米,0.35瓦每平方厘米,0.40瓦每平方厘米,0.45瓦每平方厘米,0.50瓦每平方厘米,0.60瓦每平方厘米,0.70瓦每平方厘米,0.80瓦每平方厘米,0.90瓦每平方厘米,1.00瓦每平方厘米,1.10瓦每平方厘米。在某些实施方式中,其中所述超声波技术的超声波功率密度分别为:1.15瓦每平方厘米,2.15瓦每平方厘米,3. 0瓦每平方厘米,4.0瓦每平方厘米,5.0瓦每平方厘米,6.0瓦每平方厘米,7.5瓦每平方厘米,9.0瓦每平方厘米,10.5瓦每平方厘米,12.0瓦每平方厘米。
118.更优选地,其中其中所述的一定温度下进行超声搅拌处理的搅拌速率:采用超声波技术搅拌的速率为30-550rpm。在一个实施方式中,其中所述超声波技术搅拌的速率为32rpm。在某些实施方式中,其中所述超声波技术搅拌的速率分别为35rpm,55rpm,75rpm,
95rpm,110rpm,130rpm,150rpm,180rpm,200rpm。在某些实施方式中,其中所述超声波技术搅拌的速率为分别为:185rpm,200rpm,230rpm,250rpm,300rpm,350rpm,400rpm,450rpm,500rpm,550rpm。
119.更优选地,其中所述无菌过滤器的有效面积大于10平方厘米。在一个实施方式中,其中所述无菌过滤器的有效面积为10平方厘米。在某些实施方式中,其中所述无菌过滤器的有效面积分别为:12 平方厘米,15 平方厘米,18 平方厘米,20 平方厘米,25 平方厘米,30 平方厘米,40 平方厘米,50平方厘米,55 平方厘米,80 平方厘米,100 平方厘米,125 平方厘米,150 平方厘米,200 平方厘米。
120.更优选地,其中所述无菌过滤器包含有:包括预过滤器,预连接管道和无菌连接器无菌过滤转移装置。
121.更优选地,其中所述混合设备为微流控,超流控或碰撞喷射混合器。
122.更优选地,环状mrna与递送系统进一步混合的流速为:总流速为0.1-15毫升每分钟,流速比为1/1至10/1。在一个实施方式中,其中所述总流速为0.11毫升每分钟, 流速比为1/2。在某些实施方式中,其中所述总流速为:0.12毫升每分钟,流速比为1/3。在某些实施方式中,其中所述总流速分别为:0.1毫升每分钟, 流速比为1/4。在某些实施方式中,其中所述总流速分别为:0.14毫升每分钟, 流速比为1/5。在一个实施方式中,其中所述总流速为0.115毫升每分钟, 流速比为1/6。在某些实施方式中,其中所述总流速为:0.125毫升每分钟,流速比为1/7。在某些实施方式中,其中所述总流速分别为:0.135毫升每分钟, 流速比为1/8。在某些实施方式中,其中所述总流速分别为:0.145毫升每分钟, 流速比为1/9。在某些实施方式中,其中所述总流速分别为:0.15毫升每分钟, 流速比为1/9.5。
123.更优选地,其中所述的针对ebv感染所致的鼻咽癌靶向递送的脂质体的平均粒径为90纳米。 在一个实施方式中,其中所述的针对ebv感染所致的鼻咽癌脂质体的平均粒径为50纳米。在某些实施方式中,其中所述的针对ebv感染所致的鼻咽癌脂质体的平均粒径分别为60纳米,70纳米,80纳米,100纳米,110纳米, 120纳米, 130纳米, 140纳米, 150纳米,160纳米, 170纳米,180纳米,190纳米,200纳米。在某些实施方式中,其中所述的针对ebv感染所致的鼻咽癌脂质体的平均粒径分别为210纳米,220纳米, 230纳米, 240纳米, 250纳米,260纳米,270纳米, 280纳米, 290纳米,300纳米,310纳米,320纳米, 330纳米, 340纳米, 350纳米。
124.更优选地,其中所述的针对ebv感染所致的鼻咽癌靶向递送的脂质体的载电电荷范围为30-40毫伏。在一个实施方式中,其中所述的针对ebv感染所致的鼻咽癌靶向脂质体的电荷为8毫伏。在某些实施方式中,其中所述的针对ebv感染所致的鼻咽癌的载电电荷分别为9毫伏,10毫伏,11毫伏,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。在某些实施方式中,其中所述的针对ebv感染所致的鼻咽癌的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏,35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。在某些实施方式中,其中所述的针对ebv感染所致的鼻咽癌靶向递送的脂质体的的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏, 45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
125.更优选地,其中所述的针对hbv感染所致的肝癌靶向递送的脂质体的平均粒径为150纳米。在一个实施方式中,其中所述的针对hbv感染所致的肝癌靶向脂质体的平均粒径
为50纳米。在某些实施方式中,其中所述的针对hbv感染所致的肝癌靶向脂质体的的平均粒径分别为60纳米,70纳米,80纳米,90纳米,100纳米,110纳米, 120纳米, 130纳米, 140纳米, 160纳米, 170纳米,180纳米,190纳米,200纳米, 210纳米,220纳米, 230纳米, 240纳米, 250纳米。某些实施方式中,其中所述的针对hbv感染所致的肝癌脂质体的平均粒径分别为260纳米,270纳米, 280纳米, 290纳米, 300纳米,310纳米,320纳米, 330纳米, 340纳米, 350纳米。
126.更优选地,其中所述的针对hbv感染所致的肝癌靶向递送的脂质体的载电电荷范围为30-40 毫伏。在一个实施方式中,其中所述的针对hbv感染所致的肝癌的靶向脂质体的载电电荷为8毫伏。在某些实施方式中,其中所述的针对hbv感染所致的肝癌靶向递送的脂质体的载电电荷分别为9毫伏,10毫伏,11毫伏,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。其中所述的针对hbv感染所致的肝癌靶向递送的脂质体的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏, 35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。其中所述的针对hbv感染所致的肝癌靶向递送的脂质体的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏,45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
127.更优选地,其中所述的针对hcv感染所致的肝癌靶向递送的递送系统的平均粒径为120纳米。在一个实施方式中,其中所述hcv感染所致的肝癌靶向递送的脂质体的平均粒径为50纳米。在某些实施方式中,其中所述hcv感染所致的肝癌靶向递送的脂质体的平均粒径分别为60纳米,70纳米,80纳米,90纳米,100纳米, 110纳米。在某些实施方式中,其中所述hcv感染所致的肝癌靶向递送的脂质体的平均粒径分别为130纳米, 140纳米, 150纳米, 160纳米, 170纳米,180纳米,190纳米,200纳米, 210纳米,220纳米,230纳米, 240纳米,250纳米。某些实施方式中,其中所述的针对hcv感染所致的肝癌脂质体的平均粒径分别为260纳米,270纳米,280纳米,290纳米,300纳米,310纳米,320纳米, 330纳米, 340纳米,350纳米。
128.更优选地,其中所述的针对hcv感染所致的肝癌靶向递送的脂质体的载电电荷范围为30-40 毫伏。在一个实施方式中,其中所述的针对hcv感染所致的肝癌的靶向脂质体的载电电荷为8毫伏。在某些实施方式中,其中所述的针对hcv感染所致的肝癌靶向递送的脂质体的载电电荷分别为9毫伏,10毫伏,11毫伏,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。其中所述的针对hcv感染所致的肝癌靶向递送的脂质体的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏, 35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。其中所述的针对hcv感染所致的肝癌靶向递送的脂质体的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏,45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
129.更优选地,其中所述的针对hpv感染所致的宫颈癌的靶向递送的脂质体的平均粒径为140纳米。在一个实施方式中,其中所述hpv感染所致的宫颈癌靶向递送的脂质体的平均粒径为50纳米。在某些实施方式中,其中所述hpv感染所致的宫颈癌靶向递送的脂质体的平均粒径分别为60纳米,70纳米,80纳米,90纳米,110纳米, 120纳米, 130纳米, 150纳米, 160纳米, 170纳米,180纳米,190纳米,200纳米,210纳米,220纳米,230纳米,240纳米, 250纳米。在某些实施方式中,其中所述hpv感染所致的宫颈癌靶向递送的脂质体的平均粒径分
别为260纳米,270纳米,280纳米,290纳米, 300纳米,310纳米,320纳米, 330纳米, 340纳米,350纳米。
130.更优选地,其中所述的针对hpv感染所致的宫颈癌靶向递送的脂质体的载电电荷范围为30-40毫伏。在一个实施方式中,其中所述的针对hpv感染所致的宫颈癌的靶向脂质体的载电电荷为8毫伏。在某些实施方式中,其中所述的针对hpv感染所致的宫颈癌靶向递送的脂质体的载电电荷分别为9毫伏,10毫伏,11毫伏,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。其中所述的针对hpv感染所致的宫颈癌靶向递送的脂质体的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏,35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。其中所述的针对hpv感染所致的宫颈癌靶向递送的脂质体的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏,45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
131.更优选地,其中所述的针对ebv感染所致的淋巴瘤靶向递送的脂质体的平均粒径为110纳米。在一个实施方式中,其中所述ebv感染所致的淋巴瘤靶向递送的脂质体的平均粒径为50纳米。在某些实施方式中,其中所述ebv感染所致的淋巴瘤靶向递送的脂质体的平均粒径分别为60纳米,70纳米,80纳米,90纳米,100纳米, 120纳米,130纳米,140纳米,150纳米,160纳米,170纳米,180纳米,190纳米,200纳米,210纳米,220纳米,230纳米,240纳米,250纳米。在某些实施方式中,其中所述ebv感染所致的淋巴瘤靶向递送的脂质体的平均粒径分别为260纳米,270纳米,280纳米,290纳米,300纳米,310纳米,320纳米,330纳米,340纳米,350纳米。
132.更优选地,其中所述的针对ebv感染所致的淋巴瘤靶向递送的脂质体的的载电电荷范围为30-40毫伏。在一个实施方式中,其中所述的针对ebv感染所致的淋巴瘤的靶向递送的脂质体的载电电荷为8毫伏。在某些实施方式中,其中所述的针对ebv感染所致的淋巴瘤的靶向脂质体的载电电荷分别为9毫伏,10毫伏,11毫伏,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。在某些实施方式中,其中所述的针对ebv感染所致的淋巴瘤的靶向脂质体的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏, 35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。在某些实施方式中,其中所述的针对ebv感染所致的淋巴瘤的靶向脂质体的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏, 45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
133.更优选地,其中所述的针对hhv-8感染所致的卡波西肉瘤靶向递送的脂质体的平均粒径为100纳米。 在一个实施方式中,其中所述的针对hhv-8感染所致的卡波西肉瘤靶向递送的脂质体的平均粒径为50纳米。在某些实施方式中,其中所述的针对hhv-8感染所致的卡波西肉瘤靶向递送的脂质体的平均粒径分别为60纳米,70纳米,80纳米,90纳米,110纳米,120纳米, 130纳米,140纳米,150纳米,160纳米, 170纳米,180纳米,190纳米,200纳米,210纳米,220纳米,230纳米,240纳米, 250纳米。在某些实施方式中,其中所述的针对hhv-8感染所致的卡波西肉瘤靶向递送的脂质体的平均粒径分别为260纳米,270纳米,280纳米,290纳米,300纳米,310纳米,320纳米,330纳米,340纳米,350纳米。
134.更优选地,其中所述的针对hhv-8感染所致的卡波西肉瘤靶向递送的脂质体的载电电荷范围为30-40毫伏。在一个实施方式中,其中所述的针对hhv-8感染所致的卡波西肉
瘤靶向递送的脂质体的载电电荷为为8毫伏。在某些实施方式中,其中所述的针对ebv感染所致的淋巴瘤的靶向递送的脂质体的载电电荷分别为9毫伏,10毫伏,11毫伏 ,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。在某些实施方式中,其中所述的针对ebv感染所致的淋巴瘤的靶向递送的脂质体的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏,35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。在某些实施方式中,其中所述的针对ebv感染所致的淋巴瘤的靶向递送的脂质体的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏,45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
135.更优选地,其中所述的针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的平均粒径为80纳米。在一个实施方式中,其中所述的针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的平均粒径为50纳米。在某些实施方式中,其中所述的针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的平均粒径分别为60纳米,70纳米,90纳米,100纳米,110纳米,120纳米,130纳米,140纳米,150纳米,160纳米,170纳米,180纳米,190纳米,200纳米,210纳米,220纳米,230纳米,240纳米, 250纳米。在某些实施方式中,其中所述的针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的平均粒径分别为260纳米,270纳米,280纳米,290纳米,300纳米,310纳米,320纳米,330纳米,340纳米,350纳米。
136.更优选地,其中所述的针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的载电电荷范围为30-40 毫伏。在一个实施方式中,其中所述针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的载电电荷为8毫伏。在某些实施方式中,其中所述针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的载电电荷分别为9毫伏,10毫伏,11毫伏,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。在某些实施方式中,其中所述针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏, 35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。在某些实施方式中,其中所述针对mcpyv感染所致的梅克尔癌靶向递送的脂质体的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏, 45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
137.更优选地,其中所述的针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的平均粒径为130纳米。在一个实施方式中,其中所述的针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的平均粒径为50纳米。在某些实施方式中,其中所述的针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的平均粒径分别为60纳米,70纳米,80纳米,90纳米,100纳米,110纳米,120纳米,140纳米,150纳米, 160纳米, 170纳米,180纳米,190纳米,200纳米,210纳米,220纳米,230纳米,240纳米, 250纳米。在某些实施方式中,其中所述的针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的平均粒径分别为260纳米,270纳米,280纳米,290纳米,300纳米,310纳米,320纳米,330纳米,340纳米,350纳米。
138.更优选地,其中所述的针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的载电电荷范围为30-40毫伏。在一个实施方式中,其中所述针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的载电电荷为8毫伏。在某些实施方式中,其中所述针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的载电电荷分别为9毫伏,10毫伏,
11毫伏 ,12毫伏,13毫伏,14毫伏,15毫伏,16毫伏,17毫伏,18毫伏,19毫伏,20毫伏,21毫伏,22毫伏,23毫伏,24毫伏,25毫伏,26毫伏,27毫伏,28毫伏,29毫伏。在某些实施方式中,其中所述针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的载电电荷分别为31毫伏,32毫伏,33毫伏,34毫伏, 35毫伏,36毫伏,37毫伏,38毫伏,39毫伏。在某些实施方式中,其中所述针对hltv-1感染所致的成人t细胞白血病靶向递送的脂质体的载电电荷分别为41毫伏,42毫伏,43毫伏,44毫伏, 45毫伏,46毫伏,47毫伏,48毫伏,49毫伏。
139.更进一步地,所述释放的递送系统,其是硅橡胶系统。
140.更进一步地,所述释放的递送系统,其是基于肽的系统。
141.更进一步地,所述释放的递送系统,其是水凝胶释放系统。
142.更进一步地,所述释放的递送系统,其是蜡涂层。
143.更进一步地,所述释放的递送系统,其是使用常规粘合剂和赋形剂的 压片。
144.更进一步地,所述释放的递送系统,其是部分融合的植入物。
145.更进一步地,所述释放的递送系统,其是脂质纳米颗粒。
146.更进一步地,所述释放的递送系统,其包含有式(i)结构化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物:其中: a,b,c,d,e,f,g,小时,i各自独立地为0和1;r1、r2、r3、r4、r5、r6、r7、r8和r9各自独立地为α1α
2-n-(ch2)
x-基团;x为大于或等于0的整数;α1和α
2 各自独立地为:(r-(ch2)
α-(c=o)o-(ch2)
β
)-, (r-(ch2)
α-(c=o)-(ch2)
β
)-, (r
11-(ch2)
α-o-(ch2)
β
),(r
11-(ch2)
α-(p=o)o-(ch2)
β
),(r
11-(ch2)
α-o(p=o)o-(ch2)
β
), (r
11-(ch2)
α-o(s=o)o-(ch2)
β
) , (r
11-(ch2)
α-o(o=s=o)o-(ch2)
β
), (r
11-(ch2)
α-o(n=o)o-(ch2)
β
), (r
11-(ch2)
α-o(n=o)-(ch2)
β
),(r
11-(ch2)
α-o(n=o)-(ch2)
β
),(r
11-(ch2)
α-s(c=o)-(ch2)
β
),(r
11-(ch2)
α-s-s-(ch2)
β
),(r
11-(ch2)
α-n小时(c=o)-(ch2)
β
),(r
11-(ch2)
α-nr
12 (c=o)-(ch2)
β
),(r
11-(ch2)
α-n(r
12
)o(c=o)-(ch2)
β
),(r
11-(ch2)
α-n(r
12
)o(n=o)-(ch2)
β
),(r
11-(ch2)
α-n(r
12
)o(p=o)-(ch2)
β
),(r
11-(ch2)
α-n(r
12
)o(s=o)o-(ch2)
β
),(r
11-(ch2)
α-n(r
12
)o(o=s=o)o-(ch2)
β
)中的一种;r
11
和r
12
各自独立地为氢、氧、氮、硫、磷、氰基、羟基、羧基或杂环基的一种; α和β各自独立地为大于或等于0的整数。
147.优选地,所述化合物具有式(i-1)所示的结构:
ꢀ
。
148.优选地,所述的化合物具有式(i-2)所示的结构:。
149.更进一步,所述释放的递送系统,与式(i)结构化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物可以起协同作用的脂质化合物包括阴离子脂质、中性脂质、甾醇和两亲性脂质中的一种或两种以上的组合。
150.优选地,所述与式(i)结构化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物与脂质化合物起协同作用的途径是指在合适强度的声能量或辐射能量作用下实现;优选地,所述合适强度声能量的产生方式是通过超声波技术来实现;优选地,所述合适强度辐射能量的产生方式是通过等离子体技术来实现;更优选地,所述等离子体技术为气相等离子体技术;更优选地,所述等离子体技术为液相相等离子体技术。
151.优选地,所述与式(i)结构化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物可以起协同作用的阳离子脂质包括dlindma、dodma、
dlin-mc2-mpz、dlin-kc2-dma、 dotap、c12-200、dc-chol和dotma中的一种或两种以上的组合。
152.优选地,所述与式(i)结构化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物可以起协同作用的所述阴离子脂质包括磷脂酰丝氨酸、磷脂酰肌醇、磷脂酸、磷脂酰甘油、dopg、dops 和二豆蔻酰磷脂酰甘油中的一种或两种以上的组合。
153.优选地,所述与式(i)结构化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物可以起协同作用的中性脂质包括dope、dspc、dppc、dopc、dppg、popc、pope、dppe、dmpe、dspe、和 sope中的至少一种或其经阴离子或阳离子修饰基团修饰的脂质。
154.优选地,所述与式(i)结构化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物可以起协同作用的中性脂质两亲性脂质包括peg-dmg、peg-c-dmg、peg-c14、peg-c-dma、peg-dspe、peg-pe、peg修饰的神经酰胺、peg修饰的二烷基胺、peg修饰的二酰基甘油、吐温-20、吐温-80、 peg-dpg、peg-s-dmg、daa、peg-c-domg和galnac-peg-dsg中的一种或两种以上的组合。
155.优选地,所述起协同作用的含式(i)结构化合物的脂质化合物、所述阴离子脂质、所述中性脂质、所述甾醇和所述两亲性脂质的摩尔比为(20-65):(0-20):(5-25):(25
‑ꢀ
55):(0.3-15); 其中,所述含式(i)结构化合物的脂质化合物中,根据、所述的式(i)化合物 或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物、非共价复合物或前体 药物和所述阳离子脂质的摩尔比为(1-10):(0-10)。
156.优选地,所述环形mrna与根据含式(i)结构物所述的化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物的质量比为1:(3
ꢀ‑ꢀ
40);或者,所述核酸药物与根据所述的脂质载体的质量比为1:(3
‑ꢀ
40)。
157.优选地,所述式(i)结构物所述的化合物或其药学上可接受的盐、立体异构体、互变异构体、溶剂化物、螯合物或非共价复合物与环形mrna组合形成的疫苗粒径为50-500 纳米。
158.优选地,所述环形mrna在所述疫苗制剂中的包封率大于50%。
159.更进一步地,所述包含有式(i)结构化合物的递送系统,在不需要靶向配体的情况下,可以把前面实施例中所述环形mrna递送到病毒感染所致癌症的癌细胞。
160.更进一步地,所述释放的递送系统,包含有靶向配体。
161.优选地,所述靶向配体为小分子。
162.优选地,所述靶向配体为糖类分子。
163.优选地,所述靶向配体为多肽。
164.优选地,所述靶向配体为蛋白质。
165.优选地,所述靶向配体为抗体。
166.优选地,所述靶向配体为核酸适配体。
167.更进一步地,所述核酸适配体从随机合成的不同寡核苷酸核酸分子库中筛选出来。
168.优选地,所述核酸适配体的筛选技术为指数富集配体系统进化技术。
169.优选地,所述核酸适配体的筛选步骤如下:第一步,建立一个含不同寡核苷酸构成的文库,在文库中加入病毒感染所致癌症的一种或多种癌细胞目标分子,实现二者的结合孵育;第二步,用物理方法除去未结合的适配体,分离得到靶分子-适配体复合物或混合靶分子-适配体复合物;第三步,洗脱收集结合序列,并用干扰物质对其进行反筛,排除非特异结合的竞争;第四步,将确定与靶分子或混合靶分子结合的核酸适配体进行pcr扩增并富集纯化,筛选出能够识别病毒感染所致癌症癌细胞特定靶分子或混合靶分子的核酸适配体,获得能靶向病毒感染所致癌症单一或多种癌细胞的核酸适配体, 从而建立获得广泛靶向病毒感染所致癌症的癌细胞靶标的方法。
170.优选地,所述ebv感染所致的鼻咽癌的核酸适配体选自包含以下的列表:cd109,s3,s27, s12, s5的一种及多种之间的混合物。
171.优选地,所述hbv感染所致的肝癌的核酸适配体选自包含以下的列表:10-8,ly-1,ch6, 肽适配子, zy1, tsl9a, tsl11a, as1411的一种及多种之间的混合物。
172.优选地,所述hcv感染所致的肝癌的核酸适配体选自包含以下的列表:10-8,ly-1,ch6, 肽适配子, zy1, tsl9a, tsl11a, as1411,tls3的一种及多种之间的混合物。
173.优选地,所述hpv感染所致的宫颈癌的核酸适配体选自包含以下的列表:air3a, apt, a10的一种及多种之间的混合物。
174.优选地,所述ebv感染所致的原位淋巴瘤的核酸适配体选自包含以下的列表:td05, gbi-10, wqy-9-b, u2, u8,u9,u31,gbm128. gbm131的一种及多种之间的混合物。
175.优选地,所述hltv-1感染所致的成人t细胞白血病的核酸适配体为sscgc。
176.优选地,hhv-8感染所致的卡波西肉瘤的核酸适配体为硫代磷酸酯。
177.优选地,mcpyv感染所致的梅克尔癌的核酸适配体为a10 2
´‑
氟尿嘧啶rna适配体。
178.进一步地,所述混合适配体的不同核酸适配体的具体比例和具体种类通过混合适配体与递送系统的协同性来筛选和确定。
179.更进一步地,所述疫苗组合物可起到以下作用中的一项或多项:s1. 激活cd8 t细胞对肿瘤的杀伤活性;s2.刺激cd8 t细胞的效应功能;s3.刺激cd8 t细胞分泌干扰素-r(ifn-r);s4.刺激cd8 t细胞分泌肿瘤坏死因子-a(tnf-a);s5.刺激cd8 t细胞释放细胞毒颗粒;s6.加入树突状细胞后,树突状细胞激活cd8 t细胞;s7.刺激cd8 t细胞细胞质膜上t细胞抗原受体(tcr)成簇;s8.过继激活的t细胞可抑制肿瘤生长;s9.可根据核酸序列制作多肽疫苗激活人体抗肿瘤免疫;s10.可根据核酸序列制作需要mrna疫苗激活人体抗肿瘤免疫;s11.可用作制作树突状细胞(dc)疫苗激活人体抗肿瘤免疫;s12.可用作制作dna疫苗激活人体抗肿瘤免疫;
s13.可作为免疫原激活cd8细胞后测序得到tcr序列用于tcr-t治疗。
180.更进一步地,所述疫苗组合物的制剂方式,其包括粉末、颗粒、片剂、乳剂、糖浆、气溶胶、软或硬明胶胶囊、无菌注 射溶液和无菌粉末。
181.更进一步地,所述疫苗组合物,可通过各种给药途径施用。
182.优选地,所述给药途径,其包括口服、肠外(例如栓剂、经皮、静脉、腹腔内、肌肉内、病变内、鼻、椎管,或可使用植入式装置进行持续、持续或反复释放。
183.更优选地,所述给药途径,其为通过肌肉注射或静脉注射。
184.更优选地,所述注射,其为电穿孔器注射。
185.优选地,所述给药途径,其给药次数包括所需范围内每天给药一次或多次,给药周期不受特别限制。
186.第五部分,开发平台应用库的技术方案之一。
187.本发明实施例通过开发平台的应用库还提供了一种在制备蛋白中应用的对象,其包含有本发明前述实施例所述的重组核酸分子。
188.进一步的,所述在制备蛋白中应用的对象,其包含有本发明前述实施例所述的重组表达载体。
189.进一步的,所述在制备蛋白中应用的对象,其包含有本发明前述实施例所述的线性mrna。
190.进一步的,所述在制备蛋白中应用的对象,其包含有本发明前述实施例所述的环形mrna。
191.进一步的,所述在制备蛋白中应用的对象,其包含有本发明前述实施例所述的重组宿主细胞。
192.第六部分,开发平台应用库的技术方案之二。
193.本发明实施例通过开发平台的应用库还提供了一种除了前述实施例所述疫苗之外的药物组合物,其包含有本发明前述实施例所述的病毒感染所致癌症的肿瘤新生抗原多肽。
194.进一步的,所述的药物组合物,其包含有本发明前述实施例所述的重组核酸分子。
195.进一步的,所述的药物组合物,其包含有本发明前述实施例所述的重组表达载体。
196.进一步的,所述的药物组合物,其包含有本发明前述实施例所述的线性mrna。
197.进一步的,所述的药物组合物,其包含有本发明前述实施例所述的环形mrna。
198.进一步的,所述的药物组合物,其包含有本发明前述实施例所述的重组宿主细胞。
199.进一步的,所述的药物组合物,其包含有一种或多种药学或生理学上可接受且通常不会引起过敏反应(例如,胃肠道紊乱、头晕等)或其类似反应的的载体、赋形剂或稀释剂;更进一步地,所述的载体、赋形剂和稀释剂,其可以为乳糖、葡萄糖、蔗糖、山梨醇、甘露醇、木糖醇、赤藓糖醇、麦芽糖醇、淀粉、阿拉 伯胶、海藻酸钠、明胶、磷酸钙、硅酸钙、纤维素、甲基纤维素、聚乙烯吡咯烷酮、水、甲基羟基 苯甲酸丙酯、滑石粉、硬脂酸镁和矿物油中的一种或二种或多种的合理混合物;更进一步地,所述药学上可接受的载体可以是药物组合物中除对受试者无毒的活性成分以外的成分;
更进一步地,所述药学上可接受的载体可以包括但不限于缓冲剂、赋形剂、稳定剂或防腐剂;更进一步地,所述药学上可接受的载体是指生理上相容的溶剂、分散介质、包衣、抗细菌和抗真菌剂、等渗剂和吸收延迟剂等,如盐、缓冲液、糖类、抗氧化剂、水性或非水性载体、防腐剂、润湿剂、表面活 性剂或乳化剂或其组合;更进一步地,所述药学上可接受的载体是指可以基于载体的活性和制剂的所需特性,如稳定性和/或最小氧化,通过实验确定药物组合物中的药学上可接受的量的载体。
200.进一步的,所述的药物组合物,其包含有缓冲液,如乙酸、柠檬酸、组氨酸、硼酸、甲 酸、琥珀酸、磷酸、碳酸、苹果酸、天冬氨酸、tris缓冲液、heppso、hepes、中性缓冲盐水、磷酸盐缓冲盐水等。
201.进一步的,所述的药物组合物,其包含有碳水化合物,如葡萄糖、蔗糖、甘露糖或葡聚糖、甘露醇。
202.进一步的,所述的药物组合物,其包含有蛋白质或氨基酸,如甘氨酸。
203.进一步的,所述的药物组合物,其包含有抗氧化剂。
204.进一步的,所述的药物组合物,其包含有螯合剂,如edta或谷胱甘肽。
205.进一步的,所述的药物组合物,其包含有佐剂,如氢氧化铝,磷酸铝、明矾(硫酸铝钾)、mf59、病毒体、as04[氢氧 化铝和单磷酸脂a(mpl)的混合物]、as03(dl-α-生育酚、角鲨烯和聚山梨酯80(乳化剂)的混 合物)、cpg、鞭毛蛋白、聚i:c、as01、as02、iscoms,iscommatrix等。
[0206]
进一步的,所述的药物组合物,其包含有本发明所述的菌株和/或巨噬细胞。
[0207]
进一步的,所述的药物组合物,其包含有抗细菌和抗真菌剂。
[0208]
进一步的,所述的药物组合物,其包含有防腐剂。
[0209]
进一步的,所述的药物组合物,其可以配制用于肠胃外或非肠胃外施用的多种方式。
[0210]
优选地,所述配制用于肠胃外或非肠胃外施用的多种方式,其包括通过气雾吸入、注射、输注、摄取、输血、植入或移植。
[0211]
更优选地,所述注射,其是通过皮内或皮下注射施用于受试者。
[0212]
更优选地,所述注射,其是直接注射到肿瘤、淋巴结、组织、器官或 感染部位。
[0213]
优选地,所述配制用于肠胃外或非肠胃外施用的多种方式,其包括通过静脉、鼻内、鞘内或腹膜内经动脉、皮内、皮下、肿瘤内、髓内、结节内、肌内施用于患者。
[0214]
优选地,所述配制用于肠胃外或非肠胃外施用的多种方式,其包括在一天、两天、三天、四天、五天、六天、一周、两周、三周、 一个月、五周、六周、七周、两个月、三个月、四个月、五个月、六个月以上之后重复施用的方式。
[0215]
优选地,所述配制用于肠胃外或非肠胃外施用的多种方式,其包括重复施用或长期施用。
[0216]
更优选地,所述重复施用或长期施用,其可以按相同剂量或不同剂量进行。
[0217]
更优选地,所述重复施用或长期施用,其可以按每周一次,持续6个月以上通过维持疗法施用。
[0218]
进一步的,所述的环形mrna,其可以是细胞在引入后4、5、6、7、8、9、10、11、12、13、
14、15天后的瞬时表达。
[0219]
优选地,所述瞬时表达,其是通过电穿孔将环形mrna转导到细胞中。
[0220]
进一步的,所述的环形mrna,其可以是通过本领域已知的脂质转染方法将环形mrna引入细胞。
[0221]
进一步的,所述的环形mrna,可以与其他已知试剂和疗法组合使用。
[0222]
优选地,所述组合使用是指在受试者的治疗过程中向受试者提供两种(或多种)不同的治疗。
[0223]
更优选地,所述提供两种(或多种)不同的治疗是指在受试者被诊断出患有疾病之后和在疾病治愈或消除或由于其他原因停止治疗之前提供两种以上治疗。
[0224]
更优选地,所述提供两种(或多种)不同的治疗是指当受试者开始第二种治疗时,一种治疗仍在进行中,在施用方面存在重叠。
[0225]
更优选地,所述提供两种(或多种)不同的治疗是指一种治疗的递送在另一种治疗的递送开始之前结束。
[0226]
更优选地,所述提供两种(或多种)不同的治疗是指在由于联合施用,治疗更有效。
[0227]
更优选地,所述提供两种(或多种)不同的治疗是指在较少的第二次治疗就可以看到相同的效果。
[0228]
更优选地,所述提供两种(或多种)不同的治疗是指在在较少第一治疗的情况下进行第二治疗。
[0229]
更优选地,所述提供两种(或多种)不同的治疗是指在第一治疗中观察到类似治疗效果,通过第二次治疗在更大程度上减轻了症状。
[0230]
更优选地,所述提供两种(或多种)不同的治疗是指在递送使得症状或与病症关的其他参数的减少大于在不存在另一种治疗的情况下递送一种治疗所观察到的减少。
[0231]
更优选地,所述提供两种(或多种)不同的治疗是指在两种治疗的效果可以是部分累加、完全累加或大于累加。
[0232]
更优选地,所述提供两种(或多种)不同的治疗是指在递送可以使得当递送第二种治疗 时仍然可以检测到递送的第一种治疗的效果。
[0233]
更优选地,所述提供两种(或多种)不同的治疗是指在本发明所述的药物组合物可以与外科手术、放射、化学疗法、抗体或 其他试剂组合用于治疗方案。
[0234]
更进一步地,所述的药物组合物施用的方式,其包含将组合物配制成用于输注或静脉内施用的方式。
[0235]
进一步的,所述的药物组合物, 其可以作为无菌液体制剂施用。
[0236]
更进一步地,所述的无菌液体制剂,其包含等渗水溶液、乳液、悬浮液、分散体或粘性组合物,其可以缓冲至所需的ph。
[0237]
进一步的,所述的药物组合物, 其可以是以口服给药制剂的方式施用。
[0238]
更进一步地,所述的口服给药制剂,其包括液体溶液、胶囊剂、小药囊、片剂、锭剂和糖锭,在合适的液体中的液体悬浮液粉末和乳剂。
[0239]
进一步的,所述的药物组合物,在前述实施例所述实施方式的背景下所采用的递送系统,其包括时释性、缓释性和持续释放的递送系统,使得组合物的递送发生在待治疗的部位敏化之前,并且有足够的时间引起敏化。
[0240]
更进一步地,所述的释放的递送系统,其包括基于聚合物的系统。
[0241]
优选地,所述聚合物为聚丙交酯-乙交酯、共聚草酸酯、聚酯酰胺、聚原酸酯、聚己内酯、聚羟基丁酸和聚酸酐中的一种或二种或多种混合物。
[0242]
更进一步地,所述的释放的递送系统,其包括基于非聚合物的系统。
[0243]
优选地,所述的非聚合物,其是脂质。
[0244]
更优选地,所述脂质,其是固醇,如胆固醇、胆固醇酯。
[0245]
更优选地,所述脂质,其是脂肪酸或中性脂肪,如单甘油二酯和甘油三酯。
[0246]
更进一步地,所述释放的递送系统,其是硅橡胶系统。
[0247]
更进一步地,所述释放的递送系统,其是基于肽的系统。
[0248]
更进一步地,所述释放的递送系统,其是水凝胶释放系统。
[0249]
更进一步地,所述释放的递送系统,其是蜡涂层。
[0250]
更进一步地,所述释放的递送系统,其是使用常规粘合剂和赋形剂的压片。
[0251]
更进一步地,所述释放的递送系统,其是部分融合的植入物。
[0252]
更进一步地,所述释放的递送系统,其是脂质纳米颗粒。
[0253]
更进一步地,所述释放的递送系统,其是前面疫苗实施例所述的递送系统。
[0254]
第七部分,开发平台应用库的技术方案之三。
[0255]
本发明实施例通过开发平台的应用库还提供了一种制备蛋白的方法,其包含有本发明前述实施例所述的病毒感染所致癌症的肿瘤新生抗原多肽表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的重组核酸分子表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的重组表达载体表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的线性mrna表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的环形mrna表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的重组宿主细胞表达目标蛋白的过程。
[0256]
第八部分,开发平台应用库的技术方案之四。
[0257]
本发明实施例通过开发平台的应用库还提供了一种治疗病毒感染所致癌症的方法,其包括向受试者施用本发明前述实施例所述的病毒感染所致癌症的肿瘤新生抗原多肽。
[0258]
进一步的,所述治疗病毒感染所致癌症的方法,其包括向受试者施用本发明前述实施例所述的重组核酸分子。
[0259]
进一步的,所述治疗病毒感染所致癌症的方法,其包括向受试者施用本发明前述实施例所述的重组表达载体。
[0260]
进一步的,所述治疗病毒感染所致癌症的方法,其包括向受试者施用本发明前述实施例所述的线性mrna。
[0261]
进一步的,所述治疗病毒感染所致癌症的方法,其包括向受试者施用本发明前述
实施例所述的环形mrna。
[0262]
进一步的,所述治疗病毒感染所致癌症的方法,其包括向受试者施用本发明前述实施例所述的重组宿主细胞。
[0263]
进一步的,所述治疗病毒感染所致癌症的癌包括实体瘤和非实体瘤,包括但不限于:鼻腔及鼻窦恶性肿瘤、鼻咽癌、口腔癌、喉癌、涎腺肿瘤、颅内肿瘤、甲状腺癌、舌癌、肺癌、食管癌、贲门癌、乳腺癌、纵膈肿瘤、胃癌、大肠癌、乙状结肠和直肠癌、肝癌、胰腺癌与壶腹周围癌、胆道癌、小肠恶性肿瘤、肾癌、前列腺癌、膀胱癌、睾丸恶性肿瘤、阴茎癌、子宫颈癌、子宫内膜癌、卵巢癌、恶性纤维组织细胞瘤、横纹肌肉瘤、滑膜肉瘤、皮肤恶性黑色素瘤、骨肉瘤、尤文氏肉瘤、淋巴瘤、伯基特淋巴瘤、成人t细胞白血病-淋巴瘤、卡西波肉瘤、多发性骨髓瘤、白血病等。
[0264]
第九部分,开发平台应用库的技术方案之五。
[0265]
本发明实施例通过开发平台的应用库还提供了一种试剂盒,其包含下列任一组或多组之间合理的组合物:s1. 本发明前述实施例所述的病毒感染所致癌症的肿瘤新生抗原多肽;s2.本发明前述实施例所述的重组核酸分子;s3. 本发明前述实施例所述的重组表达载体;s4. 本发明前述实施例所述的线性mrna;s5. 本发明前述实施例所述的环形mrna;s6. 本发明前述实施例所述的重组宿主细胞。
[0266]
进一步的,所述试剂盒的应用,其包括在制备诊断或治疗与病毒感染相关疾病的产品中的应用,或者在t细胞标记、检测、细胞分选或活化中的应用。
[0267]
进一步的,所述的与病毒感染相关疾病,包括但不限于:传染性单核细胞增多症、连锁淋巴细胞增生综合症、病毒性嗜血细胞综合症、口腔毛状黏膜白斑病、病毒性脑膜炎、周围神经炎、病毒性肺炎、病毒性心肌炎、鼻咽癌、霍奇金淋巴瘤、伯基特淋巴瘤、胃炎、胃溃疡、十二指溃疡、胃粘膜相关淋巴组织淋巴瘤、胃癌、肝细胞癌、淋巴上皮样肉瘤、唾液腺肿瘤、乳腺癌、胸腺 瘤、原发性渗出性淋巴瘤或b/t/nk细胞淋巴瘤。
[0268]
本发明的积极效果如下:本发明提供了一种用于病毒感染所致癌症的环形mrna疫苗开发平台。所述开发平台具有病毒感染所致癌症的肿瘤新生抗原决定簇辨寻,肿瘤新生抗原多肽制备,肿瘤新生抗原多肽t细胞激活验证,能表达病毒感染所致癌症的肿瘤新生抗原的环形mrna制备及其在t细胞中表达病毒感染所致癌症的肿瘤新生抗原的验证功能,能有效地提供环形mrna疫苗,有助于病毒感染所致癌症的治疗,为病患和社会带来病毒感染所致癌症的免疫治疗收益。此外,本发明提供的环形mrna疫苗能激活不同患者体内病毒感染所致癌症的抗肿瘤免疫反应,而且能在引起不同患者的不同肿瘤的免疫应答反应,在保证有效性的同时具有更广泛的适用性,克服了现有技术疫苗的治疗需要根据不同个体进行单独的测序及个体化的疫苗制作缺陷,大大降低了肿瘤患者的治疗成本和用药等待周期。
附图说明
[0269]
图1为用于病毒感染所致癌症的环形mrna疫苗开发平台示意图。
[0270]
图2为实施例13中克级规模环形mrna在分离纯化后的沉淀照片和干燥后的样品照片。
[0271]
图3为实施例21中环形mrna纯度检测的色谱图谱。
[0272]
图4为实施例29中环形mrna /靶向递送系统组合成的环形mrna疫苗的电子显微镜照片。
具体实施方式
[0273]
鉴于现有技术中的不足,本案发明人在长期研究和大量实践的基础上,得以提出本发明的技术方案。
[0274]
如在权利要求和说明书中所使用的,词语
ꢀ“
具有”、“包括”或“含有”是指包括在内的或开放式的,并不排除额外的、未引述的元件或方法步骤。
[0275]
如在权利要求和说明书中所使用的,术语“多肽”在本发明中为氨基酸聚合物。所述氨基酸聚合物可以是线性或分支的。进一步地,所述氨基酸聚合物包括修饰的氨基酸或由非氨基酸隔断的形式。更进一步地,所述氨基酸聚合物包括二硫键形成、糖基化、脂质化、乙酰化、磷酸化或以标记组分缀合的氨基酸聚合物。
[0276]
如本发明所使用的,术语“环形mrna”是指一种呈封闭状的环形mrna分子, 具有蛋白翻译活性。所述环形mrna分子主要由外显子、ires元件、蛋白编码区和间隔区构成。
[0277]
如本发明所使用的,术语“线性mrna”是指能够环化形成环形mrna的mrna前体, 具有蛋白翻译活性。所述线性mrna一般由线性的dna分子转录形成。
[0278]
如本发明所使用的,术语“取代、重复、缺失或添加一个或多个氨基酸”中“取代”是指用不同氨基酸置换占用一个位置的核苷酸或氨基酸,“缺失”是指去除占据某一位置的氨基酸,“插入”是指在邻接并且紧随占据位置的氨基酸之后添加氨基酸。
[0279]
如本发明所使用的,术语“保守突变”是指可正常维持蛋白质的功能的保守突变。所述保守突变包括保守置换。所述保守置换是指在置换部位为芳香族氨基酸的情况下,在 phe、trp、tyr间相互置换的突变;在置换部位为疏水性氨基酸的情况下,在leu、ile、val间 相互置换的突变;在为极性氨基酸的情况下,在gln、asn间相互置换的突变;在为碱性氨基酸的情况下,在lys、arg、his间相互置换的突变;在为酸性氨基酸的情况下,在asp、glu间相 互置换的突变;在为具有羟基的氨基酸的情况下,在ser、thr间相互置换的突变;其他被视作保守置换的置换, ala向ser或thr的置换、arg向gln、his或lys的置 换、asn向glu、gln、lys、his或asp的置换、asp向asn、glu或gln的置换、cys向ser或ala的置 换、gln向asn、glu、lys、his、asp或arg的置换、glu向gly、asn、gln、lys或asp的置换、gly向 pro的置换、his向asn、lys、gln、arg或tyr的置换、ile向leu、met、val或phe的置换、leu向 ile、met、val或phe的置换、lys向asn、glu、gln、his或arg的置换、met向ile、leu、val或phe 的置换、phe向trp、tyr、met、ile或leu的置换、ser向thr或ala的置换、thr向ser或ala的置 换、trp向phe或tyr的置换、tyr向his、phe或trp的置换、及val向met、ile或leu的置换。进一步地,所述保守突变涵盖因于基因所来源的个体差异、株、种的差异等天然产生的突变。
[0280]
如本发明所使用的,术语“序列同一性”和“同一性百分比”是指两个或更多个多核苷酸或多肽之间相同(即同一)的核苷酸或氨基酸的百分比。
[0281]
如本发明所使用的,术语“反向互补序列”是指和原始多核苷酸序列方向相反但互
补的序列。
[0282]
如本发明所使用的,术语“重组核酸分子”是指具有在自然界中不连接在一起的序列的多核苷酸。所述多核苷酸是指能够通过标准分子生物学方法识别、操纵以及恢复序列及其组分核苷酸序列,从至少在数量或浓度上分离一次的核苷酸序列衍生而来的。作为由核苷酸组成的聚合物,多核苷酸的形式可以是单独片段,也可以是更大的核苷酸序列结构的一个组成部分。多核苷酸包括dna、rna、cdna序列、表达载体、多顺反子序列重组多核苷酸和重组核酸分子。所述重组核酸分子可以通过合适的载体转化为合适的重组宿主细胞,表达产生重组多肽、重组蛋白、融合蛋白等。重组核酸分子包含编码目标多肽的编码区,和连接于所述编码区上游的ires元件。
[0283]
如在本发明所使用的,术语“重组表达载体”是指用于表达例如编码所需多肽的多核苷酸的dna结构。所述载体是指dna构建体。所述dna构建体含有合适的控制序列。所述控制序列可操作地连接dna序列,实现在合适的宿主中表达目的基因。所述重组表达载体包括启动子和增强子等对基因表达具有调控作用的遗传元素的集合、能转录成mrna并翻译成蛋白质的结构或编码序列、能适当的转录和翻译起始和终止序列的转录亚单位。所述重组表达载体可以以任何合适的方式构建。在本发明中,重组表达载体的载体可以包括质粒、病毒、噬菌体和转座子在内的任何载体。
[0284]
如在本发明所使用的,术语“抗原”是指引发免疫应答的分子。所述免疫应答是指可能涉及抗体产生和或特异性免疫细胞的活化。所述抗原包括基本上所有的蛋白质或肽在内的任何大分子。
[0285]
如在本发明所使用的,术语“宿主细胞”是指易于用包含本发明的重组核酸分子、环形mrna或重组表达载体转化、转染、转导等的任何细胞类型。
[0286]
如在本发明所使用的,术语“重组宿主细胞”是指通过转化来实现的宿主细胞,包括导入重组核酸分子、环形mrna或重组表达载体后不同于亲本细胞的宿主细胞。所述宿主细胞包括原核细胞或真核细胞,或能够导入本公开的重组核酸分子、环形mrna或重组表达载体的细胞。
[0287]
如在本发明所使用的,术语“转化、转染、转导”是指将外源性的dna导入宿主的过程。所述转化、转染、转导的方法包括任何将核酸导入细胞的方法。所述方法包括但不限于电穿孔法、磷酸钙(capo4 )沉淀法、氯化钙(cacl2 )沉淀法、微注射。法、聚乙二醇(peg)法、deae葡聚糖法、靶向递送系统法以及乙酸锂dmso法。
[0288]
如在本发明所使用的,术语“治疗”是指在罹患疾病之后,使受试者在接触本发明的菌株和/或巨噬细胞或者含有其的药物组合物后,相比受试者不接触时,使受试者的该疾病症状减轻,但并不意味着必需完全抑制受试者该疾病的症状。
[0289]
如在本发明所使用的,术语“预防”是指在罹患疾病之前,通过使受试者接触本发明的药物组合物,从而与受试者不接触时相比减轻罹患疾病后的症状,并不意味着必需完全抑制受试者患病。
[0290]
如在本发明所使用的,术语“个体”、“患者”或“受试者”包括哺乳动物。所述哺乳动物包括但不限于,家养动物,灵长类动物,兔,以及啮齿类动物。
[0291]
如在本发明所使用的,术语“小鼠”是指免疫缺陷小鼠。
[0292]
如在本发明所使用的,术语“高严格条件”是指根据标准dna印迹程序,对于长度为
(也称作离子键合)来发生复合。
[0300]
