检测结核杆菌耐药基因突变的引物及试剂盒的制作方法
本发明属于生物检测,具体涉及一种检测结核杆菌耐药基因突变的引物及试剂盒。
背景技术:
1、近年来结核病在全世界广泛流行,严重威胁着人类的生命健康。全世界已有 1/3人感染了结核分枝杆菌,现有结核患者2000万,每年新发患者约900万,每年死亡人数高达300万。随着一线药物的使用,造成结核分枝杆菌耐药菌株不断的出现和传播。耐药菌株有两种,一种是耐多药结核分枝杆菌,是指患者感染的结核杆菌在体外对异烟肼和利福平耐药;另一种是广泛耐药结核分枝杆菌,除耐多药外,还对 6种主要抗结核药物中 3种以上的二线抗结核药耐药。由于广泛耐药结核分枝杆菌几乎无法治愈,积极防控耐多药结核分枝杆菌,使其不向广泛耐药结核分枝杆菌发展成为人类最后的防线。结核病的治疗和控制必须依赖于准确、快速、可靠的检测方法,早发现,早治疗,将结核分枝杆菌扼杀在初始阶段,才能减少传染,起到有效防控作用。
2、现有研究表明,结核分枝杆菌耐药分子机制大致有三种类型:即降低细胞膜的通透性和外排泵机制,产生降解或灭活酶类,药物靶位的改变。结核杆菌无法通过质粒的介导从其他细菌获得耐药性,因此染色体介导的耐药性是mtb产生耐药的主要基础。目前对mtb耐药分子机制的研究主要集中在药物的作用靶点及其相关基因的突变上。
3、1、利福平的耐药基因:利福平是作用于结核杆菌dna依赖的rna聚合酶亚基β亚单位(rna polymerase b subunit,rpob) ,从而抑制mrna的转录。结核分枝杆菌对利福平耐药是结核病化疗失败的主要原因,并且利福平耐药性的检测是判断多重耐药结核病(mdr-tb)的标志。rpob基因是一单拷贝基因,序列高度保守,全长3543个碱基。一些研究证实,当高度保守核心区域 (rrdr)发生突变时利福平不能与rna聚合酶β亚单位结合,抑制转录,约96%~98%的利福平耐药菌株编码rna聚合酶β亚单位的rpob基因突变导致细菌对利福平耐药主要的突变集中在编码27个氨基酸的81个碱基范围内。其中,以531位ser→leu和trp转换,526位his→tyr、asp、asn和pro的突变最为常见,且以上两个位点的突变是引起高水平耐药的主要原因。除上述两位点外,513、516、518、522位点也有突变,相对531和526较少,且是引起低水平耐药的原因。除突变引起耐药外,细胞壁渗透性的改变导致药物摄入量的减少也是导致耐药的原因之一。
4、2、异烟肼的耐药基因:结核分枝杆菌对抗结核化疗药物异烟肼的耐药机制较为复杂,约92%的inh耐药菌与katg,inha和ahpc三个基因突变有关。其中katg和inha基因是主要的耐药基因。katg的丢失或突变导致其编码的过氧化氢-过氧化物酶活性丧失或下降,而inha基因突变减弱了异烟肼对分枝杆菌酸生物合成的抑制作用。
5、katg基因是过氧化氢酶-过氧化物酶的编码基因。该基因含2223个核苷酸,上游隔44个碱基与fura基因相连,下游离cmbc基因2794碱基。katg基因编码的过氧化氢-过氧化物酶一种热稳定酶,相对分子质量为80000。引起异烟肼耐药性的主要原因是katg基因的点突变、部分缺失、碱基对插入或7%~24%的完全缺失,其突变导致过氧化氢酶-过氧化物酶活性降低或丧失,阻止异烟肼转换成活性形式,从而导致耐药。katg基因突变的位点为315位ser→thr、asn、ile或者arg,463位arg→leu,还可见104arg、108his、138asn、148leu、270his、275thr、312trp、381asp密码子突变。
