1-甲基-3-二氟甲基吡唑-4-甲酸绿色合成工艺的制作方法
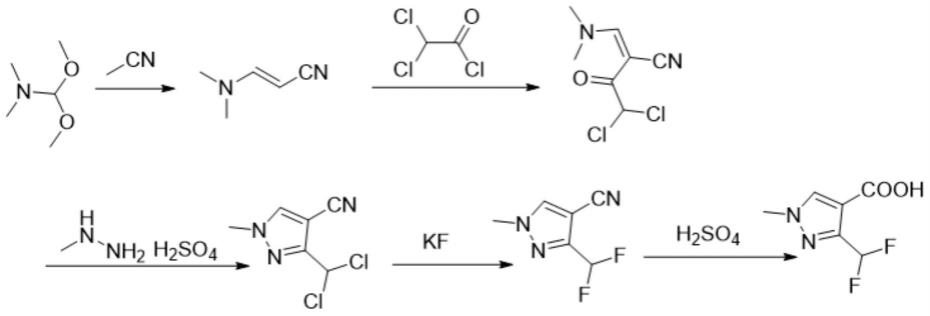
1.本发明涉及1-甲基-3-二氟甲基吡唑-4-甲酸合成领域,更具体地说,涉及1-甲基-3-二氟甲基吡唑-4-甲酸绿色合成工艺。
背景技术:
2.ep2008996介绍了以二氯乙酰氯,乙烯基醚类化合物,甲基肼等原料5步合成1-甲基-3-二氟甲基吡唑-4-甲酸的合成路线,该路线反应要求温度苛刻,后处理复杂。cn101687806对其工艺进行了改进,但对设备要求较高,氟化卤代条件苛刻。
3.us3965141介绍了一种乙腈和dmf-dma高温高压合成3-(二甲氨基)丙烯腈的合成工艺,该路线对设备要求高,反应时间长,能耗大。
4.cn204263800介绍了一种以二氟乙酰氯,3-(二甲氨基)丙烯腈和甲基肼为原料制备1-甲基-3-二氟甲基吡唑-4-甲酸的合成路线,该路线使用的二氟乙酰氯较二氯乙酰氯价格昂贵且不易得,甲基肼更为剧毒物,对环境不友好。另外该路线报导了以氰基乙酸钠和亚胺盐制备3-二甲氨基丙烯腈的合成路线,该路线三废较多并且亚胺盐的制备要求高。
5.cn106380447介绍了一种1-甲基-3-二氯甲基-4-吡唑腈氟化卤代制备1-甲基-3-二氟甲基-4-吡唑腈的合成方法,主要采用常规季铵盐(四丁基溴化铵等)作为氟化催化剂,反应温度高,反应时间长,氟化钾用量大。
技术实现要素:
6.针对现有技术中存在的问题,本发明的目的在于提供1-甲基-3-二氟甲基吡唑-4-甲酸绿色合成工艺,反应副产单一,三废少,反应条件温和,绿色环保。
7.为解决上述问题,本发明采用如下的技术方案。
8.1-甲基-3-二氟甲基吡唑-4-甲酸绿色合成工艺,包括以下步骤:
9.s1:乙腈和dmf-dma经微通道连续流反应得到3-(二甲氨基)丙烯腈;s1步骤中,优选的,所述乙腈和dmf-dma的质量比为10%-50%,优先的为33%。
10.优选的,所述微通道连续流反应采用corning g1-10fm sic高通量微通道反应器;反应温度为120-140℃,反应时间为5-10分,反应结束后降温至50℃以下,得到3-(二甲氨基)丙烯腈。
11.s2:s1步骤得到的3-(二甲氨基)丙烯腈与二氯乙酰氯反应制备2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈;
12.优选的,s2步骤中所述3-(二甲氨基)丙烯腈和二氯乙酰氯的质量比为1:1-3,优选1-2;反应体系温度为0-10℃。有机碱缚酸剂为三乙胺、吡啶或二异丙胺,优先选用三乙胺,有机碱缚酸剂和3-(二甲氨基)丙烯腈的质量比为2-5,优先选用2.5当量;疏水性反应溶剂为甲苯、二氯乙烷或二氯甲烷,优先选用甲苯,疏水性反应溶剂量为3-(二甲氨基)丙烯腈的5-20倍,优选选用10倍。
13.优选的,当有机碱缚酸剂为三乙胺,疏水性反应溶剂为甲苯时;s2步骤中所述反应
具体过程为先在3-(二甲氨基)丙烯腈中加入三乙胺和甲苯,然后滴加二氯乙酰氯,并保温搅拌反应至结束后,过滤回收三乙胺盐酸盐,滤液用水洗涤后,得到2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈的甲苯溶液待用。
14.s3:s2步骤得到的2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈再经甲基硫酸肼缩合反应得到1-甲基-3-二氯甲基-4-吡唑腈;
15.优选的,s3步骤中所述甲基硫酸肼缩合反应时,2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈和甲基硫酸肼的摩尔比为1:1-1.5,优选为1:1.1;所述缩合反应时先将步骤s2得到的2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁溶液回流分水后,再加入甲基硫酸肼升温回流反应,反应结束后,降至室温调ph至中性,选用氨水调节,分层,水相降温析晶得副产硫酸铵,有机相浓缩甲苯并析晶得到1-甲基-3-二氯甲基-4-吡唑腈。