术语“前体药物”是指在适用于患者后能够直接或间接地提供本发明的化合物的衍生化合物。特别优选的衍生化合物或前药是在施用于患者时可以提高本发明的化合物的生物利用度的化合物(例如,更易吸收入血),或者促进母体化合物向作用位点(例如,淋巴系统)递送的化合物。除非另外指出,本发明的化合物的所有前药形式都在本发明的范围之内,且各种前药形式是本领域熟知的。
[0301]
术语“各自独立地”是指结构中存在的取值范围相同或相近的至少两个基团(或环系)可以在特定情形下具有相同或不同的含义。例如,取代基x和取代基y各自独立地为氢、卤素、羟基、氰基、烷基或芳基,则当取代基x为氢时,取代基y既可以为氢,也可以为卤素、羟基、氰基、烷基或芳基;同理,当取代基y为氢时,取代基x既可以为氢,也可以为卤素、羟基、氰基、烷基或芳基。
[0302]
术语“任选”或“任选地”是指随后描述的事件或情况可能发生或可能不发生,该描述包括发生所述事件或情况和不发生所述事件或情况。
[0303]
术语“包含”和“包括”以其开放、非限制性意义使用。
[0304]
术语“递送率”是指递送到目标癌细胞中的环形mrna占到所递送的环形mrna的百分比。
[0305]
下面的实施例是对本发明的技术方案、其实施过程及原理等作进一步的解释说明。
[0306]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0307]
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0308]
本发明提供了一种病毒感染所致癌症环形mrna疫苗的开发平台(图1)。利用该开发平台的肿瘤新生抗原库,rna元件结构库,cmc工艺库,递送系统库及应用库可以有效地研发和制备用于治疗病毒感染所致癌症的环形mrna疫苗。实施例1所述的病毒感染所致癌症的抗原多肽是通过本发明所提供的开发平台的计算机辅助辨寻获得。同时,通过本发明所提供的开发平台的制备方法合成得到与实施例1特征多肽系列等价的多肽序列和能表达实施例1所述抗原多肽蛋白的环形mrna,并进行合成物质的鉴定、t细胞激活和293t细胞中的病毒感染所致癌症的肿瘤新生抗原蛋白表达性能验证。通过本发明所述的开发平台确定了病毒感染所致癌症的抗原决定簇在组织中存在表达。与此同时,检测表明编码了部分或全部毒感染所致癌症的抗原多肽的环形mrna能激活抗肿瘤免疫反应。本发明所提供的这些病毒感染所致癌症的肿瘤新生抗原多肽其可视为开发平台开发治疗病毒感染所致癌症的环形mrna疫苗的抗原决定簇。
[0309]
本发明中所述一种病毒感染所致癌症环形mrna疫苗的开发平台通过下述技术方案来实现病毒感染所致癌症环形mrna疫苗的开发。所述的技术方案中的实验包括但不限于以下实验:所述的检测环形mrna在细胞或者组织中是否存在表达可采用现有技术进行,方法包括:环形mrna高通量测序和荧光定量pcr;所述的检测环形mrna可激活免疫反应,方法包括:酶联免疫斑点实验(elispot),流式实验检测cd8t细胞因子,五聚体检测抗原特异性t细胞;所述的检测环形mrna可激活抗肿瘤免疫反应的方法包括:建立移植瘤动物模型后给动物接种circrna检测肿瘤生长情况;切片和流式观察动物肿瘤中的免疫反应情况。
[0310] 实施例1肿瘤新生抗原库中病毒感染所致癌症的肿瘤新生抗原决定簇辨寻流程
第一步,获取符合医学伦理要求的通过illumina高通量测序平台所获得病毒感染所致癌症的不同患者的二代基因测序数据;第二步,对病毒感染所致癌症的不同肿瘤的原始测序数据通过软件进行质量控制,过滤低质量数据。得到质控过滤后的干净数据;第三步,将质控之后病毒感染所致癌症的不同肿瘤的reads使用相关软件比对到人类参考基因组hg19上,生成bam格式的比对文件;第四步,将获得病毒感染所致癌症的不同肿瘤的bam格式的比对文件进行进一步处理,去除重复数据、局部重新比对和碱基质量校正分析,得到过滤之后的bam文件第五步,综合分析病毒感染所致癌症的不同肿瘤患者的体细胞突变,得到包含多个突变的vcf文件;第六步,使用vep对检测得到的突变进行多种数据库的注释,其中包括基因注释;第七步,根据所获得的体细胞突变信息,进行突变位点表位肽提取,并且相对应的提取正常野生型基因型的表位肽序列。表位肽提取使用滑窗模式,设置氨基酸长度,在突变位点上下游位置进行逐步滑窗提取包含突变氨基酸的表位肽序列,滑窗的步移长度为1;第八步,对病毒感染所致癌症的不同肿瘤的肿瘤患者的基因突变进行mhci和mhcii分子类型鉴定;第九步,对所获得的表位肽序列和hla类型,使用软件对突变表位肽亲和力进行预测;第十步,利用打分函数计算预测的病毒感染所致癌症的不同肿瘤新生抗原总得分值,对高亲和力突变表位肽进行排序,得分值大小和新生抗原可信度成呈正相关关系;第十一步,综合分析病毒感染所致癌症的不同肿瘤的新生抗原多肽可信度和出现几率,构建病毒感染所致癌症的具有seqid1-seqid61所示氨基酸序列的肿瘤新生抗原决定簇。
[0311] 实施例2肿瘤新生抗原库中病毒感染所致癌症的肿瘤新生抗原多肽的制备流程实施例2中通过开发平台合成得到的与实施例1特征多肽系列等价的病毒感染所致癌症的肿瘤新生抗原多肽采用以下方法进行制备,其具体步骤如下:第一步,使用1-5克胺树脂作为不溶性的固相载体,将其投入多肽合成仪中,并启动多肽合成仪;第二步,将一个氨基被封闭基团保护的氨基酸溶液通过蠕动泵加入到反应器中,泵速为5-10毫升每分钟,使得一个氨基被封闭基团保护的氨基酸与固相载体进行共价连接。随后,加入相应的溶剂反应脱掉氨基的保护基,将第一个氨基酸接到了固相载体上;第三步,在第二步反应完成后,通过蠕动泵把合成多肽相应序列的氨基酸溶液注入多肽合成仪,泵速为2-5毫升每分钟,并吹入氮气鼓泡混合,合成过程中采用低温等离子体辐射,功率为50瓦,提高合成速度;固相载体上的氨基酸中的氨基被封闭的第二个氨基酸的羧基通过活化反应被活化,羧基被活化的第二个氨基酸再与已接在固相载体的第一个氨基酸的氨基反应形成肽键,使得固相载体上生成一个带有保护基的氨基酸肽链;第四步,通过合成仪废液排放系统将第三步完成后的废液排出;第五步,通过合成仪的清洗管路对第四步反应后的合成半成品加入溶剂进行清洗,清洗后将废液排出;
第六步,往第五步清洗完成后的合成仪通过蠕动泵加入相关溶剂脱掉多肽合成物上氨基的保护基,泵速为10-15毫升每分钟,然后进行废液排放,在废液排放完成后进一步加入溶剂进行清洗操作;第七步,往第六步清洗完成后的合成仪通过蠕动泵加入合成多肽对应序列的合成氨基酸,设置加入量及合成温度,启动自动合成,在反应结束后,将废液排出;第八步,循环重复第四步到第七步的步骤,使肽链从c端向n端生长,直至达到所需要的肽;第九步,在第八步的工作结束后,往合成仪中通过蠕动泵加入相应的溶剂反应脱去保护基并水解肽链和固相载体之间的酯键,泵速为5-10毫升每分钟,在进行多次清洗后,获得所需要的多肽粗品; 第十步,对第九步所获得的多肽粗品通过制备色谱进行进一步纯化,并通过质谱对合成的具有seqid1-seqid61所示氨基酸序列的肿瘤新生抗原多肽进行鉴定,然后对所获得多肽溶液进行冻干保存。
[0312]
实施例3肿瘤新生抗原库中肿瘤新生抗原多肽的抗体滴度反应和在肿瘤组织中的表达验证流程实施例2合成的肿瘤新生抗原多肽的激活免疫反应潜力通过抗体滴度反应来验证,其具体步骤如下:第一步,将实施例2合成的分别具有seqid1-seqid61所示氨基酸序列的肿瘤新生抗原多肽用dmso溶解成,再用包被液稀释,每孔加100微升,加盖4摄氏度放置过夜。次日倒去孔内的液体,将板子扣放在滤纸上,除净液体;第二步,在第一步完成后的每孔加200微升/孔封闭液,室温放置1小时。倒去孔内液体,用洗涤液洗涤3次,将扳子扣放在滤纸上,除净液体;第三步,将待测血清分别稀释成1:1000、1:10000、1:100000的浓度,各100微升加至第二步完成后相应的凹孔中。同时以100微升稀释液作为空白对照。加盖,置37摄氏度孵育2小时。倒去孔内液体,用洗涤液洗涤5次;第四步,用抗体稀释液把相应的酶标抗体稀释成1:30000,100微升/孔加至第三步完成后相应的凹孔中,加盖37摄氏度孵育1小时。倒去孔内液体,用洗涤液洗涤5次;第五步,在第四步完成后的每孔加80微升tmb显色液;显色10分钟后,每孔加80微升0.16摩尔每升硫酸。通过酶标仪在450纳米进行测板。证明实施例2合成的分别具有seqid1-seqid61所示氨基酸序列的肿瘤新生抗原多肽具有激活免疫反应的潜力;第六步,制作与实施例2合成的分别具有seqid1-seqid61所示氨基酸序列的肿瘤新生抗原多肽的结合的抗体,并通过westernblot验证了这些多肽的确能够在肿瘤组织中被表达。
[0313]
实施例4rna结构元件库中能编码肿瘤新生抗原多肽的环形mrna分子构建和验证流程第一步,通过病毒感染所致癌症的环形mrna疫苗开发平台rna结构元件库,设计能编码实施例2合成的具有seqid1-seqid61所示氨基酸序列的肿瘤新生抗原多肽的环形mrna的具有seqid62-seqid65所示核苷酸序列的ires序列;第二步,构建fluc-ires-rluc双荧光素酶标记的质粒,并在质粒内分别克隆空载、
野生型ires序列和突变型ires序列。通过双荧光素酶报告实验,证明了所设计的具有seqid62-seqid65所示氨基酸序列的ires序列活性强,具有编码肿瘤新生抗原多肽的潜力;第三步,同时从病毒感染所致癌症的环形mrna疫苗开发平台的rna结构元件库根据翻译效率和环化效率的大小匹配:具有seqid66-68所示核苷酸序列的第二外显子,具有seqid69-71所示核苷酸序列的5
´
间隔区,具有seqid72-74所示核苷酸序列的3
´
间隔区,具有seqid75-77所示核苷酸序列的第一外显子;第四步,制备flag标记的第三步所设计优化的环形mrna的质粒及空载质粒,并将质粒通过lipo3000转染至小时ek293t细胞内,48小时后提取细胞中的总蛋白,通过westernblot实验,验证flag所标记的蛋白;第五步,转染同样的质粒后,通过westernbolt实验得到电泳胶,通过考马斯亮蓝染色后,将相应位置的胶条进行质谱检测。通过分析质谱检测的结果,验证了第二步所设计优化的环形mrna的确能够编码具有seqid1-seqid61所示氨基酸序列的肿瘤新生抗原多肽。
[0314] 实施例5cmc工艺库中环形mrna的克级规模合成工艺优化流程第一步,从病毒感染所致癌症环形mrna疫苗开发平台的cmc工艺库根据成本核算、制备、分离纯化的难易匹配设计环形mrna的克级规模合成工艺路线;第二步,从病毒感染所致癌症的环形mrna疫苗开发平台的rna结构元件库根据环化效率的大小设计环形mrna的线性前驱物:实施例1中具有seqid1-61所示氨基酸序列的抗原多肽编码区,具有seqid62-seqid65所示核苷酸序列的ires,具有seqid66-68所示核苷酸序列的第二外显子,具有seqid69-71所示核苷酸序列的5
´
间隔区,具有seqid72-74所示核苷酸序列的3
´
间隔区,具有seqid75-77所示核苷酸序列的第一外显子;具有seqid78-80核苷酸序列的的启动子,具有seqid81-83核苷酸序列的5
´
同源臂,具有seqid84-86核苷酸序列的3
´
内含子,具有seqid87-89所示核苷酸序列的5
´
内含子,具有seqid90-92所示核苷酸序列的3
´
同源臂,以及可用于具有seqid93-95所示核苷酸序列的质粒线性化的酶切位点;第三步,合成第二步所设计环形mrna线性前驱物的基因片段,并连接到病毒感染所致癌症环形mrna疫苗开发平台cmc工艺库匹配的载体;第四步,将合成的穿刺菌在37摄氏度,适当转速下活化一定时间,取活化菌液在37摄氏度,适当转速下培养过夜。然后,抽提质粒,测定od值和质粒提取浓度;第五步,将一定量质粒,酶(1000units),10
×
cutsmartbuffer和若干体积无酶水混合,并将混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度;第六步,将一定量质粒模板,10
×
reactionbuffer,2浓度为20毫摩尔每升的atp,浓度为20毫摩尔每升的ctp,浓度为20毫摩尔每升的utp,浓度为20毫摩尔每升的gtp,t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育一定时间,然后加入dnasei在37摄氏度消化一定时间。将所获得的混合物进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测
定mrna转录纯化后rna浓度;第七步,将一定量mrna溶液中,20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的混合溶液在55摄氏度加热一定时间。将所获得的混合物进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定;测定mrna环化效率和纯度; 第八步,将一定量环化或未环化的mrna溶液,rtprmier,primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的混合液在37摄氏度加热一定时间,随后在85摄氏度加热一定时间,最后在4摄氏度保存;第九步,将第八步所获得的一定体积逆转录溶液与适量10
×
buffer,dntp,10微摩尔每升primer-f,10微摩尔每升primer-r,taq酶,无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置一定时间,60摄氏度放置一定时间,72摄氏度放置一定时间(循环次数为40次),4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,证明已成功制备所设计的目的环形mrna。
[0315] 实施例6递送系统库中环形mrna的递送系统工艺优化流程第一步,从病毒感染所致癌症环形mrna疫苗开发平台的递送系统库根据成本核算,制备,靶向递送效率匹配设计环形mrna的递送系统配方;第二步,以病毒感染所致癌症的单一癌细胞或两种及多种癌细胞为靶标进行特异性核酸适配体筛选:首先将原始文库或次级文库在100摄氏度变性5分钟,立即冰浴10分钟。加入筛选缓冲液各组分,最终孵育体系内含有:1
×
pbs、1毫摩尔每升二氯化镁、0.1克每升的酵母trna、0.1克每升鲑鱼精dna和所需要的ssdna原始文库或次级文库。随后将预先处理好的ssdna原始文库或次级文库加入接种于96孔板的靶细胞,如果为混合靶细胞,每隔混合靶细胞数目的轮次更换靶细胞,将混合靶细胞中的靶细胞交替使用,4摄氏度孵育30分钟;吸弃上清,用缓冲液(1
×
pbs、1毫摩尔每升二氯化镁)轻柔洗涤3-4次;再加入双蒸水洗脱;以洗脱液为模板,进行不等长pcr扩增。完成4轮筛选之后,从第5轮引入消减筛选,在与靶细胞进行筛选之前将孵育体系与消减细胞在4摄氏度孵育20分钟,除去与消减细胞结合的适配体,将其余未结合的文库与靶细胞孵育,进行第5轮筛选;第三步,进行pcr扩增条件优化。pcr体系总体积为100微升,其中内含:10
×
pcr反应缓冲液10微升,dntp混合物(每种核苷酸2.5毫摩尔每升)8微升,25微摩尔每升的上游引物和下游引物各1微升,taqdna聚合酶(2.5
×
10
6
u每升)1微升。首先加入2-3微升洗脱液作为模板,设置pcr反应条件为:94摄氏度、30秒,45摄氏度、30秒,58摄氏度、30秒。从12个循环至40个循环,每隔3个循环取10微升产物,应用含7摩尔每升尿素的聚丙烯酰胺凝胶进行电泳,选取非特异性扩增少、产物量刚达到平台期的循环数作为大量扩增的条件;第四步,ssdna次级文库的制备应用7摩尔每升尿素的聚丙烯酰胺凝胶进行电泳,然后将正确大小的ssdna条带切胶,置于1.5毫升的ep管中。每管加入400微升的凝胶洗脱液(其内含有0.5摩尔每升乙酸铵,2克每升sds,1毫摩尔每升edta,37摄氏度摇床上洗脱6小时以上。将凝胶洗脱液转移到新管,加入1摩尔每升二氯化镁24微升、3摩尔每升醋酸钠(ph值5.2)40微升和预冷的无水乙醇1毫升,混匀,于-70摄氏度沉淀过夜。4摄氏度、12000rpm
离心30分钟,弃上清; 用预冷的体积分数0.75乙醇1毫升洗涤,4摄氏度、12000rpm离心30分钟;弃上清,自然晾干。双蒸水逐管润洗回收ssdna(回收的 ssdna 使用nanodrop进行质量鉴定后投入次轮筛选);第五步,克隆与测序: 将经过10轮筛选的适配体文库扩增为双链后连接t载体,转化至 感受态ecoli dh5a中,随机挑取60个适配体进行克隆测序。运用rnastructure软件进行二级结构绘图并且通过meme软件进行结构同源性分析。选择具有代表性的序列并进行fam标记,与靶标孵育后通过流式细胞术鉴定序列对靶标进行识别,最终筛选获得适配体序列;第六步,流式细胞术鉴定富集文库 将癌细胞用deme洗去死细胞,胰酶消化,离心,筛选洗涤缓冲液重悬,然后分别与用fam标记的引物扩增获得fam标记的第6轮、8轮、10轮文库,4摄氏度避光孵育10分钟,离心后加入200微升筛选洗涤缓冲液重悬。流式细胞仪检测不同轮数文库中可以结合靶标的适配体富集程度;第七步,适配体与筛选细胞结合鉴定。应用流式细胞术验证获得的适配体与筛选细胞(靶细胞与消减细胞)的结合情况。同时以fam标记的原适配体库作为对照,分别比较获得的 适配体、原适配体与不同细胞结合的荧光强度;第八步,所获得的核酸适配体的特异性检验核酸适配体。将获得的适配体分别与多种不同细胞系孵育后,用流式细胞术检测结合情况。同时用非靶细胞的人源癌细胞进行特异性鉴定;第九步,从病毒感染所致癌症的环形mrna疫苗开发平台的递送系统库根据环形mrna的递送系统配方匹配递送系统工艺路线;第十步,制备实施例5所合成的环形mrna疫苗。
[0316]
实施例7应用库中能编码肿瘤新生抗原多肽的环形mrna分子作为疫苗激活免疫反应验证流程第一步,将实施例6中采用脂质体配方工艺所制备的环形mrna疫苗,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板 内,每孔5
×
10 5
个细胞。实验过程接受动物管理会的监督;第二步,取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,树突状细胞电转10微克环形mrna进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清 洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联 免疫斑点检测仪观察培养板内inf-r斑点形成情况;第三步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的并电转10微克环形mrna树突状细胞共培养,加入brefeldin a处理,抑制细胞因子分泌。5小时后,收集 处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因 子产生情况;第四步,同样处理后的小鼠,在最后一次注射后第9天取脾细胞1
×
106个,进行细
胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的比例。
[0317]
实施例8应用库中能编码肿瘤新生抗原多肽的环形mrna分子激活t细胞抗肿瘤免疫验证流程第一步,取6-8 周的体重20克的小鼠,将适量病毒阳性且具成瘤能力的癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行器官移植,建立相应原位癌小鼠模型;第二步,将小鼠分为6组,每个组别设置3只,分别进行以下处理:第一组,空白对照组不进行任何处理;第二组进行靶向递送系统皮下注射;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和环形mrna。于第三天、第六天、 第九天和第十二天后,处死小鼠并在无菌条件下取出小鼠相应器官。观察记录小鼠相应器官的肿瘤生长情况;第三步,将肿瘤组织制作成切片或者细胞悬液并观察t细胞浸润情况。
[0318]
实施例9应用库中能编码肿瘤新生抗原多肽的环形mrna分子激活t细胞抗肿瘤免疫验证流程第一步,取6-8 周的体重20克的免疫缺陷小鼠,将7.5
×
10 4
个病毒阳性且具成瘤能力的癌细胞注射入小鼠腹腔,建立腹腔多脏器成瘤小鼠模型;第二步,将小鼠分为6组,每个组别设置3只,分别进行以下处理:第一组,空白对照组不进行任何处理;第二 组进行靶向递送系统皮下注射;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和环形mrna。于第三天、第六天、 第九天和第十二天后,处死小鼠并在无菌条件下取出小鼠腹腔所有器官。观察记录小鼠的腹腔多脏器肿瘤生长情况;第三步,将肿瘤组织制作成切片或者细胞悬液并观察t细胞浸润情况。
[0319]
实施例10应用库中能编码肿瘤新生抗原多肽的环形mrna分子激活人类t细胞的验证流程第一步,分离出病毒阳性癌症患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf (粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;第二步,6天后加入pbmc电转编码肿瘤新生抗原多肽的环形mrna分子,与培养后的t细胞按1:4的比例(个数/个数),每3天加入新的电转编码肿瘤新生抗原多肽的环形mrna分子的pbmc;第三步,在加入新的电转编码肿瘤新生抗原多肽的环形mrna分子9天后,种入包埋有inf-r抗体的elispot试剂盒的培养板内。24小时后弃去上清,pbs 清洗5次后加入一抗孵育一定时间,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况;第四步,pbmc在加入1%双抗和10%ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf和il-4(20纳克每毫升)诱导;第五步,6天后加入pbmc电转编码肿瘤新生抗原多肽的环形mrna分子,与培养后的t细胞按1:4的比例(个数/个数),每3天加入新的电转编码肿瘤新生抗原多肽的环形mrna分
子的pbmc;第六步,在加入新的电转编码肿瘤新生抗原多肽的环形mrna分子9天后,加入brefeldina处理,抑制细胞因子分泌。24小时后,收集处理过的细胞进行cd3、cd8及cd4分子染色固定后,破膜剂进行破膜,然后分别进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析cd4+、cd8+t细胞的激活情况和细胞因子产生情况。
[0320] 实施例11应用库中环形mrna分子在病毒致癌肿瘤细胞中对所编码肿瘤新生抗原多肽的表达验证流程第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入适量4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠细胞或病毒阳性原位癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液; 第四步,配置ripa裂解液,往1毫升裂解液加适量pmsf(100豪摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5*106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4度离心机,12000rpm离心一定时间;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在病毒阳性原位癌细胞中表达明显高于正常细胞。证明环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在病毒阳性原位肝癌中的表达。
[0321] 实施例12应用库中环形mrna分子在细胞中的分布验证流程第一步,应用trizol试剂提取小鼠细胞和病毒阳性原位癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠细胞和病毒阳性原位癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠细胞和病毒阳性原位癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量;第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在病毒阳性原位癌细胞中高表达,明显高于正常细胞。证明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。
[0322] 实施例13用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,具有seqid1所示氨基酸序列的抗原多肽编码区,具有seqid62所示核苷酸序列的ires,具有seqid66所示核苷酸序列的第二外显子,具有seqid69所示核苷酸序列的5
´
间隔区,具有seqid72所示核苷酸序列的3
´
间隔区,具有seqid75所示核苷酸序列的第一外显子;具有seqid78核苷酸序列的的启动子,具有seqid81核苷酸序列
的5
´
同源臂,具有seqid84核苷酸序列的3
´
内含子,具有seqid87所示核苷酸序列的5
´
内含子,具有seqid90所示核苷酸序列的3
´
同源臂,以及可用于具有seqid93所示核苷酸序列的质粒线性化的酶切位点,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(285.7纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(263.4纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2小时,然后加入dnasei在37摄氏度消化20分钟。将所获得的混合物采用硅膜离心柱法纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(373.1纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热30分钟。将所获得的混合物依次通过采用硅膜离心柱法,液相等离子体法(辐射能量为0.99焦每平方厘米,辐射10秒,机械搅拌,搅拌速度10rpm,液相介质为hepes缓冲液,气体为90%氦气和10%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(287.9纳克每微升),纯度为99.2%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(88.8%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在液相等离子体法技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体法技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量或辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;
第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。图2为实施例13中制备的克级规模环形mrna在分离纯化后的沉淀照片和干燥后的样品照片。
[0323] 实施例14实施例13所制备的环形mrna分子的靶向递送系统配方工艺第一步,将石墨烯状黑磷和脂质产品按照4.2:62.3摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dspc,dotap和dope按照摩尔比例1:27.5:33.3混合得到的脂质混合物;第二步,将氧浓度为84毫克每升的1.2%氯化钠溶液加入第一步中的混合物a中,然后在4摄氏度下结合液相等离子体技术机械搅拌7分钟,得到混合液a,其中液相等离子体电子和离子的功率密度为0.12瓦每平方厘米,机械搅拌的速率为53rpm;第三步,将amino-peg4-alcohol加入第二步获得的混合液a,然后在2摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为0.13瓦每平方厘米,机械搅拌的速率为33rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s3按照摩尔比例2:1混合得到的混合适配体;hbv感染所致的肝癌为10-8和ly-1按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为肽适配子和tls11a按照摩尔比例1:3混合得到的混合适配体;hpv感染所致的宫颈癌为air-3a和a10按照摩尔比例2:1混合得到的混合适配体;ebv感染所致的原位淋巴瘤为td05和gbi-10按照摩尔比例1:1混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为13平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过微流控设备进行进一步混合,微流控的总流速为0.1毫升每分钟,流速比为1/3.5。
[0324]
实施例15实施例13所制备的环形mrna分子作为疫苗激活免疫反应第一步,将实施例13中合成的环形mrna采用实施例14的所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs),所制得样品平均粒径153纳米,表面荷电30.9
±
0.6毫伏。此外,进一步地空白对照实验结果表明实施例13所制备的环形mrna分子采用实施例14所示的靶向递送系统所获得的样品的表面荷电远远高于实施例13所制备的环形mrna分子采用实施例14中所示的未经合理改性的任何一种或多种脂质产品与实施例14所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例14中所示的脂
质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例14所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例14所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10μg rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为1.82
±
0.2%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.33
±
0.3%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.10
±
0.03%、 0.39
±
0.04%、0.65
±
0.05%;tnf-α的量分别依次为0.09
±
0.03%、0.12
±
0.03%、0.47
±
0.07%、0.75
±
0.13%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0325]
实施例16实施例13所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例13中合成的环形mrna采用实施例14所示的所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);所
制得样品平均粒径153纳米,表面荷电30.9
±
0.6毫伏。此外,进一步地空白对照实验结果表明实施例13所制备的环形mrna分子采用实施例14所示的靶向递送系统所获得的样品的表面荷电远远高于实施例13所制备的环形mrna分子采用实施例14中所示的未经合理改性的任何一种或多种脂质产品与实施例14所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例14中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例14所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例14所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为1.95
±
0.5%,远远高于第一组-第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.05
±
0.01%、0.12
±
0.02%、 0.41
±
0.04%、0.70
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0326]
实施例17实施例13所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将7.5
×
104个hbv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行肝移植,建立原位肝癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为46立方毫米,注射第15天后的肿瘤组织大小为90立方毫米,注射第18天后的肿瘤组织大小为159立方毫米,注射第21天后的肿瘤组织大小为252立方毫米,注射第24天后的肿瘤组织大小为427立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的
肿瘤组织大小分别依次为31立方毫米、25立方毫米、20立方毫米、17立方毫米;注射第12天后的肿瘤组织大小分别依次为252立方毫米、209立方毫米、165立方毫米、132立方毫米;注射第15天后的肿瘤组织大小分别依次为567立方毫米、528立方毫米、426立方毫米、303立方毫米;注射第18天后的肿瘤组织大小分别依次为855立方毫米、801立方毫米、706立方毫米、546立方毫米;注射第21天后的肿瘤组织大小分别依次为1174立方毫米、1099立方毫米、991立方毫米、867立方毫米;注射第24天后的肿瘤组织大小分别依次为1460立方毫米、1393立方毫米、1302立方毫米、1184立方毫米)。这说明,实施例13所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例13所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.14
±
0.1%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.11
±
0.05%、0.19
±
0.06%、0.35
±
0.07%)。
[0327]
实施例18实施例13所制备的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4
(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克/毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.04
±
0.1%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.12
±
0.04%、0.20
±
0.07%),仅次于pma阳性对照组(29.88
±
3.67%)。
[0328] 实施例19实施例13所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠肝细胞或hbv阳性原位肝癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hbv阳性原位肝癌
细胞中表达明显高于正常肝细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hbv阳性原位肝癌中的表达。
[0329] 实施例20实施例13所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠肝细胞和hbv阳性原位肝癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
atactcagctatgcgtgaag-3
´
(seqid96)和5
´‑
tatatgcacgtgtagtgctagatcgg-3
´
(seqid97);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hbv阳性原位肝癌细胞中高表达,明显高于正常肝细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例13所制备的环形mrna分子采用实施例14靶向递送系统递送至hbv阳性原位肝癌细胞的递送率(85.8%)远远高于实施例14中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例14所示靶向递送系统对实施例13所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(85.8%)远远高于单独使用实施例14中所示核酸适配体或其他迄今为止文献资料所报道的hbv阳性原位肝癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例14所示靶向递送系统对实施例13所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(85.8%)远远高于用实施例14中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例14中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例13所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例13所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例13所合成的环形mrna的递送能力优良。
[0330]
实施例21用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid5所示氨基酸序列的抗原多肽编码区,具有seqid63所示核苷酸序列的ires,具有seqid67所示核苷酸序列的第二外显子,具有seqid70所示核苷酸序列的5
´
间隔区,具有seqid73所示核苷酸序列的3
´
间隔区,具有seqid76所示核苷酸序列的第一外显子;具有seqid79核苷酸序列的的启动子,具有seqid82核苷酸序列的5
´
同源臂,具有seqid85核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid91所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化2.5小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(312.1纳克每微
升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(255.8纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2小时,然后加入dnasei在36.9摄氏度消化15分钟。将所获得的混合物采用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(419.3纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在54.5摄氏度加热15分钟。将所获得的混合物依次通过采用亲和柱层析法,超声波技术(声强度为100毫瓦每平方厘米,辐射15分钟,机械搅拌,搅拌速度10rpm,液相介质为hepes缓冲液,气体为95%氦气和5%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行进一步鉴定和hplc-质谱联用分析。结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后溶液浓度(275.2纳克每微升),环形mrna纯度97.8%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(89.5%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在,超声波技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在,超声波技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度; 第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,
60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。图3为实施例20中环形mrna纯度检测的色谱图谱。
[0331] 实施例22实施例21所制备的的环形mrna分子的靶向脂质体递送系统工艺优化第一步,将黑磷量子点和脂质产品按照5.5:66.2摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为pope,dotap和dmpe按照摩尔比例1:23.3:32.7混合得到的脂质混合物;第二步,将氧浓度为103毫克每升的含1.5%氯化钙和3.5%蔗糖的溶液加入第一步中的混合物a中,然后在1摄氏度下结合液相等离子体技术机械搅拌11分钟,得到混合液a,其中液相等离子体电子和离子的功率密度为0.20瓦每平方厘米,机械搅拌的速率为70rpm;第三步,将nh2-peg2-c2-boc加入第二步获得的混合液a,然后在6摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为0.22瓦每平方厘米,机械搅拌的速率为45rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s12和s3按照摩尔比例4:1混合得到的混合适配体;hbv感染所致的肝癌为10-8和ly-1按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为as1411和ly-1按照摩尔比例2:1混合得到的混合适配体;hpv感染所致的宫颈癌为apt和a10按照摩尔比例3:1混合得到的混合适配体;ebv感染所致的原位淋巴瘤为td05和wqy-9-b按照摩尔比例2:1混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为22平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过微流控设备进行进一步混合,微流控的总流速为0.1毫升每分钟,流速比为1/5。
[0332]
实施例23实施例21所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例21中合成的环形mrna采用实施例22中所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);所制得样品平均粒径125纳米,表面荷电33.6
±
0.1毫伏。此外,进一步地空白对照实验结果表明实施例21所制备的环形mrna分子采用实施例22所示的靶向递送系统所获得的样品的表面荷电远远高于实施例21所制备的环形mrna分子采用实施例22中所示的未经合理改性的任何一种或多种脂质产品与实施例22所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例22中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例22所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例22所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制
的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10μg rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为1.93
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.41
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.06
±
0.01%、0.11
±
0.03%、 0.45
±
0.05%、0.70
±
0.06%;tnf-α的量分别依次为0.08
±
0.02%、0.13
±
0.04%、0.52
±
0.06%、0.87
±
0.15%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0333]
实施例24实施例21所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例21中合成的环形mrna采用实施例22中所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);表面荷电33.6
±
0.1毫伏。此外,进一步地空白对照实验结果表明实施例21所制备的环形mrna分子采用实施例22所示的靶向递送系统所获得的样品的表面荷电远远高于实施例21所制备的环形mrna分子采用实施例22中所示的未经合理改性的任何一种或多种脂质产品与实施例22
所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例22中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例22所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例22所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.01
±
0.6%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.06
±
0.01%、0.11
±
0.02%、 0.54
±
0.05%、0.83
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0334]
实施例25实施例21所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将1
×
10 7
个ebv阳性且具成瘤能力的鼻咽癌细胞注射入小鼠腹部侧面皮下,然后取皮下移植物来源的组织块进行鼻咽癌移植,建立原位鼻咽癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为15立方毫米,注射第15天后的肿瘤组织大小为34立方毫米,注射第18天后的肿瘤组织大小为60立方毫米,注射第21天后的肿瘤组织大小为112立方毫米,注射第24天后的肿瘤组织大小为163立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为5立方毫米、6立方毫米、8立方毫米、10立方毫米;注射第12天后的肿瘤组织大小分别依次为46立方毫米、60立方毫米、79立方毫米、93立方毫米;注射第15天后的肿瘤组织大小分别依次为112立方毫米、151立方毫米、180立方毫米、199立方毫米;注射第18天后的肿瘤组织大小分别依次为187立方毫米、249立方毫米、275立方毫米、295立方毫
米;注射第21天后的肿瘤组织大小分别依次为293立方毫米、340立方毫米、372立方毫米、399立方毫米;注射第24天后的肿瘤组织大小分别依次为389立方毫米、426立方毫米、463立方毫米、484立方毫米)。这说明,实施例21所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例21所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.43
±
0.2%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.10
±
0.03%、0.12
±
0.04%、0.25
±
0.07%、0.42
±
0.09%)。
[0335]
实施例26实施例21所制备的环形mrna分子激活人类t细胞的情况第一步,分离出hbv阳性肝癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加 入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按 说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hbv阳性肝癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;
第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.52
±
0.3%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.05%、0.24
±
0.08%),仅次于pma阳性对照组(31.49
±
2.99%)。
[0336] 实施例27实施例21所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠鼻咽部位组织细胞或ebv阳性原位鼻咽癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液; 第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在ebv阳性原位鼻咽癌细胞中表达明显高于正常小鼠鼻咽部位组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在ebv阳性原位鼻咽癌细胞中的表达。