6、inha基因是一种与分枝菌酸生物合成有关的烯酰基还原酶的编码基因。异烟肼进入菌体后,在分枝杆菌过氧化氢—过氧化物酶的作用下氧化脱氢生成亲电子形式,这种形式能与分枝菌酸生化合成途径中的烯酰基还原酶—还原型烟酰胺二核苷酸复合体结合,干扰分枝菌酸合成而发挥抗菌作用。inha基因产物为相对分子质量为32000的蛋白质。研究发现,inha基因的突变率较高的主要是基因调控区,位于启动子-15位点的碱基突变。
7、3、乙胺丁醇的耐药基因:乙胺丁醇是一种阿拉伯糖类似物,作用于分枝杆菌阿拉伯糖基转移酶,抑制阿拉伯糖基聚合入阿拉伯半乳聚糖,影响细胞壁分枝菌酸—阿拉伯半乳聚糖—蛋白聚糖复合物形成,发挥抗分枝杆菌作用。研究表明,结核分枝杆菌耐乙胺丁醇与阿拉伯糖基转移酶的编码基因。embabc操纵子突变或emb蛋白表达增高有关,该操纵子由embc、emba和embb三个基因组成,其中embb基因(尤其是306位密码子)突变是耐乙胺丁醇产生的主要分子机制。约47%~65%的耐emb菌株与embb基因突变有关。结核分枝杆菌embb基因约3246bp,编码一个糖基转移酶embb基因突变使糖基转移酶结构改变,影响了乙胺丁醇和糖基转移酶的相互作用,从而导致耐乙胺丁醇的产生。结核分枝杆菌耐乙胺丁醇分离株embb基因突变主要发生在306位密码子,其为met→val、ile、leu置换。
8、4、链霉素的耐药基因:链霉素是抗结核治疗中常用的氨基糖营类抗生类。sm主要作用于结核分枝杆菌的核糖体,诱导遗传密码的错误,抑制mrna转译的开始,干扰转译过程中的校对,从而抑制蛋白质合成。最近研究认为sm耐药是由于其核糖体s12蛋白编码基因rpsl或16srrna编码基因rrs突变所致,80%耐sm结核分枝杆菌临床分离株可见rpsl或rrs突变。rpsl基因最常见的是43位密码子lys→arg的突变,88位密码子也可发生同样突变。少数可见43位lys→thr的转变。核糖体蛋白s12的正常作用可能是维持读码过程中的一些轻微的不准确性,rpsl基因突变就会导致s12蛋白改变,从而严格要求核糖体只使用与每一密码对应的氨酰trna,更准确地表达mrna的每一个密码,抑制了sm诱导的遗传密码错误而产生耐药性。并且根据morris以及heym等的研究表明rpsl基因突变是sm耐药的主要机理。
9、目前结核分枝杆菌的耐药检测方法主要有常规检测方法和分子生物学检测方法。
10、常规检测方法,包括快速检查耐药突变的显微镜观察法、噬菌体药敏法、流式细胞术用于结核分枝杆菌药物敏感性试验、微量快速显色药敏检测法等,这些方法一般都需要借助于培养基, 在抗菌药物存在的前提下通过比例法、抗性比率法或绝对浓度法观察固体培养基上结核杆菌的生长情况。bactec460半自动血培养仪和mgit960全自动分枝杆菌培养/药敏系统目前已广泛用于实验室, 被认为是结核耐药检测的金标准。但这些方法均以细菌生长为终点判断结果,由于结核分枝杆菌生长缓慢,在得到分离培养物后仍需数天的时间方能获得结果。
11、近年来,随着分子生物学的发展,结核分枝杆菌的耐药机制及耐药的分子基础大部分已被阐明,从而为建立结核分枝杆菌耐药快速检测方法开辟了新的途径。
12、分子生物学检测方法,一般包括三步骤:样品dna的提取;针对相应耐药位点设计引物通过聚合酶链反应 (polym erase chain reaction,pcr)扩增与耐药性有关的基因片段;分析扩增产物进行耐药性判断。