16.s4:然后由s3步骤得到的1-甲基-3-二氯甲基-4-吡唑腈、n-烷基共轭离子型季铵盐类主催化剂和咪唑类离子液体助催化剂经氟化制得1-甲基-3-二氟甲基-4-吡唑腈;
17.优选的,s4步骤中所述氟化反应采用氟化钾,溶剂为环丁砜,所述1-甲基-3-二氯甲基-4-吡唑腈:氟化钾:环丁砜的摩尔比为1:2-3:2-55,优先的为1:2.2:3。
18.n-烷基共轭离子型季铵盐类主催化剂和1-甲基-3-二氯甲基-4-吡唑腈的质量比为1%-3%,优选的为1%。咪唑类离子液体催化剂和n-烷基共轭离子型季铵盐类主催化剂的质量比为10%-30%,优先的为25%。优选的,s4步骤中所述n-烷基共轭离子型季铵盐类催化剂为双-(n-双(二甲胺基)亚甲基)-氯化亚铵盐。优选的,s4步骤中所述咪唑类离子液体催化剂为1-十四基-3-甲基咪唑溴盐。
19.优选的,所述氟化反应过程升温至140-160℃,氟化反应搅拌时间为5-8小时,当hplc检测转化率达到98%后,过滤,滤液经精馏得到1-甲基-3-二氟甲基-4-吡唑腈。
20.s5:最后s4步骤制得的1-甲基-3-二氟甲基-4-吡唑腈经酸性水解,待水解完全后过滤,滤饼经水洗烘干制备1-甲基-3-二氟甲基吡唑-4-甲酸。
21.优选的,s5步骤中所述酸性水解采用70%硫酸,1-甲基-3-二氟甲基-4-吡唑腈和70%硫酸的质量比为1:2-4,反应升温为80-100℃,搅拌时间为7-10小时。
22.相比于现有技术,本发明的优点在于:
23.一、本方案总反应路线新颖,反应副产单一,三废少,反应条件温和,绿色环保。
24.二、采用微通道连续流技术制备合成3-二甲氨基丙烯腈,大幅度提高了反应效率及产物纯度和收率。
25.三、使用甲基硫酸肼进行缩合关环,避免了剧毒物甲基肼的使用,另外采用氨水中和制备副产硫酸铵,避免了大量废水及固废的产生,对环境更友好。
26.四、氟化采用n-烷基共轭离子型季铵盐为n-烷基共轭离子型季铵盐类主催化剂,咪唑类离子液体为助催化剂,大幅度提高了氟化效率,有效降低了反应时间及氟化钾的用量,收率和纯度更高。
附图说明
27.图1为本发明的实施例1的反应路线图;
28.图2为本发明的实施例1的分析报告示意图;
29.图3为本发明的微通道流程示意图;
30.图4为本发明的微通道内部结构示意图;
31.图5为本发明的使用常规催化剂的hplc谱图;
32.图6为本发明的不使用助催化剂的hplc谱图;
33.图7为本发明的主催化剂和助催化剂均加入的hplc谱图。
具体实施方式
34.实施例1:
35.s1:3-(二甲氨基)丙烯腈的合成
36.corning g1-10fm sic高通量微通道反应器,设置反应模块温度140℃,降温模块温度50℃,并分别计量泵入乙腈和dmf-dma,乙腈和dmf-dma计量比例为1:3,反应停留时间5分钟。收集反应液,并减压回收副产甲醇,得到含量98%的3-(二甲氨基)丙烯腈,收率99%。
37.s2:2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈的合成
38.四口反应瓶中加入98%的3-(二甲氨基)丙烯腈(11.9g,0.1mol),三乙胺(25g,0.25mol),甲苯(100ml)。控制反应体系温度0-10℃滴加二氯乙酰氯(19g,0.13mol),并保温搅拌反应至hplc检测转换率达到98%,过滤回收三乙胺盐酸盐,滤液用水洗涤一次后直接用于下一步,滤液hplc含量97.5%。
39.s3:1-甲基-3-二氯甲基-4-吡唑腈的合成
40.2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈的甲苯溶液回流分水后,加入甲基硫酸肼(16g,0.11mol),升温至回流反应6小时,hplc检测转化率达到98%,降至室温并用氨水调ph至中性,分层,水相降温析晶得副产硫酸铵,有机相浓缩甲苯并析晶得到含量98%的1-甲基-3-二氯甲基-4-吡唑腈,两步收率85%。
41.s4:1-甲基-3-二氟甲基-4-吡唑腈的合成
42.四口烧瓶中加入1-甲基-3-二氯甲基-4-吡唑腈(190g,1mol),氟化钾(128g,2.2mol),2克双-(n-双(二甲胺基)亚甲基)-氯化亚铵盐,0.5克1-十四基-3-甲基咪唑溴盐和350克脱水处理的环丁砜,升温至140℃,搅拌反应6小时,hplc检测转化率达到98%,过滤,滤液经精馏得到1-甲基-3-二氟甲基-4-吡唑腈152克,纯度99.5%,收率96.8%。