[0337] 实施例28实施例21所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠鼻咽部位组织细胞和ebv阳性原位鼻咽癌细胞并进行高通量测序。分析测序结果,确定小鼠鼻咽细胞和ebv阳性原位鼻咽癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠鼻咽部位细胞和ebv阳性原位鼻咽癌细胞的
rna,并列分别为:5
´‑
acgctagcgtagcagcttgccgttg-3
´
(seq id 98 )和5
´‑
cctactccgaacctaggtcgatgatgg-3
´ꢀ
(seq id 99);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在ebv阳性原位鼻咽癌细胞中高表达,明显高于正常鼻咽部位细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例21所制备的环形mrna分子采用实施例22靶向递送系统递送至ebv阳性原位鼻咽癌细胞的递送率(90.1%)远远高于实施例22中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例22所示靶向递送系统对实施例21所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(90.1%)远远高于单独使用实施例22中所示核酸适配体或其他迄今为止文献资料所报道的ebv阳性原位鼻咽癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例22所示靶向递送系统对实施例21所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(90.1%)远远高于用实施例22中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例22中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例21所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例21所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例21所合成的环形mrna的递送能力优良。
[0338]
实施例29用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seq id 15所示氨基酸序列的抗原多肽编码区,具有seq id 62所示核苷酸序列的ires,具有seq id 66所示核苷酸序列的第二外显子,具有seq id 69所示核苷酸序列的5
´
间隔区,具有seq id 72所示核苷酸序列的3
´
间隔区,具有seq id 75所示核苷酸序列的第一外显子;具有seq id 78核苷酸序列的的启动子,具有seq id 81核苷酸序列的5
´
同源臂,具有seq id 84核苷酸序列的3
´
内含子,具有seq id 87所示核苷酸序列的5
´
内含子,具有seq id 90所示核苷酸序列的3
´
同源臂,以及可用于具有seq id 93所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,218rpm活化3小时后,取活化菌液在37.1摄氏度,218rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(363.8纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37.1摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(301.2纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为
20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37.1摄氏度孵育3小时,然后加入dnasei在37.1摄氏度消化20分钟。将所获得的混合物采用尺寸排阻柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(491.2纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物依次通过采用尺寸排阻柱法,气相等离子体法(辐射能量为1.1焦耳每平方厘米,辐射15秒,机械搅拌,搅拌速度3rpm,液相介质为hepes缓冲液,气体为98%氦气和2%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后环形mrna的浓度(315.3纳克每微升),纯度为98.9%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.7%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在气相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体法技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度; 第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热17分钟,随后在85摄氏度加热8秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0339] 实施例30实施例29所制备的的环形mrna分子的靶向脂质体递送系统工艺 第一步,将黑磷纳米片和脂质产品按照6.4:70.1摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dspc,dppc,dotap和dope按照摩尔比例1:6.7:31:37.7混
合得到的脂质混合物;第二步,将氧浓度为94毫克每升的含0.7%氯化钠,5.0%硫酸镁和3.8%葡萄糖的溶液加入第一步中的混合物a中,然后在0摄氏度下结合液相等离子体技术机械搅拌17分钟,得到混合液a,其中液相等离子体的功率密度为0.31瓦每平方厘米,机械搅拌的速率为110rpm;第三步,将amino-peg4-ch2cooh加入第二步获得的混合液a,然后在11摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为0.33瓦每平方厘米,机械搅拌的速率为82rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s12按照摩尔比例2:1混合得到的混合适配体; hbv感染所致的肝癌为为ly-1和肽适配子按照摩尔比例1:3混合得到的混合适配体;hcv感染所致的肝癌为为zy-1和tls3按照摩尔比例2:3混合得到的混合适配体; hpv感染所致的宫颈癌为air-3a和as1411按照摩尔比例1:1混合得到的混合适配体; ebv感染所致的原位淋巴瘤为gbi-10和u2按照摩尔比例3:2混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc; hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为35 平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和线性mrna通过微流控设备进行进一步混合,微流控的总流速为0.14毫升每分钟, 流速比为1/7.3。
[0340]
实施例31实施例29所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例29中合成的环形mrna采用实施例30脂质体配方工艺制备的环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为80纳米,表面荷电39.0
±
0.5毫伏。此外,进一步地空白对照实验结果表明实施例29所制备的环形mrna分子采用实施例30所示的靶向递送系统所获得的样品的表面荷电远远高于实施例29所制备的环形mrna分子采用实施例30中所示的未经合理改性的任何一种或多种脂质产品与实施例29所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例30中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例30所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例30所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中。图4为实施例29中环形mrna /靶向递送系统组合成的环形mrna疫苗的电子显微镜照片;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;
第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5μg/ml)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.13
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.66
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.06
±
0.01%、0.11
±
0.03%、 0.44
±
0.05%、0.69
±
0.06%;tnf-α的量分别依次为0.08
±
0.02%、0.11
±
0.03%、0.51
±
0.08%、0.77
±
0.15%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0341]
实施例32实施例29所制备的的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例29中合成的环形mrna采用实施例30中所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为80纳米,表面荷电39.0
±
0.5毫伏。此外,进一步地空白对照实验结果表明实施例29所制备的环形mrna分子采用实施例30所示的靶向递送系统所获得的样品的表面荷电远远高于实施例29所制备的环形mrna分子采用实施例30中所示的未经合理改性的任何一种或多种脂质产品与实施例29所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例30中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例30所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例30所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处
理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.17
±
0.6%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.06
±
0.02%、0.13
±
0.03%、 0.45
±
0.05%、0.76
±
0.2%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0342]
实施例33实施例29所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将2.5
×
105个hpv阳性且具成瘤能力的小时ela细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈癌移植,建立原位宫颈癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为19立方毫米,注射第15天后的肿瘤组织大小为42立方毫米,注射第18天后的肿瘤组织大小为75立方毫米,注射第21天后的肿瘤组织大小为136立方毫米,注射第24天后的肿瘤组织大小为244立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为7立方毫米、8立方毫米、10立方毫米、12立方毫米;注射第12天后的肿瘤组织大小分别依次为46立方毫米、67立方毫米、89立方毫米、103立方毫米;注射第15天后的肿瘤组织大小分别依次为117立方毫米、160立方毫米、193立方毫米、219立方毫米;注射第18天后的肿瘤组织大小分别依次为232立方毫米、279立方毫米、317立方毫米、341立方毫米;注射第21天后的肿瘤组织大小分别依次为353立方毫米、410立方毫米、459立方毫米、492立方毫米;注射第24天后的肿瘤组织大小分别依次为458立方毫米、522立方毫米、571立方毫米、615立方毫米)。这说明,实施例29所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例29所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润
cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.32
±
0.2%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.13
±
0.06%、0.22
±
0.07%、0.40
±
0.08%)。
[0343]
实施例34实施例29所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出ebv阳性鼻咽癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按 说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出ebv阳性鼻咽癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20n纳克每毫升的il
‑ꢀ
4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,
第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.33
±
0.2%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.11
±
0.03%、0.18
±
0.06%),仅次于pma阳性对照组(30.02
±
2.97%)。
[0344] 实施例35实施例29所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠子宫颈组织细胞或hpv阳性原位宫颈癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hpv阳性原位宫颈癌细胞中表达明显高于正常子宫颈组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hpv阳性原位宫颈癌中的表达。
[0345] 实施例36实施例29所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠子宫颈组织细胞和hpv阳性原位宫颈癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠宫颈部位细胞和hpv阳性原位宫颈癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠子宫颈组织和hpv阳性原位宫颈癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
ataaactttgatcagttattagttactg-3
´
(seqid100)和5
´‑
gagagactggaacttaaggttaaattcc-3
´
(seqid101);
第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hpv阳性原位宫颈癌细胞中高表达,明显高于正常子宫颈组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例29所制备的环形mrna分子采用实施例30靶向递送系统递送至hpv阳性原位宫颈癌细胞的递送率(92.3%)远远高于实施例30中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例30所示靶向递送系统对实施例29所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(92.3%)远远高于单独使用实施例30中所示核酸适配体或其他迄今为止文献资料所报道的hpv阳性原位宫颈癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例30所示靶向递送系统对实施例29所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(92.3%)远远高于用实施例30中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例30中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例29所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例29所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例29所合成的环形mrna的递送能力优良。
[0346]
实施例37用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid20所示核苷酸序列的ires,具有seqid67所示核苷酸序列的第二外显子,具有seqid70所示核苷酸序列的5
´
间隔区,具有seqid73所示核苷酸序列的3
´
间隔区,具有seqid76所示核苷酸序列的第一外显子;具有seqid79核苷酸序列的的启动子,具有seqid80核苷酸序列的5
´
同源臂,具有seqid85核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid91所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,222rpm活化3.7小时后,取活化菌液在37.1摄氏度,222rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(235.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37.1摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(249.4纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育3小时,然后加入dnasei在37.1摄氏度消化25分
钟。将所获得的混合物采用磁分离法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(494.5纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热18分钟。将所获得的混合物依次通过采用磁分离法,超声波技术法(声强度为80毫瓦每平方厘米,辐射30分钟,机械搅拌,搅拌速度50rpm,液相介质为柠檬酸盐缓冲液,气体为90%氩气和10%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后环形mrna浓度(244.7纳克每微升),纯度为99.5%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(90.1%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在超声波技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在超声波技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37.1摄氏度加热20分钟,随后在85摄氏度加热8秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0347] 实施例38实施例37所制备的环形mrna分子的靶向脂质体递送系统工艺优化第一步,将黑磷粉末和脂质产品按照7.0:80.6摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dspc,dppc,二油酰基磷脂酰丝氨酸,磷脂酰丝氨酸和dope按照摩尔比例1:3.2:7.5:13.5:34混合得到的脂质混合物;第二步,将氧浓度为111毫克每升的含0.9%氯化钠,4.0%蔗糖和2.7%葡萄糖的溶液
加入第一步中的混合物a中,然后在8摄氏度下结合液相等离子体技术机械搅拌6分钟,得到混合液a,其中液相等离子体的功率密度为0.60瓦每平方厘米,机械搅拌的速率为125rpm;第三步,将nh2-peg2-ch2cooh加入第二步获得的混合液a,然后在-4摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为0.51瓦每平方厘米,机械搅拌的速率为140rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s27按照摩尔比例1:3混合得到的混合适配体; hbv感染所致的肝癌为zy1和tls9a按照摩尔比例1:2混合得到的混合适配体; hcv感染所致的肝癌为肽适配子和as1411按照摩尔比例1:1混合得到的混合适配体; hpv感染所致的宫颈癌为as1411和a10按照摩尔比例2:1混合得到的混合适配体;ebv感染所致的原位淋巴瘤为wqy-9-b和u8按照摩尔比例4:1混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc; hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为60 平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环状mrna通过微流控设备进行进一步混合,微流控的总流速为0.1毫升每分钟, 流速比为1/8。
[0348] 实施例39 实施例37所制备的环形mrna分子作为疫苗激活免疫反应第一步,将实施例37中合成的环形mrna采用实施例38所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为92纳米,表面荷电37.3
±
0.1毫伏。此外,进一步地空白对照实验结果表明实施例37所制备的环形mrna分子采用实施例38所示的靶向递送系统所获得的样品的表面荷电远远高于实施例37所制备的环形mrna分子采用实施例38中所示的未经合理改性的任何一种或多种脂质产品与实施例37所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例38中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例38所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例30所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟
的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150v、10ms,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldina(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为1.82
±
0.2%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),tnf-α的量为2.33
±
0.3%(tnf-α阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.10
±
0.03%、0.39
±
0.04%、0.65
±
0.05%;tnf-α的量分别依次为0.09
±
0.03%、0.12
±
0.03%、0.47
±
0.07%、0.75
±
0.13%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0349] 实施例40实施例37所制备所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例37中合成的环形mrna采用实施例38中所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为92纳米,表面荷电37.3
±
0.1毫伏。此外,进一步地空白对照实验结果表明实施例37所制备的环形mrna分子采用实施例38所示的靶向递送系统所获得的样品的表面荷电远远高于实施例37所制备的环形mrna分子采用实施例38中所示的未经合理改性的任何一种或多种脂质产品与实施例37所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例38中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例38所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例30所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组
别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为1.88
±
0.4%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.06
±
0.01%、0.13
±
0.03%、 0.47
±
0.03%、0.72
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0350] 实施例41 实施例37所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将7.5
×
10 4
个hbv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行肝移植,建立原位肝癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为42立方毫米,注射第15天后的肿瘤组织大小为87立方毫米,注射第18天后的肿瘤组织大小为155立方毫米,注射第21天后的肿瘤组织大小为246立方毫米,注射第24天后的肿瘤组织大小为418立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为32立方毫米、24立方毫米、19立方毫米、16立方毫米;注射第12天后的肿瘤组织大小分别依次为255立方毫米、211立方毫米、160立方毫米、124立方毫米;注射第15天后的肿瘤组织大小分别依次为564立方毫米、522立方毫米、420立方毫米、297立方毫米;注射第18天后的肿瘤组织大小分别依次为850立方毫米、794立方毫米、692立方毫米、533立方毫米;注射第21天后的肿瘤组织大小分别依次为1167立方毫米、1090立方毫米、978立方毫米、854立方毫米;注射第24天后的肿瘤组织大小分别依次为1455立方毫米、1376立方毫米、1270立方毫米、1143立方毫米)。这说明,实施例37所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例37所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.25
±
0.2%(ifn-γ阳性cd8+t细胞在 cd8
+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.12
±
0.06%、0.22
±
0.07%、0.36
±
0.08%)。
[0351]
实施例42实施例37所制备的环形mrna分子激活人类t细胞的情况第一步,分离出ebv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按 说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出ebv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il
‑ꢀ
4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯) 和1微克每毫升 ionomycin(离子霉素);
第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.23
±
0.2%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.05%、0.22
±
0.06%),仅次于pma阳性对照组(30.33
±
3.28%)。
[0352]
实施例43实施例37所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠肝细胞或hbv阳性原位肝癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hbv阳性原位肝癌细胞中表达明显高于正常肝细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hbv阳性原位肝癌细胞中的表达。
[0353] 实施例44实施例37所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞细胞的rna并进行高通量测序。分析测序结果,确定小鼠肝细胞和hbv阳性原位肝癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用引物所用上游引物和下游引物引物序列分别为:5
´‑
atgcggtctgacgataaatttaaccttatc-3
´
(seqid102)和5
´‑
gaattcttacagatctcccgggcaaattgg-3
´
(seqid103);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hbv阳性原位肝癌细胞中高表达,明显高于正常肝细胞。说明编码肿瘤新生抗原多肽
的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例37所制备的环形mrna分子采用实施例38靶向递送系统递送至hbv阳性原位肝癌细胞的递送率(87.2%)远远高于实施例38中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例38所示靶向递送系统对实施例37所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(87.2%)远远高于单独使用实施例38中所示核酸适配体或其他迄今为止文献资料所报道的hbv阳性原位肝癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例38所示靶向递送系统对实施例37所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(87.2%)远远高于用实施例38中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例38中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例37所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例37所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例37所合成的环形mrna的递送能力优良。
[0354] 实施例45用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid25所示氨基酸序列的抗原多肽编码区,具有seqid65所示核苷酸序列的ires,具有seqid68所示核苷酸序列的第二外显子,具有seqid71所示核苷酸序列的5
´
间隔区,具有seqid74所示核苷酸序列的3
´
间隔区,具有seqid77所示核苷酸序列的第一外显子;具有seqid80核苷酸序列的的启动子,具有seqid83核苷酸序列的5
´
同源臂,具有seqid86核苷酸序列的3
´
内含子,具有seqid89所示核苷酸序列的5
´
内含子,具有seqid92所示核苷酸序列的3
´
同源臂,以及可用于具有seqid95所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,219rpm活化3小时后,取活化菌液在36.9摄氏度,219rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(355.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(268.9纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnasei在36.9摄氏度消化20分钟。将所获得的混合物采用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(472.4纳克每微升);
第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物依次通过采用硅膜离心柱法,液相等离子体法(辐射能量为0.5j每平方厘米,辐射30秒,机械搅拌,搅拌速度30rpm,液相介质为柠檬酸盐缓冲液,气体为95%氩气和5%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率并纯化后的环形mrna浓度(289.3纳克每微升),纯度为99.0%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(88.3%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在液相等离子体法技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体法技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量或辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热20分钟,随后在85摄氏度加热8秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0355] 实施例46实施例45所制备的的环形mrna分子的靶向脂质体递送系统工艺第一步,将黑磷晶体和脂质产品按照7.8:74.2摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为daa,peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:9:27:31:35混合得到的脂质混合物;第二步,将氧浓度为120毫克每升的含1.2%氯化钠,0.8%磷酸氢二钠,3.0%蔗糖和3.2%葡萄糖的溶液加入第一步中的混合物a中,然后在4摄氏度下结合气相等离子体技术机械搅拌28分钟,得到混合液a,其中气相等离子体产生的电子和离子的能量为100电子伏
特,机械搅拌的速率为40rpm;第三步,将amino-peg4-ch2cooh和nh2-peg2-c2-boc按1:3摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在0摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为9瓦每平方厘米,机械搅拌的速率为300rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109,s27和s3按照摩尔比例1:3:2混合得到的混合适配体;hbv感染所致的肝癌为zy1,tls9a和ch6按照摩尔比例1:2:1混合得到的混合适配体;hcv感染所致的肝癌为tls9a,as1411和tls3按照摩尔比例1:1:1混合得到的混合适配体;hpv感染所致的宫颈癌为as1411和a10按照摩尔比例1:1混合得到的混合适配体;ebv感染所致的原位淋巴瘤为gbm128和u9按照摩尔比例5:4混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为96平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和线性mrna通过超流控设备进行进一步混合,超流控的总流速为0.12毫升每分钟,流速比为1/9.5。
[0356]
实施例47实施例45所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例45中合成的环形mrna采用实施例46所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为112纳米,表面荷电35.0
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例45所制备的环形mrna分子采用实施例46所示的靶向递送系统所获得的样品的表面荷电远远高于实施例45所制备的环形mrna分子采用实施例46中所示的未经合理改性的任何一种或多种脂质产品与实施例45所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例46中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例46所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例46所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏和肝脏。观察记录小鼠的肿瘤生长情况。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10%(v/v)fbs的rpmi1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一
组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为1.97
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.33
±
0.3%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.03%、0.12
±
0.04%、 0.43
±
0.05%、0.72
±
0.06%;tnf-α的量分别依次为0.07
±
0.02%、0.14
±
0.04%、0.56
±
0.08%、0.84
±
0.15%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0357]
实施例48实施例45所制备环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例45中合成的环形mrna采用实施例46中所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为112纳米,表面荷电35.0
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例45所制备的环形mrna分子采用实施例46所示的靶向递送系统所获得的样品的表面荷电远远高于实施例45所制备的环形mrna分子采用实施例46中所示的未经合理改性的任何一种或多种脂质产品与实施例45所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例46中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例46所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例46所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程
接受动物管理会的监督;第三步,取脾细胞1
×
10 6
个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.07
±
0.6%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.07
±
0.02%、0.14
±
0.03%、 0.45
±
0.06%、0.75
±
0.2%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0358] 实施例49 实施例45所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的c57bl/6小鼠,将7.5
×
10 4
个hbv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行肝移植,建立原位肝癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为12立方毫米,注射第15天后的肿瘤组织大小为30立方毫米,注射第18天后的肿瘤组织大小为59立方毫米,注射第21天后的肿瘤组织大小为106立方毫米,注射第24天后的肿瘤组织大小为157立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为12立方毫米、9立方毫米、7立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为98立方毫米、84立方毫米、63立方毫米、47立方毫米;注射第15天后的肿瘤组织大小分别依次为205立方毫米、186立方毫米、155立方毫米、116立方毫米;注射第18天后的肿瘤组织大小分别依次为302立方毫米、283立方毫米、257立方毫米、192立方毫米;注射第21天后的肿瘤组织大小分别依次为408立方毫米、379立方毫米、348立方毫米、289立方毫米;注射第24天后的肿瘤组织大小分别依次为494立方毫米、471立方毫米、430立方毫米、394立方毫米)。这说明,实施例45所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例45所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.25
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.12
±
0.04%、0.21
±
0.07%、0.41
±
0.08%)。
[0359] 实施例50 实施例45所制备的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加 入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按 说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il
‑ꢀ
4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1ml含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升 pma(豆蔻酰佛波醇乙酯) 和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体
标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.21
±
0.2%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.10
±
0.03%、0.24
±
0.08%),仅次于pma阳性对照组(30.11
±
3.03%)。
[0360]
实施例51实施例45所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠鼻咽癌组织细胞或ebv阳性原位鼻咽癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在ebv阳性原位鼻咽癌细胞中表达明显高于正常鼻咽癌组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在ebv阳性原位鼻咽癌中的表达。
[0361] 实施例52实施例45所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠鼻咽癌组织细胞或ebv阳性原位鼻咽癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠鼻咽组织细胞或ebv阳性原位鼻咽癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠鼻咽癌组织细胞或ebv阳性原位鼻咽癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
gaataactactaccatatgtgcgcatatagaac-3
´
(seqid104)和5
´‑
cctcctccaaagctaaaattaattgaactatgg-3
´
(seqid105);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在ebv阳性原位鼻咽癌细胞中高表达,明显高于正常鼻咽组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例45所制备的环形mrna分子采用实施例46靶向递送系统递送至ebv阳性原位鼻咽癌
细胞的递送率(88.5%)远远高于实施例46中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例46所示靶向递送系统对实施例45所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(88.5%)远远高于单独使用实施例46中所示核酸适配体或其他迄今为止文献资料所报道的ebv阳性原位鼻咽癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例46所示靶向递送系统对实施例45所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(88.5%)远远高于用实施例46中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例46中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例45所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例45所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例45所合成的环形mrna的递送能力优良。
[0362]
实施例53用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备 第一步,构建具有seqid30所示氨基酸序列的抗原多肽编码区,具有seqid62所示核苷酸序列的ires,具有seqid66所示核苷酸序列的第二外显子,具有seqid69所示核苷酸序列的5
´
间隔区,具有seqid72所示核苷酸序列的3
´
间隔区,具有seqid75所示核苷酸序列的第一外显子;具有seqid78核苷酸序列的的启动子,具有seqid81核苷酸序列的5
´
同源臂,具有seqid84核苷酸序列的3
´
内含子,具有seqid87所示核苷酸序列的5
´
内含子,具有seqid90所示核苷酸序列的3
´
同源臂,以及可用于具有seqid93所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(481.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(276.1纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育3小时,然后加入dnasei在36.9摄氏度消化25分钟。将所获得的混合物采用亲和柱层析法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(517.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩
尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物依次通过采用亲和柱层析法,气相等离子体法(辐射能量为0.5j每平方厘米,辐射30秒,机械搅拌,搅拌速度5rpm,液相介质为柠檬酸盐缓冲液,气体为98%氩气和2%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率并纯化后环形mrna的浓度(262.8纳克每微升),纯度为97.2%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(89.3%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在气相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在气相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热20分钟,随后在85摄氏度加热8秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为30次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0363] 实施例54实施例53所制备的环形mrna分子的靶向脂质体递送系统工艺第一步,将石墨烯状黑磷和脂质产品按照6.3:63摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dspc,dppg,dodma,otma和dope按照摩尔比例1:7:13:16.4:32.1混合得到的脂质混合物;第二步,将纯氧通入含1.5%氯化钠,3.1%氯化镁,4.0%磷酸氢二钠,3.4%蔗糖和7.1%葡萄糖溶液加入第一步中的混合物a中,然后在7摄氏度下结合气相等离子体技术机械搅拌45分钟,得到混合液a,其中气相等离子体电子和离子的功率密度为90电子伏特,机械搅拌的速率为35rpm;第三步,将amino-peg4-ch2cooh和nh2-peg4-cooh按1:3摩尔比例混合形成的混合
物加入第二步获得的混合液a,然后在-3摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为375rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s3, s12和s27按照摩尔比例1:1:1混合得到的混合适配体; hbv感染所致的肝癌为as1411, zy1和tls9a按照摩尔比例1:1:2混合得到的混合适配体; hcv感染所致的肝癌为ch6, as141和tls3按照摩尔比例1:1:3混合得到的混合适配体; hpv感染所致的宫颈癌为as1411, air-3a和a10按照摩尔比例2:1:1混合得到的混合适配体; ebv感染所致的原位淋巴瘤为gbm131和wqy-9-b按照摩尔比例5:4混合得到的混合适配体; hltv-1感染所致的成人t细胞白血病为sscgc; hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为85 平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环状mrna通过超流控设备进行进一步混合,超流控的总流速为0.15毫升每分钟, 流速比为1/5。
[0364]
实施例55实施例53所制备的环形mrna分子作为疫苗激活免疫反应第一步,将实施例53中合成的环形mrna采用实施例54所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为88纳米,表面荷电37.8
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例53所制备的环形mrna分子采用实施例54所示的靶向递送系统所获得的样品的表面荷电远远高于实施例53所制备的环形mrna分子采用实施例54中所示的未经合理改性的任何一种或多种脂质产品与实施例53所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例54中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例54所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例54所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的
环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna (即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5μg/ml)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.27
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.38
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.03%、0.11
±
0.04%、 0.42
±
0.05%、0.69
±
0.06%;tnf-α的量分别依次为0.08
±
0.02%、0.13
±
0.04%、0.51
±
0.08%、0.79
±
0.15%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0365]
实施例56实施例53所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例53中合成的环形mrna采用实施例54所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为88纳米,表面荷电37.8
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例53所制备的环形mrna分子采用实施例54所示的靶向递送系统所获得的样品的表面荷电远远高于实施例53所制备的环形mrna分子采用实施例54中所示的未经合理改性的任何一种或多种脂质产品与实施例53所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例54中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例54所示的靶向递送系统的表面荷电和粒径。与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例54所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚
体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.33
±
0.4%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.06
±
0.01%、0.14
±
0.03%、 0.46
±
0.05%、0.76
±
0.2%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0366] 实施例57 实施例53所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将2.5
×
105个hpv阳性且具成瘤能力的小时ela细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈癌移植,建立原位宫颈癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为17立方毫米,注射第15天后的肿瘤组织大小为40立方毫米,注射第18天后的肿瘤组织大小为72立方毫米,注射第21天后的肿瘤组织大小为130立方毫米,注射第24天后的肿瘤组织大小为238立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为13立方毫米、10立方毫米、7立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为109立方毫米、95立方毫米、63立方毫米、44立方毫米;注射第15天后的肿瘤组织大小分别依次为227立方毫米、198立方毫米、150立方毫米、113立方毫米;注射第18天后的肿瘤组织大小分别依次为348立方毫米、322立方毫米、272立方毫米、225立方毫米;注射第21天后的肿瘤组织大小分别依次为501立方毫米、464立方毫米、401立方毫米、339立方毫米;注射第24天后的肿瘤组织大小分别依次为626立方毫米、573立方毫米、514立方毫米、450立方毫米)。这说明,实施例53所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例53所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.41
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.03%、0.16
±
0.05%、0.24
±
0.08%、0.47
±
0.09%)。
[0367]
实施例58实施例53所制备的环形mrna分子激活人类t细胞的情况第一步,分离出hbv阳性肝癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加
入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加 入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按 说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hbv阳性肝癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0 .5微克每毫升pma(豆蔻酰佛波醇乙酯) 和1微克每毫升 ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzyme b)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人
cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.32
±
0.3%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.10
±
0.03%、0.22
±
0.06%),仅次于pma阳性对照组(31.55
±
3.01%)。
[0368] 实施例59实施例53所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠正常宫颈组织细胞或hpv阳性原位宫颈癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hpv阳性原位宫颈癌细胞中表达明显高于正常宫颈组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hpv阳性原位宫颈癌中的表达。
[0369] 实施例60实施例53所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取宫颈组织细胞或hpv阳性原位宫颈癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠宫颈组织细胞或hpv阳性原位宫颈癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠宫颈组织细胞或hpv阳性原位宫颈癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
ttgcgtggccgcgaagatgaccac-3
´
(seqid106)和5
´‑
gcaggtgtgctatagttcaattaaag-3
´
(seqid107);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hpv阳性原位宫颈癌细胞中高表达,明显高于正常宫颈组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例53所制备的环形mrna分子采用实施例54靶向递送系统递送至hpv阳性原位宫颈癌细胞的递送率(86.4%)远远高于实施例54中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系
统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例54所示靶向递送系统对实施例53所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(86.4%)远远高于单独使用实施例54中所示核酸适配体或其他迄今为止文献资料所报道的hpv阳性原位宫颈癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例54所示靶向递送系统对实施例53所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(86.4%)远远高于用实施例54中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例54中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例53所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例53所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例53所合成的环形mrna的递送能力优良。
[0370]
实施例61用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid35所示氨基酸序列的抗原多肽编码区,具有seqid64所示核苷酸序列的ires,具有seqid67所示核苷酸序列的第二外显子,具有seqid70所示核苷酸序列的5
´
间隔区,具有seqid73所示核苷酸序列的3
´
间隔区,具有seqid76所示核苷酸序列的第一外显子;具有seqid79核苷酸序列的的启动子,具有seqid82核苷酸序列的5
´
同源臂,具有seqid85核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid91所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,218rpm活化3.5小时后,取活化菌液在36.9摄氏度,218rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(321.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(310.3纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnasei在36.9摄氏度消化20分钟。将所获得的混合物采用尺寸排阻柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(590.2纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行
混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热30分钟。将所获得的混合物依次通过采用尺寸排阻柱法,超声波技术(声强度为10毫瓦每平方厘米,超声50分钟,机械搅拌,搅拌速度80rpm,液相介质为tris-edta缓冲液,气体为90%氮气和10%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(302.1纳克每微升),纯度98.3%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.2%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在超声波技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在超声波技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热20分钟,随后在85摄氏度加热9秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0371]
实施例62实施例61所制备的的环形mrna分子的靶向脂质体递送系统工艺 第一步,将黑磷量子点和脂质产品按照6:72摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为peg修饰的二烷基胺,peg修饰的二酰基甘油,peg-dpg,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:5:12:30:35混合得到的脂质混合物;第二步,将空气通入含1.2%氯化钠,3.3%磷酸氢二钠,3.7%蔗糖和4.1%葡萄糖的溶液加入第一步中的混合物a中,然后在-2摄氏度下结合液相等离子体技术机械搅拌15分钟,得到混合液a,其中液相等离子体电子和离子的功率密度为0.90瓦每平方厘米,机械搅拌的速率为210rpm;第三步,将nh2-peg2-c2-boc和nh2-peg4-cooh按1:2摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在0摄氏度下进行超声搅拌处理,得到混合液b;其中超声
波的超声功率密度为10瓦每平方厘米,机械搅拌的速率为320rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s27按照摩尔比例1:1混合得到的混合适配体; hbv感染所致的肝癌为zy1和tls9a按照摩尔比例3:2混合得到的混合适配体; hcv感染所致的肝癌为zy-1,肽适配子和as1411按照摩尔比例2:2:5混合得到的混合适配体; hpv感染所致的宫颈癌为as1411和a10按照摩尔比例2:3混合得到的混合适配体; ebv感染所致的原位淋巴瘤为u31和u2按照摩尔比例2:3混合得到的混合适配体; hltv-1感染所致的成人t细胞白血病为sscgc; hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为70 平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和线性mrna通过超流控设备进行进一步混合,超流控的总流速为0.20毫升每分钟, 流速比为1/6。
[0372] 实施例63 实施例61所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例61中合成的环形mrna采用实施例62所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为94纳米,表面荷电36.9
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例61所制备的环形mrna分子采用实施例62所示的靶向递送系统所获得的样品的表面荷电远远高于实施例61所制备的环形mrna分子采用实施例62中所示的未经合理改性的任何一种或多种脂质产品与实施例61所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例62中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例62所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例62所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏和肝脏。观察记录小鼠的肿瘤生长情况。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克/毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。
24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.22
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.56
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、 0.44
±
0.05%、0.69
±
0.06%;tnf-α的量分别依次为0.10
±
0.04%、0.15
±
0.04%、0.52
±
0.08%、0.81
±
0.15%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0373] 实施例64 实施例61所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例61中合成的环形mrna采用实施例62所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为94纳米,表面荷电36.9
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例61所制备的环形mrna分子采用实施例62所示的靶向递送系统所获得的样品的表面荷电远远高于实施例61所制备的环形mrna分子采用实施例62中所示的未经合理改性的任何一种或多种脂质产品与实施例61所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例62中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例62所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例62所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚
体进行流式染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.12
±
0.6%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.07
±
0.02%、0.13
±
0.03%、0.39
±
0.05%、0.67
±
0.2%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0374] 实施例65实施例61所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8周的体重20克的小鼠,将7.5
×
104个hbv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行肝移植,建立原位肝癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为49立方毫米,注射第15天后的肿瘤组织大小为95立方毫米,注射第18天后的肿瘤组织大小为167立方毫米,注射第21天后的肿瘤组织大小为263立方毫米,注射第24天后的肿瘤组织大小为440立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为38立方毫米、30立方毫米、23立方毫米、19立方毫米;注射第12天后的肿瘤组织大小分别依次为260立方毫米、218立方毫米、171立方毫米、136立方毫米;注射第15天后的肿瘤组织大小分别依次为579立方毫米、539立方毫米、438立方毫米、317立方毫米;注射第18天后的肿瘤组织大小分别依次为872立方毫米、816立方毫米、732立方毫米、561立方毫米;注射第21天后的肿瘤组织大小分别依次为1194立方毫米、1105立方毫米、1034立方毫米、892立方毫米;注射第24天后的肿瘤组织大小分别依次为1485立方毫米、1421立方毫米、1346立方毫米、1214立方毫米)。这说明,实施例61所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例61所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.43
±
0.4%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.06
±
0.01%、0.11
±
0.03%、0.26
±
0.06%、0.42
±
0.1%)。
[0375] 实施例66实施例61所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在
加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按 说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0 .5微克每毫升 pma(豆蔻酰佛波醇乙酯) 和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzyme b)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人
cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为1.99
±
0.1%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.06
±
0.01%、0.11
±
0.03%、0.17
±
0.05%),仅次于pma阳性对照组(30.01
±
3.15%)。
[0376] 实施例67实施例61所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠宫颈组织细胞或hpv阳性原位宫颈癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hpv阳性原位宫颈癌细胞中表达明显高于正常宫颈组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hpv阳性原位宫颈癌中的表达。
[0377] 实施例68实施例61所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠宫颈组织细胞或hpv阳性原位宫颈癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠宫颈组织细胞或hpv阳性原位宫颈癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠宫颈组织细胞或hpv阳性原位宫颈癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
caggccaccaaccttagtgtcgcctatct-3
´
(seqid108)和5
´‑
cgctaccatatgagatcagttattagtgtc-3
´
(seqid109);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hpv阳性原位宫颈癌细胞中高表达,明显高于正常宫颈组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例61所制备的环形mrna分子采用实施例62靶向递送系统递送至hpv阳性原位宫颈癌细胞的递送率(91.7%)远远高于实施例62中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系
统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例62所示靶向递送系统对实施例61所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(91.7%)远远高于单独使用实施例62中所示核酸适配体或其他迄今为止文献资料所报道的hpv阳性原位宫颈癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例62所示靶向递送系统对实施例61所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(91.7%)远远高于用实施例62中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例62中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例61所合成的环形mrna递送至hpv阳性原位宫颈癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例61所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例61所合成的环形mrna的递送能力优良。
[0378] 实施例69用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid40所示氨基酸序列的抗原多肽编码区,具有seqid65所示核苷酸序列的ires,具有seqid68所示核苷酸序列的第二外显子,具有seqid71所示核苷酸序列的5
´
间隔区,具有seqid74所示核苷酸序列的3
´
间隔区,具有seqid77所示核苷酸序列的第一外显子;具有seqid80核苷酸序列的的启动子,具有seqid83核苷酸序列的5
´
同源臂,具有seqid86核苷酸序列的3
´
内含子,具有seqid89所示核苷酸序列的5
´
内含子,具有seqid92所示核苷酸序列的3
´
同源臂,以及可用于具有seqid95所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,221rpm活化3.1小时后,取活化菌液在37.1摄氏度,221rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(312.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(246.8纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37.1摄氏度孵育2.5小时,然后加入dnasei在37.1摄氏度消化25分钟。将所获得的混合物采用磁分离法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(500.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行
混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热30分钟。将所获得的混合物依次通过采用磁分离法,液相等离子体技术(辐射能量为0.1焦每平方厘米,辐射1分钟,机械搅拌,搅拌速度50rpm,液相介质为tris-edta缓冲液,气体为95%氮气和5%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(249.7纳克每微升),纯度为96.9%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.4%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在液相等离子体法技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体法技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37.1摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为37次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0379]
实施例70实施例69所制备的环形mrna分子的靶向脂质体递送系统工艺第一步,将黑磷纳米片和脂质产品按照5.5:74.5摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dspc,dopc,磷脂酰肌醇,磷脂酸和dope按照摩尔比例1:5:11:18:32混合得到的脂质混合物;第二步,将氧浓度为20%的氩气通入含4.5%硫酸镁和5.0%葡萄糖的溶液加入第一步中的混合物a中,然后在3摄氏度下结合液相等离子体技术机械搅拌10分钟,得到混合液a,其中液相等离子体的功率密度为2.2瓦每平方厘米,机械搅拌的速率为180rpm;第三步,将amino-peg4-ch2cooh和amino-peg4-alcohol按1:1摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在3摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为190rpm;
第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s27按照摩尔比例1:1混合得到的混合适配体; hbv感染所致的肝癌为ch6和tls9a按照摩尔比例2:1混合得到的混合适配体; hcv感染所致的肝癌为ly-1, tls11a和as1411按照摩尔比例2:3:1混合得到的混合适配体; hpv感染所致的宫颈癌为as1411和apt按照摩尔比例2:1混合得到的混合适配体; ebv感染所致的原位淋巴瘤为u8, gbm131和td05按照摩尔比例1:1:2混合得到的混合适配体; hltv-1感染所致的成人t细胞白血病为sscgc; hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为120 平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和trimix mrna通过超流控设备进行进一步混合,超流控的总流速为0.25毫升每分钟, 流速比为1/6.5。
[0380] 实施例71 实施例69所制备的环形mrna分子作为疫苗激活免疫反应第一步,将实施例69中合成的环形mrna采用实施例70所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为103纳米,表面荷电35.9
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例69所制备的环形mrna分子采用实施例70所示的靶向递送系统所获得的样品的表面荷电远远高于实施例69所制备的环形mrna分子采用实施例70中所示的未经合理改性的任何一种或多种脂质产品与实施例69所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例70中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例70所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例70所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,
最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.03
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.45
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.12
±
0.04%、 0.44
±
0.05%、0.69
±
0.06%;tnf-α的量分别依次为0.10
±
0.03%、0.14
±
0.04%、0.50
±
0.08%、0.75
±
0.13%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0381]
实施例72实施例69所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例69中合成的环形mrna采用实施例70所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为103纳米,表面荷电35.9
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例69所制备的环形mrna分子采用实施例70所示的靶向递送系统所获得的样品的表面荷电远远高于实施例69所制备的环形mrna分子采用实施例70中所示的未经合理改性的任何一种或多种脂质产品与实施例69所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例70中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例70所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例70所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚
体阳性的抗原特异性cd8+t细胞量为2.11
±
0.6%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.07
±
0.02%、0.14
±
0.03%、 0.43
±
0.05%、0.69
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0382] 实施例73 实施例69所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将1
×
107个ebv阳性且具成瘤能力的鼻咽癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行鼻咽癌移植,建立原位鼻咽癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为13立方毫米,注射第15天后的肿瘤组织大小为32立方毫米,注射第18天后的肿瘤组织大小为55立方毫米,注射第21天后的肿瘤组织大小为108立方毫米,注射第24天后的肿瘤组织大小为160立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为11立方毫米、8立方毫米、7立方毫米、5立方毫米;注射第12天后的肿瘤组织大小分别依次为95立方毫米、82立方毫米、65立方毫米、48立方毫米;注射第15天后的肿瘤组织大小分别依次为203立方毫米、184立方毫米、158立方毫米、115立方毫米;注射第18天后的肿瘤组织大小分别依次为307立方毫米、281立方毫米、255立方毫米、190立方毫米;注射第21天后的肿瘤组织大小分别依次为411立方毫米、383立方毫米、352立方毫米、294立方毫米;注射第24天后的肿瘤组织大小分别依次为493立方毫米、472立方毫米、433立方毫米、392立方毫米)。这说明,实施例69所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例69所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.37
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.03%、0.13
±
0.05%、0.22
±
0.06%、0.39
±
0.07%)。
[0383] 实施例74 实施例69所制备的环形mrna分子激活人类t细胞的情况第一步,分离出ebv阳性鼻咽癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;
每3天更换细胞因子,37摄氏度、co2培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加 入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按 说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯) 和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzyme b)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.27
±
0.2%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.01%、0.12
±
0.03%、0.19
±
0.06%),仅次于pma阳性对
照组(30.50
±
3.52%)。
[0384] 实施例75实施例69所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠皮肤组织细胞或hhv-8阳性原位卡西波肉瘤癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液; 第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hhv-8阳性原位卡西波肉瘤癌细胞中表达明显高于正常皮肤组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hhv-8阳性原位卡西波肉瘤中的表达。
[0385] 实施例76实施例69所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠皮肤组织细胞和hhv-8阳性原位卡西波肉瘤癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠皮肤组织细胞和hhv-8阳性原位卡西波肉瘤癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠皮肤组织细胞和hhv-8阳性原位卡西波肉瘤癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
cgaggtatcgaatgggtggccgcgaagac-3
´
(seqid110)和5
´‑
cctactccgaacctaggtcgatgatgg-3
´
(seqid111);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hhv-8阳性原位卡西波肉瘤癌细胞中高表达,明显高于正常皮肤组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例69所制备的环形mrna分子采用实施例70靶向递送系统递送至hhv-8阳性原位卡西波肉瘤癌细胞的递送率(86.9%)远远高于实施例69中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例69所示靶向递送系统对实施例70所合成的环形mrna递送至hhv-8阳性原位卡西波肉瘤癌细胞的递送率(86.9%)远远高于单独使用实施例70中所示核酸适配体或其他迄今为止文献资
料所报道的hhv-8阳性原位卡西波肉瘤癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例70所示靶向递送系统对实施例69所合成的环形mrna递送至hhv-8阳性原位卡西波肉瘤癌细胞的递送率(86.9%)远远高于用实施例70中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例70中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例69所合成的环形mrna递送至hhv-8阳性原位卡西波肉瘤癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例69所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例69所合成的环形mrna的递送能力优良。
[0386] 实施例77用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid45所示氨基酸序列的抗原多肽编码区,具有seqid62所示核苷酸序列的ires,具有seqid66所示核苷酸序列的第二外显子,具有seqid69所示核苷酸序列的5
´
间隔区,具有seqid72所示核苷酸序列的3
´
间隔区,具有seqid75所示核苷酸序列的第一外显子;具有seqid78核苷酸序列的的启动子,具有seqid81核苷酸序列的5
´
同源臂,具有seqid84核苷酸序列的3
´
内含子,具有seqid87所示核苷酸序列的5
´
内含子,具有seqid90所示核苷酸序列的3
´
同源臂,以及可用于具有seqid93所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3.5小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(277.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(245.9纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2.5小时,然后加入dnasei在37摄氏度消化25分钟。将所获得的混合物采用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(348.5纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在56摄氏度加热20分钟。将所获得的混合物依次通过采用硅膜离心柱法,气相等离子体技术(辐射能量为0.1焦每平方厘米,辐射1分钟,机械搅拌,搅拌速度10rpm,液相介质为tris-edta缓冲液,气体为98%氮气和2%氢气的混合气体,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物
进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(254.3纳克每微升),纯度为97.7%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.2%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在气相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在气相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热25分钟,随后在85摄氏度加热12秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0387] 实施例78实施例77所制备的的环形mrna分子的靶向脂质体递送系统工艺优化第一步,将黑磷粉末和脂质产品按照4.5:75摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dspe,dlin-kc2-dma和sope按照摩尔比例1:18:36混合得到的脂质混合物;第二步,将氧浓度为25%的氦气通入含1.0%氯化钠,3.0%磷酸氢二钠和6.0%葡萄糖的溶液加入第一步中的混合物a中,然后在0摄氏度下结合液相等离子体技术机械搅拌20分钟,得到混合液a,其中液相等离子体的功率密度为1.30瓦每平方厘米,机械搅拌的速率为150rpm;第三步,将nh2-peg2-ch2cooh和amino-peg4-alcohol按1:4摩尔比例混合形成的混合物加入加入第二步获得的混合液a,然后在5摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为7瓦每平方厘米,机械搅拌的速率为190rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s3和s27按照摩尔比例1:2混合得到的混合适配体;hbv感染所致的肝癌为10-8和tls9a按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为ch6,ly-1和as1411按照摩尔比例1:2:1混合得到的混合适配
体; hpv感染所致的宫颈癌为as1411和air-3a按照摩尔比例5:1混合得到的混合适配体; ebv感染所致的原位淋巴瘤为wqy-9-b, gbm128和td05按照摩尔比例3:1:3混合得到的混合适配体; hltv-1感染所致的成人t细胞白血病为sscgc; hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为180 平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环状mrna通过超流控设备进行进一步混合,超流控的总流速为0.30毫升每分钟, 流速比为1/7。