13、1)直接测序方法 dna测序法,通过pcr方法扩增待测耐药基因,对其产物纯化之后测序,与其标准株的同一片段比较异同。目前 dna 测序是检测基因突变的金标准,不仅用于突变的筛选,而且能确定突变碱基的部位和分布。但该法操作繁琐,特别是对于多个基因的测序,需要进行多重pcr,pcr产物分别进行纯化后测序,一个测序反应只能检测一个pcr产物,多重pcr就需要多个测序反应,这样测序的通量就成倍减少。同时从技术的角度来说,多个pcr反应的孔间温度差异,反应体系的差异会导致扩增的一致性也有差异,影响测序结果。同时纯化之后的产物容易对实验室造成污染,再加上费用昂贵,因此限制了它的临床应用,故该方法多用于评价其他检测方法。
14、2)高分辨熔解曲线分析方法,通过实时监测升温过程中双链 dna荧光染料与 pcr扩增产物的结合情况。由于单核苷酸多态性位点不匹配,在双链 dna升温过程中会先解开,荧光染料从局部解链 dna 分子释放,从荧光强度与时间曲线上可以判断是否存在单核苷酸多态性位点。而且不同单核苷酸多态性位点、杂合子与否等都会影响熔解曲线的峰形,因此高分辨率熔解曲线分析能有效区分不同单核苷酸多态性位点与不同基因型。根据这个原理, 可针对相关耐药基因的的耐药位点设计引物,选用 resolight、lcg green等dna荧光染料, 进行pcr扩增及溶解曲线分析。同时对产物进行测序,根据溶解曲线的峰型,可有效区分不同的耐药位点。该技术不仅是高灵敏度、高特异性的核酸突变筛查技术,还具有简单、方便、低成本、高通量的优点,并且不损伤 dna,分析后可直接纯化测序。但是,高分辨率熔解曲线分析技术是通过碱基解链的差异来区分突变与否的,最小的碱基温差在1℃左右,因此,对样品间温度均一性要求很高,限制了该技术只能使用某些特定型号的pcr仪。另外,对pcr扩增产物的保守型要求很高,在扩增产物的区域内不能出现其它的多态性位点,否则多态性位点就会影响突变位点的误判,直接影响检测结果。
15、3)多重 pcr单链构象多态方法,根据耐药基因的相应位点设计 pcr的引物,在同一个反应体系中扩增产物,将扩增得到的产物变性,而后快速复性,使之成为具有一定空间结构的单链 dna 分子,将适量的单链 dna进行非变性聚丙烯酰胺凝胶电泳,最后通过放射自显影、银染或溴化乙锭显色分析。若单链 dna迁移率与正常对照的相比发生改变,可以判定该链构象发生改变,进而推断该 dna片段中有碱基突变。该方法简便、快速、灵敏,不需要特殊的仪器,适合临床实验的需要。但它也有不足之处。例如,只能作为一种突变检测方法,要最后确定突变的位置和类型,还需进一步测序;电泳条件要求较严格;另外,由于单链构象多态是依据点突变引起单链 dna分子立体构象的改变来实现电泳分离的,这样就可能会出现当某些位置的点突变对单链 dna分子立体构象的改变不起作用或作用很小时,再加上其他条件的影响,使聚丙烯酰胺凝胶电泳无法分辨造成漏检。
16、4)多重等位特异性 pcr检测方法,属于多重pcr与等位基因特异的组合技术,基本原理是将突变基因与正常基因不同的一个或几个碱基设计在 pcr引物的 3'端,当引物与模板完成互补时,pcr从引物 3'端开始延伸,反之,链就不能延伸。根据以上原理,可利用3'端含突变碱基的引物来检测靶 dna中有无相应突变位点。该反应系统包含两个聚合酶链扩增反应,有两对 3'端有差异的引物。一对为正常引物,另一对为 3'端突变的引物。正常引物只与正常模板互解,pcr时扩增相应的产物,而突变的引物只与突变的模板结合扩增出相应的产物,扩增完成后可直接同琼脂糖电泳技术检测分析结果。利用该系统进行基因突变检测时不仅能检出突变的纯合子,也能检出杂合子个体。因而,同一个体的 dna模板利用突变引物和正常引物均能引发扩增反应,也可利用多对突变引物和正常引物进行多重3'特异pcr, 供 dna分子多位点变化的鉴定,准确、快速、简便。但该测方法仅适用于点突变的检测。