43.实施例2:
44.s1:3-(二甲氨基)丙烯腈的合成
45.corning g1-10fm sic高通量微通道反应器,设置反应模块温度130℃,降温模块温度50℃,并分别计量泵入乙腈和dmf-dma,乙腈和dmf-dma计量比例为1:3,反应停留时间8分钟。收集反应液,并减压回收副产甲醇,得到含量98%的3-(二甲氨基)丙烯腈,收率99%。
46.s2:2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈的合成
47.四口反应瓶中加入98%的3-(二甲氨基)丙烯腈(35.7g,0.3mol),三乙胺(75g,0.75mol),甲苯(300ml)。控制反应体系温度5-10℃滴加二氯乙酰氯(57g,0.39mol),并保温搅拌反应至hplc检测转换率达到98%,过滤回收三乙胺盐酸盐,滤液用水洗涤一次后直接用于下一步,滤液hplc含量97.8%。
48.s3:1-甲基-3-二氯甲基-4-吡唑腈的合成
49.2-((二甲氨基)亚甲基)-4,4-二氯-3-羰基丁腈的甲苯溶液回流分水后,加入甲基硫酸肼(48g,0.33mol),升温至回流反应6小时,hplc检测转化率达到98%后,降至室温并用
氨水调ph至中性,分层,水相降温析晶得副产硫酸铵,有机相浓缩甲苯并析晶得到含量98%的1-甲基-3-二氯甲基-4-吡唑腈,两步收率85%。
50.s4:1-甲基-3-二氟甲基-4-吡唑腈的合成
51.四口烧瓶中加入1-甲基-3-二氯甲基-4-吡唑腈(280g,2mol),氟化钾(261g,4.5mol),4克双-(n-双(二甲胺基)亚甲基)-氯化亚铵盐,1克1-十四基-3-甲基咪唑溴盐和700克脱水处理的环丁砜,升温至145℃,搅拌反应6小时,hplc检测转化率达到98%后,过滤,滤液经精馏得到1-甲基-3-二氟甲基-4-吡唑腈302克,纯度99.5%,收率96.1%。
52.s5:1-甲基-3-二氟甲基吡唑-4-甲酸的合成
53.四口烧瓶中加入1-甲基-3-二氟甲基-4-吡唑腈(157g,1mol),70%硫酸600克,升温至90℃搅拌10小时,待水解完全后过滤,滤饼经少量水洗后,烘干得165克白色固体1-甲基-3-二氟甲基吡唑-4-甲酸,纯度99.3%,收率96%。滤液经降温析晶回收副产硫酸铵。
54.对比例1:常规方式合成3-(二甲氨基)丙烯腈
55.1l高压反应釜中,投入250克乙腈和80克dmf-dma,通入氮气置换三次。用氮气充压至反应釜压力1.0mpa。升温,于145-155℃,压力1-1.5mpa,搅拌反应56小时。降温,泄压,反应液经精馏回收乙腈及副产甲醇后得60克3-(二甲氨基)丙烯腈,含量97%,收率93%。
56.对比例2:采用常规氟化催化剂
57.四口烧瓶中加入1-甲基-3-二氯甲基-4-吡唑腈(190g,1mol),氟化钾(128g,2.2mol),10克四丁基溴化铵和350克脱水处理的环丁砜,升温至140℃,搅拌反应36小时,hplc检测转化率达到98%后,过滤,滤液经精馏得到1-甲基-3-二氟甲基-4-吡唑腈130克,纯度99.5%,收率83%。
58.对比例3:不使用助催化剂
59.四口烧瓶中加入1-甲基-3-二氯甲基-4-吡唑腈(190g,1mol),氟化钾(128g,2.2mol),2克双-(n-双(二甲胺基)亚甲基)-氯化亚铵盐和350克脱水处理的环丁砜,升温至140℃,搅拌反应20小时,hplc检测转化率达到98%后,过滤,滤液经精馏得到1-甲基-3-二氟甲基-4-吡唑腈145克,纯度99.5%,收率92.3%。
60.四口烧瓶中加入1-甲基-3-二氟甲基-4-吡唑腈(78.5g,0.5mol),70%硫酸300克,升温至100℃搅拌8小时,待水解完全后过滤,滤饼经少量水洗后,烘干得84克白色固体1-甲基-3-二氟甲基吡唑-4-甲酸,纯度99.3%,收率95.5%。滤液经降温析晶回收副产硫酸铵。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1