[0388] 实施例79 实施例77所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例77中合成的环形mrna采用实施例78所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为121纳米,表面荷电34.2
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例77所制备的环形mrna分子采用实施例78所示的靶向递送系统所获得的样品的表面荷电远远高于实施例77所制备的环形mrna分子采用实施例78中所示的未经合理改性的任何一种或多种脂质产品与实施例77所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例78中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例78所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例78所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;
第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为1.95
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.29
±
0.3%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.03%、0.12
±
0.04%、 0.40
±
0.05%、0.62
±
0.05%;tnf-α的量分别依次为0.10
±
0.03%、0.13
±
0.03%、0.45
±
0.06%、0.72
±
0.11%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0389]
实施例80实施例77所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例77中合成的环形mrna采用实施例78所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为121纳米,表面荷电34.2
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例77所制备的环形mrna分子采用实施例78所示的靶向递送系统所获得的样品的表面荷电远远高于实施例77所制备的环形mrna分子采用实施例78中所示的未经合理改性的任何一种或多种脂质产品与实施例77所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例78中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例78所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例78所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.11
±
0.6%,远远高于第一组-第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.07
±
0.02%、0.11
±
0.02%、 0.38
±
0.03%、0.67
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原
特异性cd8+t细胞。
[0390] 实施例81 实施例77所制备的的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将2.5
×
105个hpv阳性且具成瘤能力的宫颈癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈移植,建立原位宫颈癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为16立方毫米,注射第15天后的肿瘤组织大小为42立方毫米,注射第18天后的肿瘤组织大小为70立方毫米,注射第21天后的肿瘤组织大小为133立方毫米,注射第24天后的肿瘤组织大小为245立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为12立方毫米、9立方毫米、8立方毫米、7立方毫米;注射第12天后的肿瘤组织大小分别依次为112立方毫米、100立方毫米、66立方毫米、47立方毫米;注射第15天后的肿瘤组织大小分别依次为229立方毫米、204立方毫米、153立方毫米、111立方毫米;注射第18天后的肿瘤组织大小分别依次为356立方毫米、333立方毫米、287立方毫米、232立方毫米;注射第21天后的肿瘤组织大小分别依次为512立方毫米、476立方毫米、404立方毫米、346立方毫米;注射第24天后的肿瘤组织大小分别依次为638立方毫米、586立方毫米、522立方毫米、461立方毫米)。这说明,实施例77所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例77所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.42
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.03%、0.14
±
0.05%、0.23
±
0.08%、0.43
±
0.09%)。
[0391] 实施例82 实施例77所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细
胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.31
±
0.2%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、0.19
±
0.06%),仅次于pma阳性对照组(30.48
±
3.24%)。
[0392] 实施例83实施例77所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达
验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠肝细胞或hbv阳性原位肝癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液; 第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hbv阳性原位肝癌细胞中表达明显高于正常肝细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hbv阳性原位肝癌中的表达。
[0393]
实施例84实施例77所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠肝细胞和hbv阳性原位肝癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
tctagacgtaaggtcgtaccaatcacaaaaccg-3
´
(seqid112)和5
´‑
ccgggtacatcgacactaaggttggtggctaag-3
´
(seqid113);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hbv阳性原位肝癌细胞中高表达,明显高于正常肝细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例77所制备的环形mrna分子采用实施例78靶向递送系统递送至hbv阳性原位肝癌细胞的递送率(87.8%)远远高于实施例78中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例78所示靶向递送系统对实施例77所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(87.8%)远远高于单独使用实施例78中所示核酸适配体或其他迄今为止文献资料所报道的hbv阳性原位肝癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例78所示靶向递送系统对实施例77所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(87.8%)
远远高于用实施例78中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例78中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例77所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例77所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例77所合成的环形mrna的递送能力优良。
[0394]
实施例85用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid50所示氨基酸序列的抗原多肽编码区,具有seqid64所示核苷酸序列的ires,具有seqid67所示核苷酸序列的第二外显子,具有seqid70所示核苷酸序列的5
´
间隔区,具有seqid73所示核苷酸序列的3
´
间隔区,具有seqid76所示核苷酸序列的第一外显子;具有seqid79核苷酸序列的的启动子,具有seqid82核苷酸序列的5
´
同源臂,具有seqid85核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid91所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3.5小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(282.4纳克每微升); 第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(237.3纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnasei在36.9摄氏度消化20分钟。将所获得的混合物采用亲和柱层析法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(332.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在56.3摄氏度加热21分钟。将所获得的混合物依次通过采用亲和柱层析法,超声波技术(声强度为1毫瓦每平方厘米,超声70分钟,机械搅拌,搅拌速度100rpm,液相介质为苹果酸盐缓冲液,气体为空气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而
且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(223.7纳克每微升)纯度为96.3%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(85.9%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在超声波技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在超声波技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热25分钟,随后在85摄氏度加热12秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0395]
实施例86实施例85所制备的的环形mrna分子的靶向脂质体递送系统工艺第一步,将黑磷晶体和脂质产品按照4.5:75.5摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dppg,dodma和popc按照摩尔比例1:25:30混合得到的脂质混合物;第二步,将氧浓度为34%的氙气通入含1.0%氯化钠,2.4%硫酸镁,1.2%磷酸氢二钠,5.0%蔗糖和4.5%葡萄糖的溶液加入第一步中的混合物a中,然后在-2摄氏度下结合液相等离子体技术机械搅拌60分钟,得到混合液a,其中液相等离子体的功率密度为2.5瓦每平方厘米,机械搅拌的速率为400rpm;第三步,将amino-peg4-ch2cooh,nh2-peg2-c2-boc和amino-peg4-alcohol按1:2.5:3摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在3摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为550rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s12和s27按照摩尔比例1:1混合得到的混合适配体;hbv感染所致的肝癌为肽适配子和tls9a按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为肽适配子和ly-1按照摩尔比例2:1混合得到的混合适配体;hpv感染所致的宫颈癌为air-3a和a10按照摩尔比例1:1混合得到的混合适配体;
ebv感染所致的原位淋巴瘤为u2, gbi-10和td05按照摩尔比例4:1:5混合得到的混合适配体; hltv-1感染所致的成人t细胞白血病为sscgc; hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为200平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和trimix mrna通过微流控设备进行进一步混合,微流控的总流速为0.35毫升每分钟, 流速比为1/6.5。
[0396]
实施例87实施例85所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例85中合成的环形mrna采用实施例86所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为148纳米,表面荷电31.6
±
0.4毫伏。此外,进一步地空白对照实验结果表明实施例85所制备的环形mrna分子采用实施例86所示的靶向递送系统所获得的样品的表面荷电远远高于实施例85所制备的环形mrna分子采用实施例86中所示的未经合理改性的任何一种或多种脂质产品与实施例85所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例86中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例86所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例114所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状
细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.21
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.40
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.14
±
0.05%、 0.45
±
0.06%、0.71
±
0.07%;tnf-α的量分别依次为0.09
±
0.03%、0.14
±
0.05%、0.41
±
0.05%、0.74
±
0.19%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0397]
实施例88实施例85所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例85中合成的环形mrna采用实施例86所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为148纳米,表面荷电31.6
±
0.4毫伏。此外,进一步地空白对照实验结果表明实施例85所制备的环形mrna分子采用实施例86所示的靶向递送系统所获得的样品的表面荷电远远高于实施例85所制备的环形mrna分子采用实施例86中所示的未经合理改性的任何一种或多种脂质产品与实施例85所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例86中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例86所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例114所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二组,皮下注射靶向递送系统;第三组,皮下注射靶向递送系统和未修饰的线性mrna;第四组,皮下注射靶向递送系统和修饰的线性mrna;第五组,皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.35
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.08
±
0.03%、0.13
±
0.04%、 0.36
±
0.04%、0.70
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0398]
实施例89实施例85所制备的的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将7.5
×
104个hbv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行肝癌移植,建立原位肝癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为48立方毫米,注射第15天后的肿瘤组织大小为92立方毫米,注射第18天后的肿瘤组织大小为163立方毫米,注射第21天后的肿瘤组织大小为258立方毫米,注射第24天后的肿瘤组织大小为432立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为34立方毫米、27立方毫米、22立方毫米、18立方毫米;注射第12天后的肿瘤组织大小分别依次为256立方毫米、214立方毫米、169立方毫米、134立方毫米;注射第15天后的肿瘤组织大小分别依次为571立方毫米、519立方毫米、432立方毫米、310立方毫米;注射第18天后的肿瘤组织大小分别依次为864立方毫米、810立方毫米、717立方毫米、551立方毫米;注射第21天后的肿瘤组织大小分别依次为1185立方毫米、1093立方毫米、1008立方毫米、974立方毫米;注射第24天后的肿瘤组织大小分别依次为1476立方毫米、1408立方毫米、1329立方毫米、1197立方毫米)。这说明,实施例85所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例85所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.61
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.06
±
0.02%、0.12
±
0.04%、0.21
±
0.06%、0.39
±
0.06%)。
[0399]
实施例90实施例85所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出hbv阳性肝癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;
第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hbv阳性肝癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.19
±
0.2%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.11
±
0.03%、0.17
±
0.05%),仅次于pma阳性对照组(29.89
±
3.05%)。
[0400]
实施例91实施例85所制备的的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证
第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠鼻咽组织细胞或ebv阳性原位鼻咽癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽ebv阳性原位鼻咽癌细胞中表达明显高于正常鼻咽组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在ebv阳性原位鼻咽癌中的表达。
[0401] 实施例92实施例85所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠皮肤组织细胞或mcpyv阳性原位梅克尔癌细胞的rna并进行高通量测序。分析测序结果,确定皮肤组织细胞和mcpyv阳性原位梅克尔癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠皮肤组织细胞和mcpyv阳性原位梅克尔癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
ttaaattattgatctatagcacacctcattc-3
´
(seqid114)和5
´‑
gtgtctgttatcgtcctagttcaattaatcgg-3
´
(seqid115);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在mcpyv阳性原位梅克尔癌细胞中高表达,明显高于正常皮肤组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例85所制备的环形mrna分子采用实施例86靶向递送系统递送至mcpyv阳性原位梅克尔癌细胞的递送率(88.6%)远远高于实施例86中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例86所示靶向递送系统对实施例85所合成的环形mrna递送至mcpyv阳性原位梅克尔癌细胞的递送率(88.6%)远远高于单独使用实施例86中所示核酸适配体或其他迄今为止文献资料所报道的mcpyv阳性原位梅克尔癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例86所示靶向递送系统对实施例85所合成的环形mrna递送至mcpyv阳性原位梅克尔癌细胞的递送率(88.6%)远远高于用实施例86中所示核酸适配体或
其他迄今为止文献资料所报道的其他核酸适配体与实施例86中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例85所合成的环形mrna递送至mcpyv阳性原位梅克尔癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例85所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例85所合成的环形mrna的递送能力优良。
[0402]
实施例93用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid55所示氨基酸序列的抗原多肽编码区,具有seqid65所示核苷酸序列的ires,具有seqid68所示核苷酸序列的第二外显子,具有seqid71所示核苷酸序列的5
´
间隔区,具有seqid74所示核苷酸序列的3
´
间隔区,具有seqid77所示核苷酸序列的第一外显子;具有seqid80核苷酸序列的的启动子,具有seqid83核苷酸序列的5
´
同源臂,具有seqid86核苷酸序列的3
´
内含子,具有seqid89所示核苷酸序列的5
´
内含子,具有seqid92所示核苷酸序列的3
´
同源臂,以及可用于具有seqid95所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,220rpm活化3小时后,取活化菌液在37.1摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(330.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37.1摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(320纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37.1摄氏度孵育2.2小时,然后加入dnasei在37.1摄氏度消化24分钟。将所获得的混合物采用尺寸排阻柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(584.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热30分钟。将所获得的混合物依次通过采用尺寸排阻柱法,液相等离子体技术(辐射能量为1毫焦每平方厘米,辐射5分钟,机械搅拌,搅拌速度100rpm,液相介质为苹果酸盐缓冲液,气体为空气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定
mrna环化效率和纯化后的环形mrna浓度(300.4纳克每微升)纯度为99.1%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.8%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在液相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37.1摄氏度加热20分钟,随后在84摄氏度加热9秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0403] 实施例94实施例93所制备的环形mrna分子的靶向脂质体递送系统工艺优化第一步,将石墨烯状黑磷和脂质产品按照7:70摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为dspc,反式dope,dotap,otma和dope按照摩尔比例1:3:20:12:36混合得到的脂质混合物;第二步,将氧浓度为100毫克每升的含1.2%氯化钠,2.8%氯化镁,3.5%磷酸氢二钾,6.0%蔗糖和4.2%葡萄糖的溶液加入第一步中的混合物a中,然后在4摄氏度下结合气相等离子体技术机械搅拌150分钟,得到混合液a,其中气相等离子体电子和离子的能量为100ev,机械搅拌的速率为40rpm;第三步,将amino-peg4-ch2cooh,nh2-peg2-c2-boc和nh2peg5cooh按1:1:1摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在8摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为400rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s27按照摩尔比例1:2混合得到的混合适配体;hbv感染所致的肝癌为as1411和tls9a按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为10-8和tls11a按照摩尔比例1:3混合得到的混合适配体;hpv感染所致的宫颈癌为as1411和a10按照摩尔比例3:2混合得到的混合适配体;ebv感染所致的原位淋巴瘤为u8,gbm-128和td05按照摩尔比例1:1:3混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸
酯;mcpyv感染所致的梅克尔癌为a10 2
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为100 平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和线性mrna通过微流控设备进行进一步混合,微流控的总流速为0.40毫升每分钟, 流速比为1/4。
[0404]
实施例95实施例93所制备的环形mrna分子作为疫苗激活免疫反应第一步,将实施例93中合成的环形mrna采用实施例94所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为143纳米,表面荷电32.0
±
0.5毫伏。此外,进一步地空白对照实验结果表明实施例93所制备的环形mrna分子采用实施例94所示的靶向递送系统所获得的样品的表面荷电远远高于实施例93所制备的环形mrna分子采用实施例94中所示的未经合理改性的任何一种或多种脂质产品与实施例93所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例94中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例94所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例94所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿
瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.17
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.48
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.11
±
0.03%、 0.36
±
0.04%、0.57
±
0.05%;tnf-α的量分别依次为0.10
±
0.02%、0.14
±
0.04%、0.41
±
0.05%、0.65
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0405]
实施例96实施例93所制备的的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例93中合成的环形mrna采用实施例94中所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为143纳米,表面荷电32.0
±
0.5毫伏。此外,进一步地空白对照实验结果表明实施例93所制备的环形mrna分子采用实施例94所示的靶向递送系统所获得的样品的表面荷电远远高于实施例93所制备的环形mrna分子采用实施例94中所示的未经合理改性的任何一种或多种脂质产品与实施例93所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例94中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例94所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例94所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.34
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.09
±
0.03%、0.13
±
0.03%、 0.43
±
0.05%、0.71
±
0.2%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0406] 实施例97 实施例93所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生
长第一步,取30只6-8周的体重20克的小鼠,将1
×
107个ebv阳性且具成瘤能力的鼻咽癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行鼻咽癌移植,建立原位鼻咽癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为12立方毫米,注射第15天后的肿瘤组织大小为30立方毫米,注射第18天后的肿瘤组织大小为51立方毫米,注射第21天后的肿瘤组织大小为104立方毫米,注射第24天后的肿瘤组织大小为155立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为10立方毫米、9立方毫米、7立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为102立方毫米、93立方毫米、70立方毫米、49立方毫米;注射第15天后的肿瘤组织大小分别依次为209立方毫米、197立方毫米、165立方毫米、120立方毫米;注射第18天后的肿瘤组织大小分别依次为314立方毫米、294立方毫米、263立方毫米、205立方毫米;注射第21天后的肿瘤组织大小分别依次为418立方毫米、389立方毫米、359立方毫米、311立方毫米;注射第24天后的肿瘤组织大小分别依次为505立方毫米、483立方毫米、441立方毫米、402立方毫米)。这说明,实施例93所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例93所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.67
±
0.6%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.12
±
0.03%、0.25
±
0.05%、0.49
±
0.07%)。
[0407] 实施例98实施例93所制备的环形mrna分子激活人类t细胞的情况第一步,分离出ebv阳性鼻咽癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗(青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰
的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出ebv阳性鼻咽癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.56
±
0.4%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.11
±
0.03%、0.21
±
0.07%),仅次于pma阳性对照组(29.77
±
3.09%)。
[0408] 实施例99实施例93所制备的的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进
行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠肝细胞或hcv阳性原位肝癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hcv阳性原位肝癌细胞中表达明显高于正常肝细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hcv阳性原位肝癌中的表达。
[0409]
实施例100实施例93所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠肝细胞和hcv阳性原位肝癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠肝细胞和hcv阳性原位肝癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠肝细胞和hcv阳性原位肝癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
ccaccagtattgtgaaatgtagactgc-3
´
(seqid116)和5
´‑
aaaccagtattgtgaaatgtagacttc-3
´
(seqid117);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hcv阳性原位肝癌细胞中高表达,明显高于正常肝细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例93所制备的环形mrna分子采用实施例94靶向递送系统递送至hcv阳性原位肝癌细胞的递送率(85.1%)远远高于实施例94中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例94所示靶向递送系统对实施例93所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(85.1%)远远高于单独使用实施例94中所示核酸适配体或其他迄今为止文献资料所报道的hcv阳性原位肝癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例94所示靶向递送系统对实施例93所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(85.1%)远远高于用实施例94中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例94中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文
献资料迄今所报道的脂质体组合合成的递送系统对实施例93所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例93所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例93所合成的环形mrna的递送能力优良。
[0410] 实施例101用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid60所示氨基酸序列的抗原多肽编码区,具有seqid62所示核苷酸序列的ires,具有seqid66所示核苷酸序列的第二外显子,具有seqid69所示核苷酸序列的5
´
间隔区,具有seqid72所示核苷酸序列的3
´
间隔区,具有seqid75所示核苷酸序列的第一外显子;具有seqid78核苷酸序列的的启动子,具有seqid81核苷酸序列的5
´
同源臂,具有seqid84核苷酸序列的3
´
内含子,具有seqid87所示核苷酸序列的5
´
内含子,具有seqid90所示核苷酸序列的3
´
同源臂,以及可用于具有seqid93所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(273.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(245.6纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnasei在36.9摄氏度消化25分钟。将所获得的混合物采用磁分离法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(307.8纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物依次通过采用磁分离法,气相等离子体技术(辐射能量为50毫焦每平方厘米,辐射10分钟,机械搅拌,搅拌速度15rpm,液相介质为苹果酸盐缓冲液,气体为空气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(255.5纳克每微升)纯度为95.9%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成
环形mrna的纯度(86.7%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在气相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在气相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在84摄氏度加热9秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0411] 实施例102实施例101所制备的环形mrna分子的靶向脂质体递送系统工艺第一步,将石墨烯状黑磷和脂质产品按照6:81摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为peg修饰的二烷基胺,peg-pe,peg修饰的神经酰胺,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:5:12:24:30混合得到的脂质混合物;第二步,将空气通入含2.53%氯化钾和5.1%蔗糖的溶液加入第一步中的混合物a中,然后在-4摄氏度下结合气相等离子体技术机械搅拌120分钟,得到混合液a,其中气相等离子体电子和离子的能量为80电子伏特,机械搅拌的速率为30rpm;第三步,将amino-peg4-alcohol,nh2-peg2-c2-boc,amino-peg4-ch2cooh,和nh2-peg10-cooh按2:2:2:1摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在10摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为3瓦每平方厘米,机械搅拌的速率为375rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s5,s12和s27按照摩尔比例1:2:3混合得到的混合适配体;hbv感染所致的肝癌为ch6:zy1和tls9a按照摩尔比例1:1:2混合得到的混合适配体;hcv感染所致的肝癌为ly-1:肽适配子和as1411按照摩尔比例2:1:1混合得到的混合适配体;hpv感染所致的宫颈癌为air-3a,as1411和a10按照摩尔比例2:3:1混合得到的混合适配体;ebv感染所致的原位淋巴瘤为u9和td05按照摩尔比例1:4混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;
第五步,将第四步中的混合液c通过有效面积为125平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为0.75毫升每分钟,流速比为1/4。
[0412]
实施例103实施例101所制备的环形mrna分子作为疫苗激活免疫反应第一步,将实施例101中合成的环形mrna采用实施例102所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为115纳米,表面荷电34.7
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例101所制备的环形mrna分子采用实施例102所示的靶向递送系统所获得的样品的表面荷电远远高于实施例101所制备的环形mrna分子采用实施例102中所示的未经合理改性的任何一种或多种脂质产品与实施例101所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例102中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例102所示的靶向递送系统的表面荷电和粒径;第二步,将6-8周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10%(v/v)fbs的rpmi1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldina(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细
胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.61
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.89
±
0.6%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.02%、0.13
±
0.04%、 0.42
±
0.06%、0.67
±
0.07%;tnf-α的量分别依次为0.12
±
0.03%、0.14
±
0.03%、0.49
±
0.07%、0.76
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0413] 实施例104 实施例101所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例101中合成的环形mrna采用实施例102所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为115纳米,表面荷电34.7
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例101所制备的环形mrna分子采用实施例102所示的靶向递送系统所获得的样品的表面荷电远远高于实施例101所制备的环形mrna分子采用实施例102中所示的未经合理改性的任何一种或多种脂质产品与实施例101所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例102中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例102所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例102所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.40
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.08
±
0.02%、0.13
±
0.03%、 0.41
±
0.04%、0.70
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0414]
实施例105实施例101所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将2.5
×
105个hpv阳性且具成瘤能力的宫颈癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈移植,建立原位宫颈癌小鼠模型;
第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为14立方毫米,注射第15天后的肿瘤组织大小为43立方毫米,注射第18天后的肿瘤组织大小为67立方毫米,注射第21天后的肿瘤组织大小为129立方毫米,注射第24天后的肿瘤组织大小为237立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为13立方毫米、9立方毫米、7立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为117立方毫米、102立方毫米、64立方毫米、45立方毫米;注射第15天后的肿瘤组织大小分别依次为234立方毫米、201立方毫米、150立方毫米、108立方毫米;注射第18天后的肿瘤组织大小分别依次为359立方毫米、329立方毫米、279立方毫米、227立方毫米;注射第21天后的肿瘤组织大小分别依次为520立方毫米、476立方毫米、396立方毫米、338立方毫米;注射第24天后的肿瘤组织大小分别依次为644立方毫米、597立方毫米、513立方毫米、457立方毫米)。这说明,实施例101所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例101所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.65
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.12
±
0.03%、0.21
±
0.06%、0.38
±
0.07%)。
[0415] 实施例106 实施例101所制备的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加
入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯) 和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzyme b)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.47
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.12
±
0.03%、0.21
±