17、5)分子信标法,是以 pcr为基础,在pcr过程中引入荧光标记探针,可以对 pcr反应过程随时进行监测并进行精确定量。该荧光标记探针为一茎环状结构的寡核苷酸探针,环状结构与靶序列互补,茎环结构即探针两臂由互补序列构成,这一部分与靶序列毫无关系。两臂的末端分别有荧光集团和猝灭集团,当靶核酸存在时,茎部结构结合靶序列而打开,荧光基团和淬灭基团分开,产生荧光,如序列不互补,即使存在一个碱基的差异,也会影响杂交,探针又恢复茎环结构,猝灭集团仍产生作用,则无荧光产生。该方法适于检测单个位点突变,一个突变位点一个反应体系,检测结果清晰明了,判读方便。但是对于多基因多位点的突变,该方法只能采用多管平行来检测,在技术上没有优势。而且对于突变位点临近的检测,需要兼顾至少两个以上的突变,探针设计难度较大,突变位点临近的检测效果不理想,也有可能错过新发生的突变,所以该方法在多基因多位点突变的检测中不是很普及。
18、6)基因芯片技术,是将特异的探针固定于玻璃片上,与已经标记荧光的 pcr 扩增产物杂交,如果扩增产物与探针序列完全匹配则结合较为牢固,杂交信号较强;如果有单个或多个碱基错配则信号较弱。通过比较有突变和没有突变的探针的杂交信号得到结果。该技术用于检测分子突变,不仅可以准确地确定突变位点和突变类型,更主要的是它的快速高效性是目前其他方法无法比拟的,并且它可以同时检测多个基因乃至整个基因组的突变。基因芯片能够快速、准确、高效地针对结核杆菌对异烟肼和利福平等多种药物产生的耐药性进行一次性的检测和筛选,在一次反应中进行多种信息的平行分析。应用该技术有利于结核病治疗方案的合理安排,提高结核病的防治水平。目前该技术还存在一定的不足和缺憾:需昂贵的仪器设备,检测成本较高,芯片制备比较复杂,样品的准备与标记又较为繁琐。
19、基于现有多重 pcr检测方法存在的不足,以及在已公开的专利文献中,如cn112011633a、cn110499377a等均没有涉及有关利福平、异烟肼、链霉素、乙胺丁醇四种药物的耐药基因同时进行检查的相关技术,改进检测结核杆菌耐药基因突变的方法非常必要。
技术实现思路
1、本发明的目的是提供一种检测结核杆菌耐药基因突变的引物及试剂盒。旨在解决现有技术中多重pcr检测方法复杂,灵敏度低,检测结果存在偏差等技术问题。
2、本发明的目的通过下述技术方案实现:
3、一种检测结核杆菌耐药基因突变的引物,包括以下用于检测四种抗结核病药物耐药基因突变的5对引物,所述5对引物的核苷酸序列如seq id no.1-10所示。
4、所述四种抗结核病药物为利福平、异烟肼、链霉素、乙胺丁醇四种一线抗结核病药。
5、进一步地,所述5对引物及其对应的检测位点如下:
6、第1对引物为rpob-f1/rpob-r1:其核苷酸序列如seq id no.1-2所示,用于检测利福平rpob耐药基因组上耐药突变位点中的531、526、522、516、513位点;
7、第2对引物为katg-f1/katg-r1:其核苷酸序列如seq id no.3-4所示,用于检测异烟肼katg耐药基因组上耐药突变位点中的315位点;
8、第3对引物为inha-f1/inha-r1:其核苷酸序列如seq id no.5-6所示,用于检测异烟肼inha耐药基因组上耐药突变位点中的15位点;
9、第4对引物为rpsl-f1/rpsl-r1:其核苷酸序列如seq id no.7-8所示,用于检测链霉素rpsl耐药基因组上耐药突变位点中的43、88位点,和/或检测链霉素its耐药基因组上耐药突变位点中的513、516位点;
10、第5对引物为embb-f1/embb-r1:其核苷酸序列如seq id no.9-10所示,用于检测乙胺丁醇embb耐药基因组上耐药突变位点中的306位点。
11、进一步地,所述5对引物中的所有上游引物(f-引物)的5’端连接一条相同的通用引物(mtb-f1)序列acttgagcatggcacgctgc;5对引物中的所有下游引物(r-引物)的5’端都连接另一条相同的通用引物(mtb-r1)序列ctacgtgaggtcgcgatcgc。
12、所述5对引物中的所有上游引物的核苷酸序列包括seq id no.1、seq id no.3、seq id no.5、seqid no.7、seq id no.9。
13、所述5对引物中的所有下游引物的核苷酸序列包括seq id no.2、seq id no.4、seq id no.6、seqid no.8、seq id no.10。
14、本发明结核分枝杆菌耐药突变位点检测的扩增引物和探针的具体核苷酸序列分别如下表1、表2所示。
15、
16、
17、 进一步地,本发明的核酸膜条上的核酸探针排列顺序如表3所示:
18、
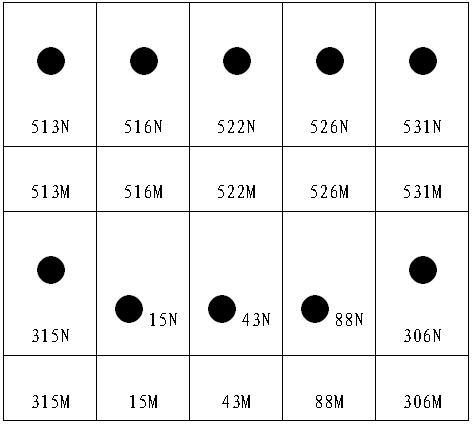
19、说明:1)n代表该位点为野生型,m代表该位点是突变型。2)数字分别代表不同耐药基因的突变氨基酸位点。3)根据不同位点的野生型或突变型显色情况判断结核分枝杆菌对某种抗生素的耐药与否。
20、本发明还提供了用于检测结核杆菌耐药基因突变的试剂盒,本发明试剂盒依据核酸分子杂交的基本原理,采用聚合酶链式反应(pcr)产物和固定于膜基因芯片上的核酸探针进行固液相的核酸杂交技术,通过过氧化物酶催化底物显色的方式显示核酸杂交结果。膜基因芯片由尼龙膜和固定在其上的探针阵列构成,膜芯片的开发具有操作简便、成本低廉等明显优势。
21、本发明用于检测结核杆菌耐药基因突变的试剂盒除包括上述5对引物外,还包括dna提取试剂、pcr反应液、杂交反应试剂、孵育液和pcr显色液。
22、进一步地,所述pcr反应液中各试剂的用量如下:
23、
24、进一步地,所述杂交反应试剂包括a液:1×ssc,0.1%sds,ph 7.4; b液:0.5×ssc,0.1%sds,ph 7.4;c液:0.1m柠檬酸钠,ph 5.4。
25、进一步地,所述孵育液为由a液(1×ssc,0.1%sds,ph 7.4)与过氧化物酶(pod)按体积比2000:1配制得到。
26、进一步地,所述pcr显色液的组成成分为:c液(0.1m柠檬酸钠,ph 5.4)19ml,tmb1ml,30% h2o2,2μl。
27、本发明用于检测结核杆菌耐药基因突变的试剂盒的使用方法如下:
28、1、样本获取:留取深咳痰标本3-5ml。
29、2、样本处理
30、1)收集痰标本,加入等量的4%的naoh静置处理30分钟。
31、2)采用痰样本提取试剂(磁珠法)提取dna,备用。
32、3、pcr扩增
33、取出pcr反应液,加入提取的痰样本dna作为pcr扩增模板;同时,以阳性对照dna和裂解液为模板设置一个阳性对照和一个阴性对照,然后进行扩增。
34、4、杂交
35、将标有样本编号的膜条放入杂交盒中,加入a液于48℃进行预热后,将pcr扩增产物在100℃加热后加入对应膜条编号的a液中,48℃杂交1小时;同时,将适量b液预热至48℃。
36、5、洗膜
37、弃去a液,加入预热的b液,于48℃轻摇洗涤。
38、6、孵育