0.07%),仅次于pma阳性对照组(29.32
±
2.99%)。
[0416]
实施例107实施例101所制备的的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步, 将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行; 第二步, 每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠正常皮肤组织细胞或mcpyv阳性原位梅克尔癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离
心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液; 第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在mcpyv阳性原位梅克尔癌细中表达明显高于正常皮肤组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在mcpyv阳性原位梅克尔癌细中的表达。
[0417] 实施例108实施例101所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠鼻咽组织细胞或ebv阳性原位鼻咽癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠鼻咽组织细胞或ebv阳性原位鼻咽癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠鼻咽组织细胞或ebv阳性原位鼻咽癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
ctgacataaaccacgaccctacgg-3
´
(seqid118)和5
´‑
agaatcccaaggaggcggagac-3
´
(seqid119);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在ebv阳性原位鼻咽癌细胞中高表达,明显高于正常鼻咽组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例101所制备的环形mrna分子采用实施例102靶向递送系统递送至ebv阳性原位鼻咽癌细胞的递送率(89.4%)远远高于实施例102中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例102所示靶向递送系统对实施例101所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(89.4%)远远高于单独使用实施例102中所示核酸适配体或其他迄今为止文献资料所报道的ebv阳性原位鼻咽癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例102所示靶向递送系统对实施例101所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(89.4%)远远高于用实施例102中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例102中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例101所合成的环形mrna递送至ebv阳性原位鼻咽癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例101所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例101所合成
的环形mrna的递送能力优良。
[0418] 实施例109用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid3所示氨基酸序列的抗原多肽编码区,具有seqid62所示核苷酸序列的ires,具有seqid65所示核苷酸序列的第二外显子,具有seqid71所示核苷酸序列的5’间隔区,具有seqid72所示核苷酸序列的3’间隔区,具有seqid76所示核苷酸序列的第一外显子;具有seqid80核苷酸序列的的启动子,具有seqid81核苷酸序列的5
´
同源臂,具有seqid85核苷酸序列的3
´
内含子,具有seqid89所示核苷酸序列的5
´
内含子,具有seqid92所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,218rpm活化3小时后,取活化菌液在37摄氏度,218rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(402.4纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(305.9纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2.5小时,然后加入dnasei在37摄氏度消化21分钟。将所获得的混合物采用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(335.2纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物依次通过采用硅膜离心柱法,超声波技术(声强度为500微瓦每平方厘米,超声80分钟,机械搅拌,搅拌速度15rpm,液相介质为组氨酸盐缓冲液,气体为氦气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(297.6纳克每微升)纯度为96.5%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.4%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在超声波技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在超声波技术处理后,再进行磷酸酶处理及rna酶r处理,在随
后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存; 第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为37次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0419] 实施例110实施例109所制备的环形mrna分子的靶向脂质体递送系统工艺第一步,将石墨烯状黑磷和脂质产品按照6:66摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为peg-c-dmg,peg-c-dma,磷脂酰甘油,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:7.5:17:28:35混合得到的脂质混合物;第二步,将氧浓度为28%的氩气通入含1.2%氯化钠,3.5%氯化钾和6.8%蔗糖的溶液加入第一步中的混合物a中,然后在-6摄氏度下结合气相等离子体技术机械搅拌90分钟,得到混合液a,其中气相等离子体电子和离子的能量为75ev,机械搅拌的速率为25rpm;第三步,将amino-peg4-alcohol,nh2-peg2-c2-boc和nh2-peg12-cooh按1:3:2摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在1摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为2瓦每平方厘米,机械搅拌的速率为325rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s3按照摩尔比例1:1混合得到的混合适配体;hbv感染所致的肝癌为zy1和10-8按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为肽适配子和as1411按照摩尔比例3:1混合得到的混合适配体;hpv感染所致的宫颈癌为air-3a和a10按照摩尔比例2:3混合得到的混合适配体;ebv感染所致的原位淋巴瘤为u31和td05按照摩尔比例1:6混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为140平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为0.9毫升每分钟,流速比为1/5。
[0420] 实施例111 实施例109所制备的环形mrna分子作为疫苗激活免疫反应第一步,将实施例109中合成的环形mrna采用实施例110所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为92纳米,表面荷电37.3
±
0.1毫伏。此外,进一步地空白对照实验结果表明实施例109所制备的环形mrna分子采用实施例110所示的靶向递送系统所获得的样品的表面荷电远远高于实施例109所制备的环形mrna分子采用实施例110中所示的未经合理改性的任何一种或多种脂质产品与实施例109所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例110中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例110所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例110所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.13
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量
为2.41
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.13
±
0.03%、 0.38
±
0.04%、0.60
±
0.05%;tnf-α的量分别依次为0.09
±
0.02%、0.12
±
0.03%、0.43
±
0.05%、0.68
±
0.10%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0421] 实施例112 实施例109所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例109中合成的环形mrna采用所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为92纳米,表面荷电37.3
±
0.1毫伏。此外,进一步地空白对照实验结果表明实施例109所制备的环形mrna分子采用实施例110所示的靶向递送系统所获得的样品的表面荷电远远高于实施例109所制备的环形mrna分子采用实施例110中所示的未经合理改性的任何一种或多种脂质产品与实施例109所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例110中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例110所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例110所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.41
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.09
±
0.03%、0.14
±
0.04%、 0.42
±
0.04%、0.70
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0422] 实施例113 实施例109所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将7.5
×
104个hpv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行肝癌移植,建立原位肝癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向
递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为52立方毫米,注射第15天后的肿瘤组织大小为90立方毫米,注射第18天后的肿瘤组织大小为176立方毫米,注射第21天后的肿瘤组织大小为251立方毫米,注射第24天后的肿瘤组织大小为435立方毫米)比第一组~第四组生长缓慢的多 (注射第9天后的肿瘤组织大小分别依次为36立方毫米、28立方毫米、23立方毫米、19立方毫米;注射第12天后的肿瘤组织大小分别依次为263立方毫米、221立方毫米、174立方毫米、138立方毫米;注射第15天后的肿瘤组织大小分别依次为589立方毫米、533立方毫米、440立方毫米、321立方毫米;注射第18天后的肿瘤组织大小分别依次为871立方毫米、819立方毫米、722立方毫米、564立方毫米;注射第21天后的肿瘤组织大小分别依次为1182立方毫米、1104立方毫米、1017立方毫米、895立方毫米;注射第24天后的肿瘤组织大小分别依次为1483立方毫米、1420立方毫米、1341立方毫米、1202立方毫米)。这说明,实施例97所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例97所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8 t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激 肿瘤浸润cd8 t细胞产生细胞因子ifn-r的量为1.77
±
0.6%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.12
±
0.03%、0.24
±
0.06%、0.40
±
0.07%)。
[0423] 实施例114 实施例109所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗 (青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的 gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组, cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t 细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的 t细胞转移到包被人ifn-γ抗体的ifn-γ elispot 96孔板中共培养,24 小时后弃去上清,pbs清洗5次后加入生物素化的检测一
抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.55
±
0.5%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.11
±
0.03%、0.18
±
0.04%),仅次于pma阳性对照组(29.91
±
3.33%)。
[0424] 实施例115实施例109所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠淋巴组织细胞或ebv阳性原位淋巴瘤细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;
ꢀ
第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在ebv阳性原位淋巴瘤细胞中表达明显高于正常淋巴组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在ebv阳性原位淋巴瘤中的表达。
[0425] 实施例116实施例109所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠淋巴组织细胞和ebv阳性原位淋巴瘤细胞的rna并进行高通量测序。分析测序结果,确定小鼠淋巴组织细胞和ebv阳性原位鼻咽癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠淋巴组织细胞和ebv阳性原位淋巴瘤细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
gcctccactgtcgtctccac-3
´
(seqid120)和5
´‑
atcaacatccactggtcctt-3
´
(seqid121);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在ebv阳性原位淋巴瘤细胞中高表达,明显高于正常淋巴组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例109所制备的环形mrna分子采用实施例110靶向递送系统递送至ebv阳性原位淋巴瘤细胞的递送率(90.3%)远远高于实施例110中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例110所示靶向递送系统对实施例109所合成的环形mrna递送至ebv阳性原位淋巴瘤细胞的递送率(90.3%)远远高于单独使用实施例110中所示核酸适配体或其他迄今为止文献资料所报道的ebv阳性原位淋巴瘤细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例110所示靶向递送系统对实施例109所合成的环形mrna递送至ebv阳性原位淋巴瘤细胞的递送率(90.3%)远远高于用实施例110中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例110中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例109所合成的环形mrna递送至ebv阳性原位淋巴瘤细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例109所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例109所合成的环形mrna的递送能力优良。
[0426] 实施例117用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid12所示氨基酸序列的抗原多肽编码区,具有seqid62所示核苷酸序列的ires,具有seqid68所示核苷酸序列的第二外显子,具有seqid71所示核苷酸序列的5’间隔区,具有seqid73所示核苷酸序列的3’间隔区,具有seqid76所示核苷酸序列的第一外显子;具有seqid78核苷酸序列的的启动子,具有seqid81核苷酸序列的5’同源臂,具有seqid85核苷酸序列的3’内含子,具有seqid87所示核苷酸序列的5’内含子,具有seqid91所示核苷酸序列的3’同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.8摄氏度,220rpm活化3.5小时后,取活化菌液在36.8摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(292.6纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.8摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(247.8纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.8摄氏度孵育2.5小时,然后加入dnasei在36.8摄氏度消化25分钟。将所获得的混合物采用亲和柱层析法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(515.9纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热30分钟。将所获得的混合物依次通过采用亲和柱层析法,液相等离子体技术(辐射能量为500微焦每平方厘米,辐射20分钟,机械搅拌,搅拌速度200rpm,液相介质为组氨酸盐缓冲液,气体为氦气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(251.4纳克每微升)纯度为98.8%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(88.7%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在液相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体技术处理后,再进行磷酸酶处理
及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.8摄氏度加热20分钟,随后在85摄氏度加热9秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0427] 实施例118递送系统库中环形mrna的靶向脂质体递送系统工艺优化第一步,将石墨烯状黑磷和脂质产品按照4:80摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为daa,peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:5:15:25:30混合得到的脂质混合物;第二步,将氧浓度为35%的氦气通入含4.0%磷酸二氢钠和4.0%蔗糖的溶液加入第一步中的混合物a中,然后在10摄氏度下结合气相等离子体技术机械搅拌70分钟,得到混合液a,其中气相等离子体电子和离子的能量为60ev,机械搅拌的速率为20rpm;第三步,将amino-peg4-alcohol,amino-peg4-ch2cooh和nh2-peg3-cooh按3:1:1摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在12摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为1瓦每平方厘米,机械搅拌的速率为250rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s3,s12和s27按照摩尔比例1:3:1混合得到的混合适配体;hbv感染所致的肝癌为ly1,肽适配子和tls11a按照摩尔比例1:2:1混合得到的混合适配体;hcv感染所致的肝癌为zy1,tls9a和as1411按照摩尔比例1:1:1混合得到的混合适配体;hpv感染所致的宫颈癌为as1411和a10按照摩尔比例4:1混合得到的混合适配体;ebv感染所致的原位淋巴瘤为gbm128和td05按照摩尔比例5:2混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为110平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为1毫升每分钟,流速比为1/9。
[0428] 实施例119实施例117所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例117中合成的环形mrna采用实施例118所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为124纳米,表面荷电33.8
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例117所制备的环形mrna分子采用实施例118所示的靶向递送系统所获得的样品的表面荷电远远高于实施例117所制备的环形mrna分子采用实施例118中所示的未经合理改性的任何一种或多种脂质产品与实施例117所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例118中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例118所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例118所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10%(v/v)fbs的rpmi1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldina(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.14
±
0.3%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),tnf-α的量
为2.42
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、 0.44
±
0.06%、0.67
±
0.07%;tnf-α的量分别依次为0.11
±
0.03%、0.15
±
0.05%、0.41
±
0.05%、0.73
±
0.09%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0429]
实施例120实施例117所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例117中合成的环形mrna采用实施例118所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为124纳米,表面荷电33.8
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例117所制备的环形mrna分子采用实施例118所示的靶向递送系统所获得的样品的表面荷电远远高于实施例117所制备的环形mrna分子采用实施例118中所示的未经合理改性的任何一种或多种脂质产品与实施例117所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例118中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例118所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例118所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.52
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.09
±
0.03%、0.13
±
0.03%、 0.41
±
0.04%、0.68
±
0.09%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0430]
实施例121实施例117所制备的的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将1
×
107个hpv阳性且具成瘤能力的鼻咽癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行鼻咽癌移植,建立原位鼻咽癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向
递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为14立方毫米,注射第15天后的肿瘤组织大小为30立方毫米,注射第18天后的肿瘤组织大小为52立方毫米,注射第21天后的肿瘤组织大小为105立方毫米,注射第24天后的肿瘤组织大小为158立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为11立方毫米、9立方毫米、7立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为92立方毫米、84立方毫米、66立方毫米、47立方毫米;注射第15天后的肿瘤组织大小分别依次为210立方毫米、181立方毫米、160立方毫米、112立方毫米;注射第18天后的肿瘤组织大小分别依次为306立方毫米、289立方毫米、252立方毫米、186立方毫米;注射第21天后的肿瘤组织大小分别依次为417立方毫米、394立方毫米、363立方毫米、287立方毫米;注射第24天后的肿瘤组织大小分别依次为493立方毫米、481立方毫米、448立方毫米、389立方毫米)。这说明,实施例117所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例117所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.77
±
0.5%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.13
±
0.04%、0.25
±
0.07%、0.41
±
0.08%)。
[0431] 实施例122实施例117所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗(青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一
抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.63
±
0.5%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.06
±
0.02%、0.11
±
0.03%、0.21
±
0.07%),仅次于pma阳性对照组(30.01
±
3.07%)。
[0432]
实施例123实施例117所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠肝细胞或hbv阳性原位肝癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇
匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hbv阳性原位肝癌细胞中表达明显高于正常肝细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hbv阳性原位肝癌中的表达。
[0433] 实施例124实施例117所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠肝细胞和hbv阳性原位肝癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠肝细胞和hbv阳性原位肝癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
acgtaagccgcggtcgacata-3
´
(seqid122)和5
´‑
gtagtccctgtccgtcagta-3
´
(seqid123);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hbv阳性原位肝癌细胞中高表达,明显高于正常肝细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例117所制备的环形mrna分子采用实施例118靶向递送系统递送至hbv阳性原位肝癌细胞的递送率(88.8%)远远高于实施例118中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例118所示靶向递送系统对实施例117所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(88.8%)远远高于单独使用实施例118中所示核酸适配体或其他迄今为止文献资料所报道的hbv阳性原位肝癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例118所示靶向递送系统对实施例117所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(88.8%)远远高于用实施例118中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例118中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例117所合成的环形mrna递送至hbv阳性原位肝癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例117所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例117所合成的环形mrna的递送能力优良。
[0434] 实施例125用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备
第一步,构建具有seq id 22所示氨基酸序列的抗原多肽编码区,具有seq id 63所示核苷酸序列的ires,具有seq id 67所示核苷酸序列的第二外显子,具有seq id 71所示核苷酸序列的5
´
间隔区,具有seq id 74所示核苷酸序列的3
´
间隔区,具有seq id 77所示核苷酸序列的第一外显子;具有seq id 79核苷酸序列的的启动子,具有seq id 82核苷酸序列的5
´
同源臂,具有seq id 85核苷酸序列的3
´
内含子,具有seq id 88所示核苷酸序列的5
´
内含子,具有seq id 92所示核苷酸序列的3
´
同源臂,以及可用于具有seq id 95所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,225rpm活化3小时后,取活化菌液在36.9摄氏度,225rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(294.6纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(265.7纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnase i在36.9摄氏度消化25分钟。将所获得的混合物采用尺寸排阻柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(515.2纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物依次通过采用尺寸排阻柱法,气相等离子体技术(辐射能量为500微焦每平方厘米,辐射20分钟,机械搅拌,搅拌速度20rpm,液相介质为组氨酸盐缓冲液,气体为氦气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(288.1纳克每微升),纯度为99.0%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(88.4%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在气相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在气相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质
的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热25分钟,随后在85摄氏度加热11秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0435] 实施例126实施例125所制备的的环形mrna分子的靶向脂质体递送系统工艺第一步,将石墨烯状黑磷和脂质产品按照8.6:61摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为反式dope,otma和popg按照摩尔比例1:18:32混合得到的脂质混合物;第二步,将氧浓度为42%的氙气通入含6.5%氯化钙和2.5%葡萄糖的溶液加入第一步中的混合物a中,然后在4摄氏度下结合气相等离子体技术机械搅拌55分钟,得到混合液a,其中气相等离子体电子和离子的能量为55ev,机械搅拌的速率为16rpm;第三步,将amino-peg4-alcohol,nh2-peg2-c2-boc和nh2-peg2-cooh按2:1:1摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在0摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为0.75瓦每平方厘米,机械搅拌的速率为225rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s5按照摩尔比例1:1混合得到的混合适配体;hbv感染所致的肝癌为tls11a和tls9a按照摩尔比例3:2混合得到的混合适配体;hcv感染所致的肝癌为肽适配子和as1411按照摩尔比例2:1混合得到的混合适配体;hpv感染所致的宫颈癌为as1411和a10按照摩尔比例3:4混合得到的混合适配体;ebv感染所致的原位淋巴瘤为gbm131和gbi-10按照摩尔比例1:1混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为170平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为1.25毫升每分钟,流速比为1/5。
[0436]
实施例127实施例125所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例125中合成的环形mrna采用实施例126所示的脂质体配方工艺制
备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为97纳米,表面荷电36.6
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例125所制备的环形mrna分子采用实施例126所示的靶向递送系统所获得的样品的表面荷电远远高于实施例125所制备的环形mrna分子采用实施例126中所示的未经合理改性的任何一种或多种脂质产品与实施例125所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例126中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例126所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例126所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.07
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.31
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、 0.38
±
0.05%、0.67
±
0.06%;
tnf-α的量分别依次为0.11
±
0.03%、0.14
±
0.04%、0.43
±
0.06%、0.74
±
0.12%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0437]
实施例128实施例125所制备环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例125中合成的环形mrna采用实施例126所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为97纳米,表面荷电36.6
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例125所制备的环形mrna分子采用实施例126所示的靶向递送系统所获得的样品的表面荷电远远高于实施例125所制备的环形mrna分子采用实施例126中所示的未经合理改性的任何一种或多种脂质产品与实施例125所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例126中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例126所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例126所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.25
±
0.5%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.08
±
0.03%、0.13
±
0.03%、 0.34
±
0.04%、0.69
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0438]
实施例129实施例125所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将2.5
×
105个hpv阳性且具成瘤能力的宫颈癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈移植,建立原位宫颈癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;
第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为14立方毫米,注射第15天后的肿瘤组织大小为44立方毫米,注射第18天后的肿瘤组织大小为68立方毫米,注射第21天后的肿瘤组织大小为135立方毫米,注射第24天后的肿瘤组织大小为250立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为12立方毫米、10立方毫米、8立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为115立方毫米、104立方毫米、70立方毫米、49立方毫米;注射第15天后的肿瘤组织大小分别依次为236立方毫米、209立方毫米、158立方毫米、117立方毫米;注射第18天后的肿瘤组织大小分别依次为378立方毫米、348立方毫米、294立方毫米、226立方毫米;注射第21天后的肿瘤组织大小分别依次为520立方毫米、487立方毫米、412立方毫米、339立方毫米;注射第24天后的肿瘤组织大小分别依次为645立方毫米、599立方毫米、530立方毫米、453立方毫米)。这说明,实施例125所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例125所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.73
±
0.7%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.15
±
0.06%、0.25
±
0.08%、0.47
±
0.09%)。
[0439] 实施例130实施例125所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出ebv阳性鼻咽癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗(青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按
说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫 斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0 .5微克每毫升pma(豆蔻酰佛波醇乙酯) 和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzyme b)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.55
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.11
±
0.03%、0.18
±
0.05%),仅次于pma阳性对照组(29.79
±
3.00%)。
[0440]
实施例131实施例125所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步, 将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步, 每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠肝细胞或hcv阳性原位肝癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加 10 微升 pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入
100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在hcv阳性原位肝癌细胞中表达明显高于正常肝细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hcv阳性原位肝癌中的表达。
[0441] 实施例132实施例125所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠肝细胞和hcv阳性原位肝癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠肝细胞和hcv阳性原位肝癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠肝细胞和hcv阳性原位肝癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
tcacactcgctactcgtgcact-3
´
(seqid124)和5
´‑
cttccgacctaacacgac-3
´
(seqid125);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hcv阳性原位肝癌细胞中高表达,明显高于正常肝细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例125所制备的环形mrna分子采用实施例126靶向递送系统递送至hcv阳性原位肝癌细胞的递送率(89.5%)远远高于实施例126中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例126所示靶向递送系统对实施例125所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(89.5%)远远高于单独使用实施例126中所示核酸适配体或其他迄今为止文献资料所报道的hcv阳性原位肝癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例126所示靶向递送系统对实施例125所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(89.5%)远远高于用实施例126中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例126中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例125所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例125所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例125所合成的环形mrna的递送能力优良。
[0442] 实施例133用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid37所示氨基酸序列的抗原多肽编码区,具有seqid64所示核苷酸序列的ires,具有seqid67所示核苷酸序列的第二外显子,具有seqid69所
示核苷酸序列的5
´
间隔区,具有seqid73所示核苷酸序列的3
´
间隔区,具有seqid76所示核苷酸序列的第一外显子;具有seqid79核苷酸序列的的启动子,具有seqid82核苷酸序列的5
´