39、弃去b液,加入由a液与过氧化物酶(pod)配制成的孵育液,室温轻摇进行孵育。
40、7、显色
41、用a液室温轻摇洗涤后,再用c液室温洗膜,然后将膜条浸泡于显色液中避光显色5-10分钟即可观察结果,检测后得到的核酸膜条结果示意图参阅图1、图2或图3。
42、图1显示痰样检测结果为:利福平、异烟肼、链霉素和乙胺丁醇都敏感;
43、图2显示痰样检测结果为:利福平耐药(531m)、异烟肼敏感、链霉素和乙胺丁醇敏感;
44、图3显示痰样检测结果为:利福平耐药(531m)、异烟肼耐药(315m)、链霉素耐药(43m)和乙胺丁醇耐药(306m)。
45、相较于现有技术,本发明的优点及有益效果在于:
46、(1)本发明采用将每对耐药基因的引物和通用引物进行巧妙设计和配套运用,解决了多重pcr的扩增因各引物的扩增效率不同而带来的扩增产物有差异的普遍性问题。为了提高扩增效率,减少扩增试剂的平行次数,目前对于多基因的扩增,普遍采用多重pcr,但是多重pcr中由于多对引物在一个反应体系中同时扩增,必然会竞争酶和其它扩增原材料,扩增效率高、结合能力强的引物对必然会优先占用酶和原材料,优先扩增;扩增效率低的引物对没有充分扩增,如此以来,多重pcr的扩增产物量差异较大,扩增结果不一致,结果也会有很大的偏差。本发明在各不同基因的特异引物对5’端统一增加一对扩增效率高的通用引物,通过设计特异引物和通用引物的碱基数差异来控制扩增退火温度,设计的特异引物碱基数多,退火温度高,在pcr扩增的第一个循环时优选扩增。而设计的通用引物碱基数少,退火温度低,在pcr扩增的第二个循环时优先扩增,如此,相当于对于各耐药基因进行第二次扩增放大,通过这二次放大,各耐药基因均能达到扩增饱和状态,最终的扩增产物趋向一致。
47、(2)本发明采用这种二次扩增的技术,还能够提高扩增灵敏度。在第一个扩增循环中,浓度极低的基因由于多重pcr的竞争,扩增效率低,扩增放大的次数有限,但是即便如此,相对于原来的极低浓度,基因也是有了几十倍的增加,在第二个扩增循环中,这些极低浓度的基因增加了几十倍后作为第二个扩增循环的模板,经过二次扩增后均可以与其它高浓度的基因一起进入一个扩增平台期,与其它基因的扩增产物无明显区别,产物更均一。
48、(3)本发明同时检测4种抗结核药物对应的5个耐药基因,这5个耐药基因在结核分枝杆菌基因组内的分布较分散,采用一对引物同时扩增5个目的基因是难以实现的,因此,本发明采用多重pcr的设计方法,分别设计各耐药基因的引物和探针,缩短目的基因的扩增片段,提高扩增和杂交效率。由于结核分枝杆菌基因组分子量较大,达到了4.4mb,目的基因在基因组中暴露在外的概率较小,因此,引物与目的基因序列结合时受到空间位阻效应影响较大,结合较困难。因此,本发明中引物设计难度较大,在研发和试验过程中,经过了大量的摸索和试验,才优选出扩增效率高的引物序列。
49、(4)本发明采用的是单管五重pcr扩增体系,即在一个pcr扩增体系中同时含有五对耐药基因的引物,五重pcr扩增体系的优化难度较大,在试验过程中需要从两种pcr开始摸索,两种pcr稳定后再摸索三重pcr、四重pcr到五重pcr这么一步步的优化摸索过来的。五对引物之间容易出现互相抑制的情况,导致某两对引物不能共存于一个反应体系,需要重新调整和设计引物对;多对引物在扩增过程中互相竞争酶等扩增原材料,导致扩增不均一,形成引物二聚体从而降低引物的有效浓度,影响扩增效率。本发明经过多次的摸索和比较,优选出最佳引物对,通过调整引物的用量,实现了五重pcr扩增体系也能均一高效的扩增。
- 还没有人留言评论。精彩留言会获得点赞!