同源臂,具有seqid85核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid91所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3.5小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(393.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(310.4纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2.5小时,然后加入dnasei在37摄氏度消化20分钟。将所获得的混合物采用磁分离法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(338.7纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热30分钟。将所获得的混合物依次通过采用磁分离法,超声波技术(声强度为500微瓦每平方厘米,超声100分钟,机械搅拌,搅拌速度300rpm,液相介质为天冬氨酸盐缓冲液,气体为氩气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(278.3纳克每微升),纯度为98.3%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.6%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在超声波技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在超声波技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;
第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在84.5摄氏度加热15秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为38次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0443] 实施例134实施例133所制备的的环形mrna分子的靶向脂质体递送系统工艺第一步,将石墨烯状黑磷和脂质产品按照8.6:65摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为daa,peg-c-domg,galnac-peg-dsg,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:6:13:20:40混合得到的脂质混合物;第二步,将氧浓度为120毫克每升的含1.0%氯化钠,6.3%氯化镁和5.5%葡萄糖的溶液加入第一步中的混合物a中,然后在-7摄氏度下结合超声波技术机械搅拌180分钟,得到混合液a,其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为700rpm;第三步,将amino-peg4-alcohol,nh2-peg2-c2-boc和nh2-peg7-cooh按2:1:3摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在-4摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为550rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s3按照摩尔比例3:1混合得到的混合适配体;hbv感染所致的肝癌为ly1和tls11a按照摩尔比例3:2混合得到的混合适配体;hcv感染所致的肝癌为tls10a和ch6按照摩尔比例2:1混合得到的混合适配体;hpv感染所致的宫颈癌为as1411和air-3a按照摩尔比例5:3混合得到的混合适配体;ebv感染所致的原位淋巴瘤为u2,u8和td05按照摩尔比例1:1:4混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为180平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为1.5毫升每分钟,流速比为1/5。
[0444] 实施例135实施例133所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例133中合成的环形mrna采用实施例134所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为108纳米,表面荷电37.2
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例133所制备的环形mrna分子采用实施例134所示的靶向递送系统所获得的样品的表面荷电远远高于实施例133所制备的环形mrna分子采用实施例134中所示的未经合理改性的任何一种
或多种脂质产品与实施例133所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例134中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例134所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例134所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.27
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.55
±
0.4%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、 0.39
±
0.05%、0.65
±
0.05%;tnf-α的量分别依次为0.120.03%、0.15
±
0.03%、0.48
±
0.05%、0.77
±
0.12%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0445]
实施例136实施例133所制备的的环形mrna分子皮下注射可刺激产生抗原特异性t细胞
第一步,将实施例133中合成的环形mrna采用实施例134所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为108纳米,表面荷电37.2
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例133所制备的环形mrna分子采用实施例134所示的靶向递送系统所获得的样品的表面荷电远远高于实施例133所制备的环形mrna分子采用实施例134中所示的未经合理改性的任何一种或多种脂质产品与实施例133所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例134中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例134所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例134所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.52
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.08
±
0.03%、0.12
±
0.03%、 0.42
±
0.04%、0.71
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0446]
实施例137实施例133所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将7.5
×
104个hbv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈移植,建立原位宫颈癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后
的肿瘤组织大小为44立方毫米,注射第15天后的肿瘤组织大小为92立方毫米,注射第18天后的肿瘤组织大小为163立方毫米,注射第21天后的肿瘤组织大小为257立方毫米,注射第24天后的肿瘤组织大小为433立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为33立方毫米、26立方毫米、21立方毫米、16立方毫米;注射第12天后的肿瘤组织大小分别依次为256立方毫米、217立方毫米、172立方毫米、136立方毫米;注射第15天后的肿瘤组织大小分别依次为579立方毫米、534立方毫米、420立方毫米、311立方毫米;注射第18天后的肿瘤组织大小分别依次为878立方毫米、815立方毫米、709立方毫米、542立方毫米;注射第21天后的肿瘤组织大小分别依次为1192立方毫米、1108立方毫米、993立方毫米、857立方毫米;注射第24天后的肿瘤组织大小分别依次为1471立方毫米、1402立方毫米、1324立方毫米、1183立方毫米)。这说明,实施例133所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例133所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.67
±
0.54%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、0.25
±
0.07%、0.42
±
0.08%)。
[0447] 实施例138实施例133所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗(青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;
第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.78
±
0.4%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.12
±
0.03%、0.22
±
0.07%),仅次于pma阳性对照组(29.91
±
3.19%)。
[0448] 实施例139实施例133所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠淋巴细胞或小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;
第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞中表达明显高于正常淋巴细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞中的表达。
[0449] 实施例140实施例133所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠淋巴细胞和小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞的rna并进行高通量测序。分析测序结果,确定小鼠淋巴细胞和小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠淋巴细胞和小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
agttcgtatgcactaccgaa-3
´
(seqid126)和5
´‑
atcaacatccactggtcctt-3
´
(seqid127);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞中高表达,明显高于正常淋巴细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例133所制备的环形mrna分子采用实施例134靶向递送系统递送至小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞的递送率(87.4%)远远高于实施例134中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例134所示靶向递送系统对实施例133所合成的环形mrna递送至小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞的递送率(87.4%)远远高于单独使用实施例134中所示核酸适配体或其他迄今为止文献资料所报道的小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例134所示靶向递送系统对实施例133所合成的环形mrna递送至小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞的递送率(87.4%)远远高于用实施例134中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例134中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例133所合成的环形mrna递送至小时tlv-1阳性淋巴细胞原位恶性肿瘤细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例133所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例133所合成的环形mrna的递送能力优良。
[0450] 实施例141用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备 第一步,构建具有seqid53所示氨基酸序列的抗原多肽编码区,具有seqid63所示核苷酸序列的ires,具有seqid68所示核苷酸序列的第二外显子,具有seqid70所示核苷酸序列的5
´
间隔区,具有seqid73所示核苷酸序列的3
´
间隔区,具有seqid77所示核苷酸序列的第一外显子;具有seqid79核苷酸序列的的启动子,具有seqid83核苷
酸序列的5
´
同源臂,具有seqid86核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid92所示核苷酸序列的3
´
同源臂,以及可用于具有seqid93所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,222rpm活化3小时后,取活化菌液在36.9摄氏度,222rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(321.3纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(265.5纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2小时,然后加入dnasei在36.9摄氏度消化25分钟。将所获得的混合物采用磁分离法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(470.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物依次通过采用磁分离法,液相等离子体技术(辐射能量为100微焦每平方厘米,辐射30分钟,机械搅拌,搅拌速度250rpm,液相介质为天冬氨酸盐缓冲液,气体为氩气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(240.2纳克每微升),纯度为98.6%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(89.0%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在液相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升
primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热25分钟,随后在85摄氏度加热11秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0451] 实施例142实施例141所制备的的环形mrna分子的靶向脂质体递送系统工艺第一步,将石墨烯状黑磷和脂质产品按照8.6:78摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为反式dope,dc-chol和popg按照摩尔比例1:20:35混合得到的脂质混合物;第二步,将纯氧通入含1.0%氯化钠,1.5%氯化钾,3.2%磷酸氢二钾,9.0%蔗糖和8.1%葡萄糖的溶液加入第一步中的混合物a中,然后在-3摄氏度下结合超声波技术机械搅拌150分钟,得到混合液a,其中超声波的超声功率密度为8瓦每平方厘米,机械搅拌的速率为610rpm;第三步,将amino-peg4-alcohol,nh2-peg2-c2-boc,amino-peg4-ch2cooh和nh2-peg8-cooh按3:2:1:2摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在-2摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为9瓦每平方厘米,机械搅拌的速率为300rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s3按照摩尔比例1:2混合得到的混合适配体;hbv感染所致的肝癌为as1411和tls-11a按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为10-8和as1411按照摩尔比例2:1混合得到的混合适配体;hpv感染所致的宫颈癌为as1411和air-3a按照摩尔比例3:7混合得到的混合适配体;ebv感染所致的原位淋巴瘤为u9,wqy-9-b和td05按照摩尔比例1:3:3混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为90平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为2毫升每分钟,流速比为1/9。
[0452]
实施例143实施例141所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例141中合成的环形mrna采用实施例142中所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为133纳米,表面荷电33.9
±
0.5毫伏。此外,进一步地空白对照实验结果表明实施例141所制备的环形mrna分子采用实施例142所示的靶向递送系统所获得的样品的表面荷电
远远高于实施例141所制备的环形mrna分子采用实施例142中所示的未经合理改性的任何一种或多种脂质产品与实施例141所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例142中所示的脂质产品中同时引入合理强度的,声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例142所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例142所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二组,皮下注射靶向递送系统;第三组,皮下注射靶向递送系统和未修饰的线性mrna;第四组,皮下注射靶向递送系统和修饰的线性mrna;第五组,皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.17
±
0.5%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.42
±
0.6%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、 0.37
±
0.04%、0.58
±
0.04%;tnf-α的量分别依次为0.11
±
0.04%、0.15
±
0.04%、0.49
±
0.07%、0.69
±
0.10%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0453]
实施例144实施例141所制备的的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例69中合成的环形mrna采用所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为133纳米,表面荷电33.9
±
0.5毫伏。此外,进一步地空白对照实验结果表明实施例141所制备的环形mrna分子采用实施例142所示的靶向递送系统所获得的样品的表面荷电远远高于实施例141所制备的环形mrna分子采用实施例142中所示的未经合理改性的任何一种或多种脂质产品与实施例141所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例142中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例142所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例142所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.50
±
0.5%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.08
±
0.02%、0.13
±
0.03%、 0.41
±
0.04%、0.69
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0454] 实施例145 实施例141所制备的的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将1
×
107个hpv阳性且具成瘤能力的鼻咽癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行鼻咽癌移植,建立原位鼻咽癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形
成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为14立方毫米,注射第15天后的肿瘤组织大小为36立方毫米,注射第18天后的肿瘤组织大小为65立方毫米,注射第21天后的肿瘤组织大小为107立方毫米,注射第24天后的肿瘤组织大小为159立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为11立方毫米、9立方毫米、7立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为97立方毫米、81立方毫米、64立方毫米、49立方毫米;注射第15天后的肿瘤组织大小分别依次为190立方毫米、174立方毫米、148立方毫米、107立方毫米;注射第18天后的肿瘤组织大小分别依次为288立方毫米、270立方毫米、246立方毫米、107立方毫米;注射第21天后的肿瘤组织大小分别依次为393立方毫米、365立方毫米、337立方毫米、298立方毫米;注射第24天后的肿瘤组织大小分别依次为489立方毫米、472立方毫米、435立方毫米、382立方毫米)。这说明,实施例141所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例141所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.83
±
0.61%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.11
±
0.03%、0.23
±
0.05%、0.39
±
0.07%)。
[0455] 实施例146实施例141所制备的环形mrna分子激活人类t细胞的情况第一步,分离出hpv阳性宫颈癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗(青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成
的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hpv阳性宫颈癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.63
±
0.4%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.12
±
0.03%、0.23
±
0.07%),仅次于pma阳性对照组(29.77
±
2.95%)。
[0456] 实施例147实施例141所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠内皮细胞或hhv-8阳性原位卡西波肉瘤癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解
时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心 25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,western blot实验证明环形mrna分子所编码的多肽在hhv-8阳性原位卡西波肉瘤癌细胞中表达明显高于正常内皮细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hhv-8阳性原位卡西波肉瘤中的表达。
[0457] 实施例148 实施例141所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠内皮细胞和 hhv-8阳性原位卡西波肉瘤癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠内皮细胞和 hhv-8阳性原位卡西波肉瘤癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠内皮细胞和 hhv-8阳性原位卡西波肉瘤癌的rna,并 且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
caaatatccacatttggaatttgat-3
´
(seq id 128)和5
´‑ꢀ
caccacgtctaccctccgaa-3
´ꢀ
(seq id 129);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hhv-8阳性原位卡西波肉瘤细胞中高表达,明显高于内皮细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例141所制备的环形mrna分子采用实施例142靶向递送系统递送至hhv-8阳性原位卡西波肉瘤细胞的递送率(91.2%)远远高于实施例142中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例142所示靶向递送系统对实施例141所合成的环形mrna递送至hhv-8阳性原位卡西波肉瘤细胞的递送率(91.2%)远远高于单独使用实施例142中所示核酸适配体或其他迄今为止文献资料所报道的hhv-8阳性原位卡西波肉瘤细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例142所示靶向递送系统对实施例141所合成的环形mrna递送至hhv-8阳性原位卡西波肉瘤细胞的递送率(91.2%)远远高于用实施例142中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例142中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例141所合成的环形mrna递送至hhv-8阳性原位卡西波肉瘤细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例141所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例141所合成的环形mrna的递送能力优良。
[0458] 实施例149 用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seq id 57所示氨基酸序列的抗原多肽编码区,具有seq id 62所示核苷酸序列的ires,具有seq id 68所示核苷酸序列的第二外显子,具有seq id 70所示核苷酸序列的5
´
间隔区,具有seq id 73所示核苷酸序列的3
´
间隔区,具有seq id 76所示核苷酸序列的第一外显子;具有seq id 80核苷酸序列的的启动子,具有seq id 82核苷
酸序列的5
´
同源臂,具有seqid84核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid91所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,220rpm活化3.5小时后,取活化菌液在37.1摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(274.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37.1摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(221.3纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37.1摄氏度孵育2.5小时,然后加入dnasei在37.1摄氏度消化30分钟。将所获得的混合物采用亲和柱层析法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(326.6纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热30分钟。将所获得的混合物依次通过采用亲和柱层析法,气相等离子体技术(辐射能量为100微焦每平方厘米,辐射30分钟,机械搅拌,搅拌速度250rpm,液相介质为heppso缓冲液,气体为氧气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和小时plc分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(238.6纳克每微升),纯度为99.5%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(88.4%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在气相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在气相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;
第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0459] 实施例150实施例149所制备的的环形mrna分子的靶向脂质体递送系统工艺 第一步,将石墨烯状黑磷和脂质产品按照8.0:81摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为反式dope,磷脂酰肌醇,磷脂酸和dope按照摩尔比例1:8:10:15:33混合得到的脂质混合物;第二步,将空气通入含0.9%氯化钠,1.4%磷酸二氢钠,1.9%磷酸二氢钾,3.6%蔗糖和5.4%葡萄糖的溶液加入第一步中的混合物a中,然后在0摄氏度下结合气相等离子体技术机械搅拌120分钟,得到混合液a,其中超声波的超声功率密度为10瓦每平方厘米,机械搅拌的速率为480rpm;第三步,将nh2-peg2-c2-boc,amino-peg4-ch2cooh,和nh2-peg5-coo小时按3:2:1摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在-4摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为520rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s12,cd109和s27按照摩尔比例2:1:3混合得到的混合适配体;hbv感染所致的肝癌为10-8和tls9a按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为ch6,肽适配子和as1411按照摩尔比例1:1:1混合得到的混合适配体;hpv感染所致的宫颈癌为as1411,apt和a10按照摩尔比例1:1:1混合得到的混合适配体;ebv感染所致的原位淋巴瘤为gbm128,gbm131和u31按照摩尔比例1:1:1混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为80平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为3毫升每分钟,流速比为1/5。
[0460]
实施例151实施例149所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例149中合成的环形mrna采用实施例150所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为128纳米,表面荷电33.3
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例149
所制备的环形mrna分子采用实施例150所示的靶向递送系统所获得的样品的表面荷电远远高于实施例149所制备的环形mrna分子采用实施例150中所示的未经合理改性的任何一种或多种脂质产品与实施例149所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例150中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例150所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例150所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.10
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.45
±
0.5%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.03%、0.13
±
0.04%、 0.38
±
0.05%、0.65
±
0.06%;tnf-α的量分别依次为0.11
±
0.04%、0.15
±
0.04%、0.51
±
0.06%、0.75
±
0.12%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫
苗。
[0461]
实施例152实施例149所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例149中合成的环形mrna采用实施例150所示的所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为128纳米,表面荷电33.3
±
0.2毫伏。此外,进一步地空白对照实验结果表明实施例149所制备的环形mrna分子采用实施例150所示的靶向递送系统所获得的样品的表面荷电远远高于实施例149所制备的环形mrna分子采用实施例150中所示的未经合理改性的任何一种或多种脂质产品与实施例149所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例150中所示的脂质产品中同时引入合理强度的声能量,辐射能量和自我牺牲型的辅助剂黑磷,可以调控实施例150所示的靶向递送系统的表面荷电和粒径;与此同时,声能量,辐射能量和自我牺牲型的辅助剂黑磷在实施例150所示的靶向递送系统的表面荷电和粒径调控中的具体作用机制的研究还在进行中;第二步,将6-8周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.42
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.09
±
0.03%、0.13
±
0.03%、0.39
±
0.04%、0.58
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0462] 实施例153实施例149所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8周的体重20克的小鼠,将2.5
×
105个hpv阳性且具成瘤能力的宫颈癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈移植,建立原位宫颈癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4的pbs;第二组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实
验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为16立方毫米,注射第15天后的肿瘤组织大小为39立方毫米,注射第18天后的肿瘤组织大小为72立方毫米,注射第21天后的肿瘤组织大小为133立方毫米,注射第24天后的肿瘤组织大小为248立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为13立方毫米、11立方毫米、7立方毫米、6立方毫米;注射第12天后的肿瘤组织大小分别依次为108立方毫米、94立方毫米、63立方毫米、49立方毫米;注射第15天后的肿瘤组织大小分别依次为210立方毫米、199立方毫米、156立方毫米、112立方毫米;注射第18天后的肿瘤组织大小分别依次为353立方毫米、314立方毫米、275立方毫米、228立方毫米;注射第21天后的肿瘤组织大小分别依次为499立方毫米、450立方毫米、404立方毫米、345立方毫米;注射第24天后的肿瘤组织大小分别依次为624立方毫米、572立方毫米、516立方毫米、453立方毫米)。这说明,实施例149所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例149所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.83
±
0.60%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.11
±
0.03%、0.27
±
0.06%、0.38
±
0.07%)。
[0463] 实施例154实施例149所制备的的环形mrna分子激活人类t细胞的情况第一步,分离出hbv阳性肝癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗(青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培
养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活人类t细胞的免疫反应,可作为人类疫苗;第四步,通过淋巴细胞分离液分离出hbv阳性肝癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血 清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯) 和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzyme b)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.52
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.11
±
0.03%、0.20
±
0.07%),仅次于pma阳性对照组(29.57
±
2.55%)。
[0464] 实施例155 实施例149所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步, 将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步, 每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠淋巴细胞或ebv阳性原位淋巴瘤细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加 10 微升 pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;
第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在ebv阳性原位淋巴瘤细胞中表达明显高于正常淋巴细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在ebv阳性原位淋巴瘤中的表达。
[0465] 实施例156实施例149所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠淋巴细胞和ebv阳性原位淋巴瘤细胞的rna并进行高通量测序。分析测序结果,确定小鼠淋巴细胞和ebv阳性原位淋巴瘤细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠淋巴细胞和ebv阳性原位淋巴瘤细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
gcgtctaccacgtcggtgtcacc-3
´
(seqid130)和5
´‑
gattccagtacctattgtc-3
´
(seqid131);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在ebv阳性原位淋巴瘤细胞中高表达,明显高于正常淋巴细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例149所制备的环形mrna分子采用实施例150靶向递送系统递送至ebv阳性原位淋巴瘤细胞的递送率(92.6%)远远高于实施例150中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例150所示靶向递送系统对实施例149所合成的环形mrna递送至ebv阳性原位淋巴瘤细胞的递送率(85.8%)远远高于单独使用实施例150中所示核酸适配体或其他迄今为止文献资料所报道的ebv阳性原位淋巴瘤细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例150所示靶向递送系统对实施例149所合成的环形mrna递送至ebv阳性原位淋巴瘤细胞的递送率(92.6%)远远高于用实施例150中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例150中所示未经合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例149所合成的环形mrna递送至ebv阳性原位淋巴瘤细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例149所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例149所合成的环形mrna的递送能力优良。
[0466] 实施例157环形mrna的制备及在293t细胞的蛋白表达验证第一步,构建具有seqid8所示氨基酸序列的抗原多肽编码区,具有seqid64所示核苷酸序列的ires,具有seqid67所示核苷酸序列的第二外显子,具有seqid70所示核苷酸序列的5’间隔区,具有seqid73所示核苷酸序列的3’间隔区,具有seqid76所示
核苷酸序列的第一外显子;具有seqid79核苷酸序列的的启动子,具有seqid82核苷酸序列的5’同源臂,具有seqid85核苷酸序列的3’内含子,具有seqid88所示核苷酸序列的5’内含子,具有seqid91所示核苷酸序列的3’同源臂,以及可用于具有seqid95所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3.5小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(289.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(242.1纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2.5小时,然后加入dnasei在37摄氏度消化30分钟。将所获得的混合物采用尺寸排阻柱法法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(389.5纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热25分钟。将所获得的混合物依次通过采用尺寸排阻柱法,超声波技术(声强度为1微焦每平方厘米,超声120分钟,机械搅拌,搅拌速度500rpm,液相介质为pbs缓冲液,气体为氨气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(298.4纳克每微升),纯度为98.0%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(87.8%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在超声波技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在超声波技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的声能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;
第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热25分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为38次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0467]
实施例158实施例157所制备的的环形mrna分子的靶向脂质体递送系统工艺优化第一步,将结构式(i)化合物和脂质产品按照7:76摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为peg-dspe,peg-pe,peg修饰的神经酰胺,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:5:15:30:33混合得到的脂质混合物;第二步,将氧浓度为50%的氩气通入含0.8%氯化钠,2.8%氯化钙,5.0%磷酸二氢钠,5.0%蔗糖和8.0%葡萄糖的溶液加入第一步中的混合物a中,然后在-10摄氏度下结合超声波技术机械搅拌210分钟,得到混合液a,其中超声波的超声功率密度为8瓦每平方厘米,机械搅拌的速率为400rpm;第三步,将amino-peg4-alcohol,amino-peg4-ch2cooh和nh2-peg7-cooh按5:2:1摩尔比例混合形成的混合物加入第二步获得的混合液a,然后在-4摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为12瓦每平方厘米,机械搅拌的速率为300rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为s3和s27按照摩尔比例1:2混合得到的混合适配体;hbv感染所致的肝癌为ly1和10-8按照摩尔比例1:3混合得到的混合适配体;hcv感染所致的肝癌为td05和as1411按照摩尔比例2:1混合得到的混合适配体;hpv感染所致的宫颈癌为air3a和a10按照摩尔比例1:8混合得到的混合适配体;ebv感染所致的原位淋巴瘤为wqy-9-b,gbm128和gbm131按照摩尔比例2:2:1混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为180平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为4毫升每分钟,流速比为1/4。
[0468] 实施例159实施例157所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例157中合成的环形mrna采用实施例158所示的所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为139纳米,表面荷电32.4
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施
例157所制备的环形mrna分子采用实施例158所示的靶向递送系统所获得的样品的表面荷电远远高于实施例157所制备的环形mrna分子采用实施例158中所示的未经合理改性的任何一种或多种脂质产品与实施例157所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例158中所示的脂质产品中同时引入合理强度的声能量和结构式(i-1)有机化合物,可以调控实施例158所示的靶向递送系统的表面荷电和粒径。与此同时,声能量和结构式(i-1)有机化合物在实施例158所示的靶向递送系统的表面荷电和粒径调控中的作用机制还在研究中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.14
±
0.3%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.42
±
0.5%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.10
±
0.03%、 0.38
±
0.04%、0.58
±
0.04%;tnf-α的量分别依次为0.11
±
0.03%、0.14
±
0.04%、0.48
±
0.07%、0.69
±
0.1)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0469] 实施例160 实施例157所制备的环形mrna分子皮下注射可刺激产生抗原特异性t
细胞第一步,将实施例157中合成的环形mrna采用实施例158中所示的所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为139纳米,表面荷电32.4
±
0.3毫伏。此外,进一步地空白对照实验结果表明实施例157所制备的环形mrna分子采用实施例158所示的靶向递送系统所获得的样品的表面荷电远远高于实施例157所制备的环形mrna分子采用实施例158中所示的未经合理改性的任何一种或多种脂质产品与实施例157所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例158中所示的脂质产品中同时引入合理强度的声能量和结构式(i-1)有机化合物,可以调控实施例158所示的靶向递送系统的表面荷电和粒径。与此同时,声能量和结构式(i-1)有机化合物在实施例158所示的靶向递送系统的表面荷电和粒径调控中的作用机制还在研究中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二组,皮下注射靶向递送系统;第三组,皮下注射靶向递送系统和未修饰的线性mrna;第四组,皮下注射靶向递送系统和修饰的线性mrna;第五组,皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.05
±
0.4%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.08
±
0.03%、0.12
±
0.03%、 0.41
±
0.04%、0.59
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0470]
实施例161实施例157所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将7.5
×
104个hbv阳性且具成瘤能力的肝癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行肝癌移植,建立原位肝癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后
的肿瘤组织大小为48立方毫米,注射第15天后的肿瘤组织大小为95立方毫米,注射第18天后的肿瘤组织大小为150立方毫米,注射第21天后的肿瘤组织大小为241立方毫米,注射第24天后的肿瘤组织大小为422立方毫米)比第一组~第四组生长缓慢的多(注射第9天后的肿瘤组织大小分别依次为30立方毫米、26立方毫米、20立方毫米、18立方毫米;注射第12天后的肿瘤组织大小分别依次为249立方毫米、203立方毫米、157立方毫米、134立方毫米;注射第15天后的肿瘤组织大小分别依次为560立方毫米、515立方毫米、419立方毫米、297立方毫米;注射第18天后的肿瘤组织大小分别依次为847立方毫米、784立方毫米、698立方毫米、542立方毫米;注射第21天后的肿瘤组织大小分别依次为1182立方毫米、1103立方毫米、980立方毫米、855立方毫米;注射第24天后的肿瘤组织大小分别依次为1468立方毫米、1386立方毫米、1294立方毫米、1169立方毫米)。这说明,实施例157所制备的能编码肿瘤新生抗原多肽的环形mrna通过皮下注射可抑制小鼠肿瘤的生长;第四步,与此同时,将第三步中的肿瘤组织制作成单细胞悬液,检测免疫细胞的情况。结果表明,实施例157所制备的能编码肿瘤新生抗原多肽的环形mrna可以刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r。因为第五组转能编码肿瘤新生抗原多肽的环形mrna刺激肿瘤浸润cd8t细胞产生细胞因子ifn-r的量为1.58
±
0.41%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.07
±
0.02%、0.11
±
0.03%、0.23
±
0.05%、0.37
±
0.07%)。
[0471] 实施例162实施例157所制备的环形mrna分子激活人类t细胞的情况第一步,分离出ebv阳性鼻咽癌患者的pbmc(外周血单个核细胞)及t细胞。pbmc在加入1%双抗(青霉素和链霉素)和10%(v/v)ab血型的人血清的dmem培养基中培养,并通过50纳克每毫升的gm-csf(粒细胞-巨噬细胞集落刺激因子)和il-4(20纳克每毫升)诱导;每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第二步,分为5组,每个组别分别为:第一组,空白对照组,即pbs组;第二组,未修饰的线性mrna组;第三组,修饰的线性mrna组;第四组,能编码肿瘤新生抗原多肽的环形mrna组;第五组,cd3-2阳性对照组。每个组分别设置3次实验。其中,第二到第四组每5
×
104个dc细胞电转10微克rna;第三步,在培养6天后的t细胞中,按4:1的比例(t细胞个数/dc细胞个数)每3天加入含新的电转rna的dc,9天后将dc细胞和培养后的t细胞转移到包被人ifn-γ抗体的ifn-γelispot96孔板中共培养,24小时后弃去上清,pbs清洗5次后加入生物素化的检测一抗孵育2小时,pbs清洗5次后加入hrp标记二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。cd3-2阳性对照组在培养6天后的t细胞中,按4:1的细胞个数比例(t细胞个数/dc细胞个数)每3天加入dc后于elispot板孔中再加入cd3-2,cd3-2每次加入的浓度按说明书浓度加入,其余操作步骤同第二到第四组。然后,通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况。结果表明第四组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点大大多于第一到第三组,仅次于cd3-2阳性对照组。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激活人类t细胞的免疫反应,可作为人类疫苗;
第四步,通过淋巴细胞分离液分离出ebv阳性鼻咽癌患者外周血中的pbmc(外周血单个核细胞)及t细胞。磁珠分选得到的pbmc细胞重悬于含1%双抗、10%(v/v)ab血型人血清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细胞。磁珠分选得到的cd8t细胞重悬于含10%v/vab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldina(在细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基继续培养6小时,抑制细胞因子的分泌;该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0.5微克每毫升pma(豆蔻酰佛波醇乙酯)和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗ifn-γ、tnf-α、granzymeb、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzymeb)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.72
±
0.4%(ifn-γ阳性cd8+t细胞在cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.08
±
0.02%、0.10
±
0.03%、0.21
±
0.07%),仅次于pma阳性对照组(30.02
±
3.33%)。
[0472] 实施例163实施例157所制备的的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步,将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步,每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠皮肤组织细胞或mcpyv阳性原位梅克尔癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液; 第四步,配置ripa裂解液,往1毫升裂解液加10微升pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;
第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;第九步,westernblot实验证明环形mrna分子所编码的多肽在mcpyv阳性原位梅克尔癌细胞中表达明显高于正常皮肤组织细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在mcpyv阳性原位梅克尔癌中的表达。
[0473] 实施例164实施例157所制备的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠皮肤组织细胞或mcpyv阳性原位梅克尔癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠小鼠皮肤组织细胞和mcpyv阳性原位梅克尔癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠皮肤组织细胞和mcpyv阳性原位梅克尔癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
acgtgtcgtctgtgttgttg-3
´
(seqid132)和5
´‑
ggccccgcctacgtgaaa-3
´
(seqid133);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在mcpyv阳性原位梅克尔癌细胞中高表达,明显高于正常皮肤组织细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力。此外,进一步地空白对照实验结果表明实施例157所制备的环形mrna分子采用实施例158靶向递送系统递送至mcpyv阳性原位梅克尔癌细胞的递送率(86.7%)远远高于实施例158中所示未加入结构式(i-1)有机化合物进行合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例158所示靶向递送系统对实施例157所合成的环形mrna递送至mcpyv阳性原位梅克尔癌细胞的递送率(86.7%)远远高于单独使用实施例158中所示核酸适配体或其他迄今为止文献资料所报道的mcpyv阳性原位梅克尔癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例158所示靶向递送系统对实施例13所合成的环形mrna递送至mcpyv阳性原位梅克尔癌细胞的递送率(86.7%)远远高于用实施例158中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例158中所示未加入结构式(i-1)有机化合物进行合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例157所合成的环形mrna递送至mcpyv阳性原位梅克尔癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例157所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例157所合成的环形mrna的递送能力优良。加入结构式(i-1)有机化合物进行合理改性增强实施例158靶向递送系统递的递送率的机制正在进行进一步研究。
[0474] 实施例165用于病毒阳性癌症治疗的能编码肿瘤新生抗原多肽的环形mrna制备第一步,构建具有seqid27所示氨基酸序列的抗原多肽编码区,具有seqid65所示核苷酸序列的ires,具有seqid66所示核苷酸序列的第二外显子,具有seqid71所示核苷酸序列的5
´
间隔区,具有seqid72所示核苷酸序列的3
´
间隔区,具有seqid77所示核苷酸序列的第一外显子;具有seq id 80核苷酸序列的的启动子,具有seq id 82核苷酸
序列的5
´
同源臂,具有seqid85核苷酸序列的3
´
内含子,具有seqid88所示核苷酸序列的5
´
内含子,具有seqid91所示核苷酸序列的3
´
同源臂,以及可用于具有seqid94所示核苷酸序列的质粒线性化的酶切位点的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(359.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units),50微升10
×
cutsmartbuffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(290.3纳克每微升);第四步,将1微克质粒模板,2微升10
×
reactionbuffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp,2微升t7rnapolymerasemix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnasei在36.9摄氏度消化25分钟。将所获得的混合物采用磁分离法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(507.6纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的trishcl,10毫摩尔每升的二氯化镁及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热25分钟。将所获得的混合物依次通过采用磁分离法,液相等离子体技术(辐射能量为1微焦每平方厘米,辐射60分钟,机械搅拌,搅拌速度300rpm,液相介质为pbs缓冲液,气体为氨气,气体流速为1毫升每分钟),磷酸酶处理及rna酶r处理相结合进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环形mrna大小进行鉴定和hplc-质谱联用分析。分析结果表明纯化得到的环形mrna完全达到当初设计的要求。这不仅表明已成功制备所需的环形mrna,而且也说明所制备的环形mrna在分离纯化处理中没有遭受到损伤。与此同时,测定mrna环化效率和纯化后的环形mrna浓度(288.3纳克每微升),纯度为98.9%,高于采用文献报道的高效液相色谱尺寸排阻柱,磷酸酶处理及rna酶r处理相结合的方法分离纯化本实施例所合成环形mrna的纯度(88.5%)。在进一步地空白对照实验中,实验结果显示本实施例合成的环形mrna样品中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的高效液相色谱峰在液相等离子体技术处理后并没有发生明显的变化。但是,本实施例合成的环形mrna样品在液相等离子体技术处理后,再进行磷酸酶处理及rna酶r处理,在随后进行的高效液相色谱分析谱图中已几乎看不到其合成过程中残留的内含子片段、带切口的线性rna、线性和环状连接以及由体外转录和剪接反应产生的杂质的杂质峰。这些实验结果表明,含有所需纯化的环形mrna的液相介质中引入合理强度的辐射能量,可以提高杂质的磷酸酶处理及rna酶r处理活性,从而在不损伤所需纯化的环形mrna目标产物的情况下,大幅度提高环形mrna的分离纯化纯度;第六步,将1微克环化或未环化的mrna溶液,4微升rtprmier,1微升
primerscriptrtenzyemixi和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热25分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,且整个序列符合所设计的目标环形mrna的要求。这进一步表明已成功制备环形mrna,而且再次证明所制备的环形mrna在分离纯化处理中没有遭受到损伤。
[0475] 实施例166实施例165所制备的的环形mrna分子的靶向脂质体递送系统工艺第一步,将结构式(i-2)和脂质产品按照8:64摩尔比例进行混合,得到混合物a,其中混合物a中的脂质产品为peg-dspe,peg-pe,peg修饰的神经酰胺,二豆蔻酰磷脂酰甘油和dope按照摩尔比例1:7:18:32:35混合得到的脂质混合物;第二步,将氧浓度为48%的氦气通入含7.5%氯化钙溶液加入第一步中的混合物a中,然后在2摄氏度下结合超声波技术机械搅拌240分钟,得到混合液a,其中超声波的超声功率密度为6瓦每平方厘米,机械搅拌的速率为450rpm;第三步,将amino-peg4-alcohol加入第二步获得的混合液a,然后在2摄氏度下进行超声搅拌处理,得到混合液b;其中超声波的超声功率密度为10瓦每平方厘米,机械搅拌的速率为300rpm;第四步,将核酸适配体(ebv感染所致的鼻咽癌为cd109和s3按照摩尔比例3:2混合得到的混合适配体;hbv感染所致的肝癌为tls11a和tls9a按照摩尔比例1:1混合得到的混合适配体;hcv感染所致的肝癌为肽适配子和td05按照摩尔比例1:1混合得到的混合适配体;hpv感染所致的宫颈癌为as1411,air-3a和a10按照摩尔比例2:3:1混合得到的混合适配体;bv感染所致的原位淋巴瘤为u9,u31和td05按照摩尔比例1:1:3混合得到的混合适配体;hltv-1感染所致的成人t细胞白血病为sscgc;hhv-8感染所致的卡波西肉瘤为硫代磷酸酯;mcpyv感染所致的梅克尔癌为a102
´‑
氟尿嘧啶rna适配体)加入第三步中的混合液b中,混合均匀得到混合液c;第五步,将第四步中的混合液c通过有效面积为140平方厘米的无菌过滤器过滤得到混合液d;第六步,将混合液d和环形mrna通过碰撞喷射混合器进行进一步混合,碰撞喷射混合器的总流速为5毫升每分钟,流速比为1/6。
[0476]
实施例167实施例165所制备的的环形mrna分子作为疫苗激活免疫反应第一步,将实施例165中合成的环形mrna采用实施例166中所示的所示的脂质体配方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升pbs);平均粒径为101纳米,表面荷电37.7
±
0.4毫伏。此外,进一步地空白对照实验结果表明实施例165所制备的环形mrna分子采用实施例166所示的靶向递送系统所获得的样品的表面荷电远远高于实施例165所制备的环形mrna分子采用实施例166中所示的未经合理改性的任何一种或多种脂质产品与实施例165所示的核酸适配体组合成的递送系统所获得的
样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例166中所示的脂质产品中同时引入合理强度的声能量和结构式(i-2)有机化合物,可以调控实施例166所示的靶向递送系统的表面荷电和粒径。与此同时,声能量和结构式(i-2)有机化合物在实施例166所示的靶向递送系统的表面荷电和粒径调控中的作用机制还在研究中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后3天,处死小鼠并在无菌条件下取出小鼠脾脏。实验过程接受动物管理会的监督;第三步,分离出小鼠原代脾细胞,并种入包埋有inf-r抗体的elispot试剂盒的培养板内,每孔5
×
105个细胞。取同源小鼠的骨髓细胞,在加入1%双抗(青霉素和链霉素)和10% (v/v)fbs的rpmi 1640培养基,并通过20纳克每毫升的gm-csf诱导,8天后可获得成熟的骨髓来源的树突状细胞。在elispot小孔的脾细胞中按10:1的比例加入树突状细胞,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中进行刺激。24小时后弃去上清,pbs清洗5次后加入一抗孵育2小时,pbs清洗5次后加入二抗孵育1小时,最后加入tmb显色液,待斑点出现后去离子水冲洗。通过酶联免疫斑点检测仪观察培养板内inf-r斑点形成情况(一抗、二抗以及tmb显色液均为elispot试剂盒组分)。结果表明第五组转能编码肿瘤新生抗原多肽的环形mrna形成的斑点最多;第四步,取同样处理后的小鼠脾细胞1
×
106个按10:1的比例与骨髓来源的树突状细胞共培养,第一组和第二组加pbs,其余3组按相应小鼠分组每5
×
104个树突状细胞电转10微克 rna(即第三组转未修饰的线性mrna、第四组转修饰的线性mrna、第五组转能编码肿瘤新生抗原多肽的环形mrna),电转的条件为150伏特、10毫秒,将电转后的树突细胞加入脾细胞中,然后加入brefeldin a(在细胞混合液中的浓度为5微克每毫升)处理,抑制细胞因子分泌。5小时后,收集处理过的细胞进行细胞活性抗体,cd3和cd8分子染色,固定后,破膜剂进行破膜,然后分别 进行ifn-γ、tnf-α及il-2的染色。进行流式细胞仪检测,分析脾细胞中cd8+细胞的细胞因子产生情况。其中,第五组转能编码肿瘤新生抗原多肽的环形mrna的 ifn-γ量为2.35
±
0.4%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),tnf-α的量为2.69
±
0.5%(tnf-α阳性cd8+t细胞在cd8+t细胞中的 百分比),远远高于第一组~第四组(ifn-γ的量分别依次为0.09
±
0.03%、0.13
±
0.04%、 0.42
±
0.06%、0.58
±
0.07%;tnf-α的量分别依次为0.11
±
0.03%、0.15
±
0.04%、0.48
±
0.06%、0.70
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以激 活小鼠cd8+t细胞的免疫反应,可作为疫苗。
[0477]
实施例168实施例165所制备的环形mrna分子皮下注射可刺激产生抗原特异性t细胞第一步,将实施例165中合成的环形mrna采用实施例166中所示的所示的脂质体配
方工艺制备环形mrna疫苗(组分为:20微克环形mrna,40微克靶向递送系统和200微升 pbs);平均粒径为101纳米,表面荷电37.7
±
0.4毫伏。此外,进一步地空白对照实验结果表明实施例165所制备的环形mrna分子采用实施例166所示的靶向递送系统所获得的样品的表面荷电远远高于实施例165所制备的环形mrna分子采用实施例166中所示的未经合理改性的任何一种或多种脂质产品与实施例165所示的核酸适配体组合成的递送系统所获得的样品的表面荷电(平均粒径大于350纳米,小于20毫伏)。以上实验结果表明:对实施例166中所示的脂质产品中同时引入合理强度的声能量和结构式(i-2)有机化合物,可以调控实施例166所示的靶向递送系统的表面荷电和粒径。与此同时,声能量和结构式(i-2)有机化合物在实施例166所示的靶向递送系统的表面荷电和粒径调控中的作用机制还在研究中;第二步,将6-8 周的体重20克的小鼠分为5组,每个组别设置3只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂,于第三天、第六天、 第九天和第十二天对小鼠行皮下注射,在最后一次注射后9天,处死小鼠并在无菌条件下取出小鼠脾脏,实验过程接受动物管理会的监督;第三步,取脾细胞1
×
106个,进行细胞死活,cd3和cd8抗体表面标记,然后用五聚体进行流式 染色,最后通过流式检测检测cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞的产生情况。其中,第五组能编码肿瘤新生抗原多肽的环形mrna的cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量为2.72
±
0.7%,远远高于第一组~第四组(cd8阳性细胞中五聚体阳性的抗原特异性cd8+t细胞量分别依次为0.09
±
0.03%、0.14
±
0.03%、 0.40
±
0.04%、0.70
±
0.1%)。这说明,能编码肿瘤新生抗原多肽的环形mrna可以刺激产生抗原特异性cd8+t细胞。
[0478]
实施例169实施例165所制备的环形mrna分子通过皮下注射可抑制小鼠肿瘤的生长第一步,取30只6-8 周的体重20克的小鼠,将2.5
×
105个hpv阳性且具成瘤能力的宫颈癌细胞注射入小鼠腹侧部皮下,然后取皮下移植物来源的组织块进行宫颈移植,建立原位宫颈癌小鼠模型;第二步,将小鼠分为5组,每个组别设置6只,分别进行以下处理:第一组,空白对照组注射0.1摩尔每升、ph7.4 的pbs;第二 组皮下注射靶向递送系统;第三组皮下注射靶向递送系统和未修饰的线性mrna;第四组皮下注射靶向递送系统和修饰的线性mrna;第五组皮下注射靶向递送系统和能编码肿瘤新生抗原多肽的环形mrna;各组别的量为一剂注射剂,每次每只小鼠注射一剂;第三步,9天后处死每组的其中一只小鼠,并在无菌条件下取出小鼠肿瘤组织,实验过程接受动物管理会的监督。随后,每隔3天处死小鼠,并在无菌条件下取出小鼠体内形成的肿瘤组织,观察肿瘤组织生产情况。结果表明,小鼠在皮下注射能编码肿瘤新生抗原多肽的环形mrna后,观察到肿瘤组织(注射第9天后的肿瘤组织为肉眼不可见,注射第12天后的肿瘤组织大小为14立方毫米,注射第15天后的肿瘤组织大小为36立方毫米,注射第18天后的肿瘤组织大小为69立方毫米,注射第21天后的肿瘤组织大小为135立方毫米,注射第24
清的dmem完全培养基中,加入适当浓度的gm-csf(在培养基中的浓度50纳克每毫升)与il-4(在培养基中的浓度20纳克每毫升)诱导,每3天更换细胞因子,37摄氏度、二氧化碳培养箱中培养6天,得到dc细 胞。磁珠分选得到的cd8 t细胞重悬于含10%v/v ab血型的人血清的dmem完全培养基中,加入相应浓度的il-2(在培养基中的浓度20纳克每毫升),37摄氏度、二氧化碳培养箱中培养,得到培养后的t 细胞;第五步,将dc细胞电转10微克rna后和培养后的t细胞(1
×
106个)按照1:4的数量 比例在24孔板中共培养;每3天更换细胞因子(终浓度为50纳克每毫升的gm-csf和20纳克每毫升的il-4,以及加入相同数量的电转10微克rna的dc);9天后接着加入brefeldin a(在 细胞混合液中的浓度5微克每毫升),每孔加入1毫升含10%(v/v)ab血型人血清的dmem完全培养基 继续培养6小时,抑制细胞因子的分泌; 该步实验中还设置了pbs组(第二组)、未修饰的线性mrna组(第三组)、修饰的线性mrna组(第四组)和pma组(第五组)。其中,第一,第三和第四组每5
×
104个dc细胞分别电转相应的10微克rna;第五组加入0 .5微克每毫升pma(豆蔻酰佛波醇乙酯) 和1微克每毫升ionomycin(离子霉素);第六步,培养结束后收集细胞,进行细胞死活,cd3和cd8抗体表面标记,然后用ic固定液室温固定30分钟,用破膜剂对细胞膜打孔后加抗 ifn-γ、tnf-α、granzyme b、抗体标记细胞内相应蛋白,最后通过流式检测检测cd8阳性细胞中相应蛋白(ifn-γ、tnf-α和granzyme b)的表达水平。实验结果表明,能编码肿瘤新生抗原多肽的环形mrna可以激活人cd8+t细胞的免疫反应。其中,第一组能编码肿瘤新生抗原多肽的环形mrna的ifn-γ量为2.81
±
0.6%(ifn-γ阳性cd8+t细胞在 cd8+t细胞中的百分比),远远高于第二组~第四组(ifn-γ的量分别依次为0.10
±
0.03%、0.14
±
0.04%、0.21
±
0.06%),仅次于pma阳性对照组(29.82
±
3.03%)。
[0480]
实施例171实施例165所制备的环形mrna分子对所编码肿瘤新生抗原多肽的表达验证第一步, 将细胞培养板,置于冰上,用吸引器吸除上清液。所有操作均在冰上进行;第二步, 每孔细胞(六孔板)中加入1-2毫升4摄氏度预冷的pbs溶液。平放轻轻摇动十秒钟清洗小鼠肝细胞或hcv阳性原位肝癌细胞,然后吸除洗液,共清洗洗细胞三次(目的是去除剩余的上清液),最后一次不吸除上清液;第三步,用刮勺将细胞轻轻刮下,接着用移液器将细胞和上清液转移至1.5毫升离心管中,置入4摄氏度离心机(提前开离心机预冷),离心1.5分钟,弃去上清液;第四步,配置ripa裂解液,往1毫升裂解液加 10 微升 pmsf(100毫摩尔每升),摇匀置于冰上;第五步,将配置好的裂解液加入第三步获得的细胞沉淀中,每5
×
106个细胞加入100微升的裂解液,然后吹打重悬,使细胞充分裂解;第六步,用超声仪进一步裂解细胞:将细胞放置于冰上进行超声,功率为15%,裂解时间为1分钟(超声2秒,休息4秒),当超声一次后,离心管仍比较浑浊,然后间隔3分钟再次超声;第七步,超声结束后,将细胞于4摄氏度离心机,12000rpm离心 25分钟;第八步,将离心后的上清分装转移至1.5毫升离心管,保存于-80摄氏度备用;
第九步,westernblot实验证明环形mrna分子所编码的多肽在hcv阳性原位肝癌细胞中表达明显高于正常肝细胞。证明了环形mrna分子具有编码多肽的功能;也证明了环形mrna分子所编码的多肽在hcv阳性原位肝癌中的表达; 实施例172实施例165所制备的的环形mrna分子在细胞中的分布第一步,应用trizol试剂提取小鼠肝细胞和hcv阳性原位肝癌细胞的rna并进行高通量测序。分析测序结果,确定小鼠肝细胞和hcv阳性原位肝癌细胞均存在前述实施例设计的环形mrna分子;第二步,应用trizol试剂提取小鼠肝细胞和hcv阳性原位肝癌细胞的rna,并且进行荧光定量pcr验证环形mrna分子的表达量,所用上游引物和下游引物引物序列分别为:5
´‑
agccttgtgcaamaagtccagca-3
´
(seqid134)和5
´‑
gagtctaccacgtcggtgtcatc-3
´
(seqid135);第三步,反应条件为环形mrna荧光定量pcr试剂盒所推荐实验条件。结果表明环形mrna在hcv阳性原位肝癌细胞中高表达,明显高于正常肝细胞。说明编码肿瘤新生抗原多肽的环形mrna分子具有加强的靶向递送能力此外,进一步地空白对照实验结果表明实施例165所制备的环形mrna分子采用实施例166靶向递送系统递送至hcv阳性原位肝癌细胞的递送率(87.9%)远远高于实施例166中所示未经加入结构式(i-2)有机化合物进行合理改性的任何一种或多种脂质产品混合合成的递送系统的递送率(均小于30%),及采用文献资料迄今所报道的脂质体递送系统的递送率(均小于30%)。同时,进一步地空白对照实验结果表明实施例166所示靶向递送系统对实施例165所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(87.9%)远远高于单独使用实施例166中所示核酸适配体或其他迄今为止文献资料所报道的hcv阳性原位肝癌细胞核酸适配体的靶向递送率(均小于35%)。随后,再进一步地空白对照实验结果表明实施例166所示靶向递送系统对实施例165所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(87.9%)远远高于用实施例166中所示核酸适配体或其他迄今为止文献资料所报道的其他核酸适配体与实施例166中所示未经加入结构式(i-2)有机化合物进行合理改性的任何一种或多种脂质产品混合合成的递送系统或文献资料迄今所报道的脂质体组合合成的递送系统对实施例165所合成的环形mrna递送至hcv阳性原位肝癌细胞的递送率(均小于45%)。以上实验结果表明:核酸适配体与脂质体递送系统之间对实施例165所合成的环形mrna的靶向递送存在协同作用。此外,只有经过合理匹配的核酸适配体与脂质体组合合成的递送系统才具有优良的靶向递送能力。与此同时,本发明递送系统库所匹配的递送系统对实施例165所合成的环形mrna的递送能力优良。加入结构式(i-2)有机化合物进行合理改性增强实施例166靶向递送系统递的递送率的机制正在进行进一步研究。
[0481] 实施例173实施例13所制备的环形mrna分子用于梅克尔癌患者预后实时荧光定量检测试剂盒的制备制备实施例13所制备的环形mrna分子用于梅克尔癌患者预后20次反应的实时荧光定量检测试剂盒。具体组分为:异丙醇20毫升、三氯甲烷20毫升、rna稳定溶液10毫升、trizol10毫升、无酶水2毫升、逆转录缓冲溶液40毫升、逆转录随机引物10微升、三磷酸碱基脱氧核苷酸20微升、rna酶抑制剂2微升、逆转录酶2微升、premixextaq2微升、实施例5所制备的的环形mrna分子特异性引物6微升(上游引物seqid96:5
´‑
atactcagctatgcgtgaag-3
´
;下游引物seq id 97:5
´‑
tatatgcacgtgtagtgctagatcgg-3
´
);gapd小时特异性引物6微升(上游引物 seq id 136:5
´‑
ggagcgagatccctccaaaat-3
´
;下游引物seq id 137:5
´‑ꢀ
ggctgttgtcatacttctcatgg-3
´
)。
[0482]
实施例174 梅克尔癌组织中实施例13所制备的环形mrna分子的检测第一步,梅克尔癌组织的保存:收集待测梅克尔癌组织存放于盛有rna稳定溶液的冻存管中,放至
‑ꢀ
80摄氏度冰箱备用;第二步,研钵用洗涤剂清洗后浸泡于3%的双氧水中4小时以上,冲刷数次,用双 蒸水洗一次,用锡箔纸覆盖研钵置于180摄氏度干燥箱中干烤6小时以上;第三步,rna抽提:在研钵中先加入液氮预冷,取冻存管中保存的组织,加入液氮快速研磨, 直到将组织磨成粉末状加入1毫升的trizol进一步研磨成液体状后移至tube管,每1毫升含 trizol的裂解液添加200微升预冷的氯仿,震荡约15-30秒进行混匀,4摄氏度,12 000rpm离心15分钟,取上层水相以1:1的比例加入预冷的异丙醇中,上下颠倒混匀数次,室温静置10分钟,4 摄氏度,12000rpm离心10分钟,弃上清,加入500微升用depc水配置的75%乙醇,洗涤rna沉淀。然 后4摄氏度、7500rpm离心5分钟,重复洗涤两次,最后用移液枪尽量弃去上清,室温干燥10-15分钟,加入10-20微升decp水,保存于-80摄氏度;第四步,实施例13所制备的环形mrna分子进行二步逆转录:所述逆转录第一步反应的反应体系总体积12微升,所述第一步逆转录反应的反应体系包含1微升随机引物,2微克rna模板及适量depc水,所述第一步逆转录反应的反应条件为:65摄氏度,5分钟;所述第二步逆转录反应的反应体系总体积20微升,所述第一步逆转录反应的反应体系包含4微升逆转录缓冲液,3微升三磷酸碱基脱氧核苷酸,1微升rna酶抑制剂,1微升逆转录酶,及12微升所述逆转录第一步反应的产物,所述第二步逆转录反应的反应程序为:25摄氏度、5分钟,42摄氏度、60分钟,70摄氏度、5分钟。待反应 结束后,-20摄氏度保存产物备用;第五步,依据qpcr试剂盒的实验说明手册操作,在实时荧光定量pcr仪进行实时荧光定量pcr反应:先将逆转录反应产物稀释10倍,反应体系总体积为20微升。实时荧光定量pcr反应条件为:95 摄氏度、3分钟,40个循环,95摄氏度、10秒,60摄氏度、30秒;第六步,在第五步反应完成后,与内参基因gapdh标准化,以δct值显示目标基因的相对表达值,判定基因的表达差异并计算p-value值。
[0483]
结果表明, 实施例13所制备的环形mrna分子与正常组织中的表达相比,梅克尔癌患者中的表达明显上调,差异有显著性。与20例正常上皮组织(用做对照)相比,实施例13所制备的环形mrna分子在40例梅克尔癌组织中明显高表达。在梅克尔癌组中实施例13所制备的环形mrna分子表达水平大约是正常组织的2倍,两组数据的差异具有统计学意义(p=0.0187)。所以,实施例13所制备的环形mrna分子在梅克尔癌组织中高表达,即实施例13所制备的环形mrna分子对梅克尔癌的发生发展可能具有重要的生物学功能。
[0484] 实施例175 梅克尔癌患者的预后分析对45例梅克尔癌患者进行了电话随访,详细询问了他们的首发时间、治疗情况、有无复发、有无再患其他疾病、复发及死亡时间等,并登记了生存时间和状态,并对梅克尔癌组织中实施例13所制备的环形mrna分子的表达与病人的生存时间和状态进行 的生存分析,发现实施例13所制备的环形mrna分子表达水平不同的梅克尔癌患者中预后存在明 显差异,对其进行kaplan-meier生存分析,分析结果表明:28例梅克尔癌组织中高表达实施例
5所制备的环形mrna分子的患者平均生存时间明显短于17例实施例13所制备的环形mrna分子表达较低的患者,两组数据的差异具有统计学意义(p=0.0088)。说明实施例13所制备的环形mrna分子是一个与梅克尔癌预后相关的分子标记,表明梅克尔癌高表达的患者预后较低表达的患者预后差,实施例13所制备的环形mrna的高表达是梅克尔癌患者预后风险预测的独立危险因素。
[0485] 实施例176实施例29所制备的环形mrna分子用于卡西波肉瘤患者预后实时荧光定量检测试剂盒的制备制备实施例29所制备的环形mrna分子用于卡西波肉瘤患者预后10次反应的实时荧光定量检测试剂盒。具体组分为:异丙醇25毫升、三氯甲烷25毫升、rna稳定溶液15毫升、trizol15毫升、无酶水3毫升、逆转录缓冲溶液25毫升、逆转录随机引物5微升、三磷酸碱基脱氧核苷酸15微升、rna酶抑制剂1微升、逆转录酶1微升、premixextaq1微升、实施例27所制备的的环形mrna分子特异性引物3微升(上游引物seqid100:5
´‑
ataaactttgatcagttattagttactg-3
´
;下游引物seqid101:5
´‑
gagagactggaacttaaggttaaattcc-3
´
);gapd小时特异性引物6微升(上游引物seqid136:5
´‑
ggagcgagatccctccaaaat-3
´
;下游引物seqid137:5
´‑
ggctgttgtcatacttctcatgg-3
´
)。
[0486] 实施例177卡西波肉瘤组织中实施例29所制备的环形mrna分子的检测第一步,收集待测卡西波肉瘤组织存放于盛有rna稳定溶液的冻存管中,放至-80摄氏度冰箱备用;第二步,取10毫克左右组织加入1毫升trizol,用匀浆机匀浆;离心15分钟,12000rpm,取上清;向上清中加入200微升氯仿,上下用力颠倒混匀半分钟,静置3分钟;第三步,在4摄氏度,12000rpm离心15分钟,取上清到新的ep管内;加入等体积的异丙醇,混匀,静置10分钟后,4摄氏度,12000rpm离心10分钟;第四步,小心去掉上清,加入1毫升75%乙醇,上下颠倒,使沉淀块重悬起;在4摄氏度,12000rpm离心10分钟,小心去掉上清,尽量吸干管壁的液体,若沉淀松动可再次离心。晾干约15分钟,至管壁无液体;第五步,加入适量体积(20-30微升)的depc水溶解rna,58摄氏度水浴10分钟;第六步,cdna逆转录:所述逆转录第一步反应的反应体系总体积15微升,所述第一步逆转录反应的反应体系包含1微升随机引物,2微克rna模板适量depc水,所述第一步逆转录反应的反应条件为:70摄氏度,5分钟;冰上,5分钟;所述第二步逆转录反应的反应体系总体积25微升,所述第一步逆转录反应的反应体系包含5微升逆转录缓冲液,3微升三磷酸碱基脱氧核苷酸,1微升rna酶抑制剂,1微升逆转录酶,及15微升所述逆转录第一步反应的产物,所述第二步逆转录反应的反应程序为:30摄氏度、2分钟,42摄氏度、45分钟,72摄氏度、8分钟。待反应结束后,-20摄氏度保存产物备用;第七步,实时荧光定量pcr反应:反应管中分别加入2微升10
×
buffer,2微升dntp,1微升正向引物,1微升反向引物,1微升cdna模板,0.2微升taq酶,加水至20微升。pcr反应条件如下:94摄氏度,5分钟预变性;94摄氏度,30秒变性;55摄氏度,30秒退火;72摄氏度,30秒延伸;35个循环;第八步,在第七步反应完成后,与内参基因gapdh标准化,以δct值显示目标基因
的相对表达值,判定基因的表达差异并计算p-value值。结果表明,实施例29所制备的环形mrna分子与正常组织中的表达相比,卡西波肉瘤患者中的表达明显上调,差异有显著性。与15例正常上皮组织(用做对照)相比,实施例29所制备的环形mrna分子在25例卡西波肉瘤组织中明显高表达。在卡西波肉瘤组中实施例29所制备的环形mrna分子表达水平大约是正常组织的2.3倍,两组数据的差异具有统计学意义(p=0.0131)。所以,实施例29所制备的环形mrna分子在卡西波肉瘤组织中高表达,即实施例29所制备的环形mrna分子对卡西波肉瘤的发生发展可能具有重要的生物学功能。
[0487] 实施例178卡西波肉瘤患者的预后分析对35例卡西波肉瘤患者进行了电话随访,详细询问了他们的首发时间、治疗情况、有无复发、有无再患其他疾病、复发及死亡时间等,并登记了生存时间和状态,并对卡西波肉瘤组织中实施例29所制备的环形mrna分子的表达与病人的生存时间和状态进行的生存分析,发现实施例29所制备的环形mrna分子表达水平不同的卡西波肉瘤患者中预后存在明显差异,对其进行kaplan-meier生存分析,分析结果表明:23例卡西波肉瘤组织中高表达实施例37所制备的环形mrna分子的患者平均生存时间明显短于12例实施例29所制备的环形mrna分子表达较低的患者,两组数据的差异具有统计学意义(p=0.0105)。说明实施例29所制备的环形mrna分子是一个与卡西波肉瘤预后相关的分子标记,表明卡西波肉瘤高表达的患者预后较低表达的患者预后差,实施例29所制备的环形mrna的高表达是卡西波肉瘤患者预后风险预测的独立危险因素。
[0488] 实施例179实施例37所制备的环形mrna分子用于htlv-1所致淋巴细胞恶性肿瘤患者预后实时荧光定量检测试剂盒的制备制备实施例37所制备的环形mrna分子用于htlv-1所致淋巴细胞恶性肿瘤患者预后30次反应的实时荧光定量检测试剂盒。具体组分为:异丙醇30毫升、三氯甲烷30毫升、rna稳定溶液20毫升、trizol20毫升、无酶水3毫升、逆转录缓冲溶液20毫升、逆转录随机引物15微升、三磷酸碱基脱氧核苷酸15微升、rna酶抑制剂3微升、逆转录酶3微升、premixextaq3微升、实施例37所制备的的环形mrna分子特异性引物9微升(上游引物seqid102:5
´‑
atgcggtctgacgataaatttaaccttatc-3
´
;下游引物seqid103:5
´‑
gaattcttacagatctcccgggcaaattgg-3
´
);gapd小时特异性引物9微升(上游引物seqid136:5
´‑
ggagcgagatccctccaaaat-3
´
;下游引物seqid137:5
´‑
ggctgttgtcatacttctcatgg-3
´
)。
[0489] 实施例180htlv-1所致淋巴细胞恶性肿瘤样本中实施例37所制备的环形mrna分子的检测第一步,收集待测htlv-1所致淋巴细胞恶性肿瘤样本存放于盛有rna稳定溶液的冻存管中,放至-80摄氏度冰箱备用;第二步,提取细胞rna时,先离心沉淀细胞,每5
×
106个细胞加1毫升trizol后,反复用枪吹打或剧烈震荡以裂解细胞;随后转入ep管中,在15-30摄氏度下放置5分钟;第三步,在上述ep管中,按照每1毫升trizol后加0.2毫升氯仿的量加入氯仿,盖上ep管盖子,在手中用力震荡15秒,在室温下(15-30摄氏度)放置2-3分钟后,12000rpm(2-8摄氏度)离心15分钟;第四步,去上层水相置于新ep管中,按照每1毫升trizol加0.5毫升异丙醇的量加
入异丙醇,在室温下(15-30摄氏度)放置10分钟后,12000rpm(2-8摄氏度)离心10分钟;第五步,弃上清,按照每1毫升trizol加1毫升75%乙醇进行洗涤,涡旋混合,7500rpm(2-8摄氏度)离心5分钟,弃上清。待反应结束后,-20摄氏度保存产物备用;第六步,依据qpcr试剂盒的实验说明手册操作,在实时荧光定量pcr仪进行实时荧光定量pcr反应:先将逆转录反应产物稀释10倍,反应体系总体积为20微升。实时荧光定量pcr反应条件为:95摄氏度,预变性10分钟,然后以95摄氏度变性15秒、60摄氏度退火30秒、72摄氏度延伸30秒,共进行38个循环;第七步,在第六步反应完成后,与内参基因gapdh标准化,以δct值显示目标基因的相对表达值,判定基因的表达并计算p-value值。结果表明,实施例差异37所制备的环形mrna分子与正常组织中的表达相比,htlv-1所致淋巴细胞恶性肿瘤患者中的表达明显下调,差异有显著性。与25例正常上皮组织(用做对照)相比,实施例37所制备的环形mrna分子在40例htlv-1所致淋巴细胞恶性肿瘤样本中明显低表达。在htlv-1所致淋巴细胞恶性肿瘤组中实施例37所制备的环形mrna分子表达水平大约是正常组织的50%,两组数据的差异具有统计学意义(p=0.0187)。所以,实施例37所制备的环形mrna分子在htlv-1所致淋巴细胞恶性肿瘤中高表达,即实施例37所制备的环形mrna分子对htlv-1所致淋巴细胞恶性肿瘤的发生发展可能具有重要的生物学功能。
[0490] 实施例181小时tlv-1所致淋巴细胞恶性肿瘤患者的预后分析对40例htlv-1所致淋巴细胞恶性肿瘤患者进行了电话随访,详细询问了他们的首发时间、治疗情况、有无复发、有无再患其他疾病、复发及死亡时间等,并登记了生存时间和状态,并对htlv-1所致淋巴细胞恶性肿瘤中实施例37所制备的环形mrna分子的表达与病人的生存时间和状态进行的生存分析,发现实施例37所制备的环形mrna分子表达水平不同的htlv-1所致淋巴细胞恶性肿瘤患者中预后存在明显差异,对其进行kaplan-meier生存分析,分析结果表明:27例htlv-1所致淋巴细胞恶性肿瘤中高表达实施例12所制备的环形mrna分子的患者平均生存时间明显长于13例实施例37所制备的环形mrna分子表达较低的患者,两组数据的差异具有统计学意义(p=0.0072)。说明实施例37所制备的环形mrna分子是一个与htlv-1所致淋巴细胞恶性肿瘤预后相关的分子标记,表明htlv-1所致淋巴细胞恶性肿瘤高表达的患者预后较低表达的患者预后好,实施例37所制备的环形mrna的低表达是htlv-1所致淋巴细胞恶性肿瘤患者预后风险预测的独立危险因素。
[0491] 实施例182递送系统库中结构式(i-1)化合物的合成第一步,将2,7-二羟基-9小时-芴-9-酮(d小时fl),无水碳酸钾,碘化钠和2-(2-(2-氯乙氧基)乙氧基)乙烷-1-醇和二甲基甲酰胺按一定比例混合,将反应混合物为在30瓦功率的液相等离子体场中反应2小时;第二步,在第一步反应完后,向反应中混合物中加入水,随后用二氯甲烷提取三次。将有机层用无水硫酸钠干燥,然后在减压蒸馏去除有机溶剂得粗产品;第三步,通过硅胶柱色谱法(洗脱液:97v/v%二氯甲烷+3使用v/v%甲醇)获得结构式(i-1)化合物。产率:67%-82%。1小时核磁共振谱为(500兆赫,cdcl3):δ7.29
–
7.27(m,2小时),7.17(d,j=2.4赫兹,2小时),6.98(dd,j=8.1,2.5赫兹,2小时),4.29-4.01(m,4h),3.94-3.81(m,4h)、3.79-3.67(m,4h)、3.66-3.51(m,4h)、1.79(p,2h)。
13
c核磁共振谱为(126兆赫,cdcl3)δ193.62、159.13、137.70、135.92、121.14、120.58、110.33、72.47、70.86、
70.37、69.61、67.91,61.78。esi高分辨质谱给出的分子量475.20,与计算值一致。
[0492] 实施例183递送系统库中结构式(i-2)化合物的合成第一步,将实施例184制得的结构式(i-1)化合物在30毫升离子液体中搅拌,然后通入氢气在0摄氏度下于50瓦功率的液相等离子体场中反应1小时;第二步,将混合物在冰水浴中放置,同时缓慢加入n小时4cl和hcl(1摩尔每升)的饱和水溶液,然后静置2小时;第三步,减压蒸馏去除有机溶剂,得粗产品,用乙醇提取二甲基亚砜提取产物。产率:70%-88%。1h核磁共振谱为(500兆赫,cdcl3):δ7.33-7.26(m,2h),7.15(d,j=2.4赫兹,2h),7.01(dd,j=8.1,2.5赫兹,2h),5.36(d,j=4.4赫兹,1h),4.31-4.04(m,4小时),3.97-3.84(m,4h)、3.77-3.66(m,4h)、3.64-3.48(m,4h)、1.77(p,2h)。
13
c核磁共振谱为(126兆赫,cdcl3)δ194.55、157.09、136.99、134.72、120.95、119.37、109.22、72.48、70.35、70.28、69.59、67.86,61.53。esi高分辨质谱给出的分子量477.20,与计算值一致。
[0493]
以上所述的仅是本发明的一些实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明的创造构思的前提下,还可以做出其它变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1