一种甾体衍生物及其制备方法和应用、抗肿瘤药物组合物
1.本发明属于药物合成技术领域,具体涉及一种甾体衍生物及其制备方法和应用、抗肿瘤药物组合物。
背景技术:
2.癌症是具有高度致死性的恶性疾病,是影响公众健康的全球性问题。随着人口老龄化的加速,老年人比例逐步攀升,癌症发病人数逐年增加且呈现年轻化趋势。因此,开发研究疗效优异、耐药性弱、靶向性好、副作用小且价格低廉的抗癌药物显得尤为迫切。
3.天然产物具有丰富的结构及化学多样性,它们是药物及药物先导化合物的重要来源,据统计约80%的药物都源于天然产物或与天然产物相关的衍生物。甾体类化合物广泛存在于多种动植物的次级代谢产物中,许多的甾体化合物都表现出多种独特的生理活性。而且多种天然甾体化合物被开发为药物。通过甾体分子骨架的修饰与改变或者引入某些复杂的官能团可以大幅度的提高化合物的药效、减少对人体的毒副作用,并能改变化合物作用性质,使其专属作用性变得单一。
4.鉴于甾体类化合物所具有的特殊且广泛的生物活性,对甾体化合物结构的修饰倍受研究者的追捧,其中骨架中c-17位杂环的引入使得甾体与杂环的结合得以实现,该类衍生物表现出独特的生物活性,如抑菌、抗炎、抗抑郁、抗氧化等活性。目前,将甾体类化合物用于抗肿瘤细胞的应用较少,而且目前发现的甾体类化合物的抗肿瘤细胞活性不佳。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种甾体衍生物及其制备方法和应用、抗肿瘤药物组合物。本发明提供的甾体衍生物表现出良好的肿瘤细胞抑制活性,有望成为制备抗肿瘤药物的活性成分。
6.为了实现上述目的,本发明提供了以下技术方案:
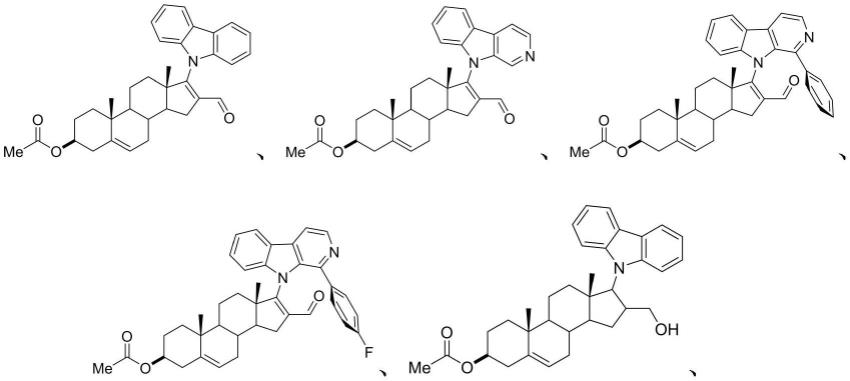
7.本发明提供了一种甾体衍生物,包括β-咔啉基甾体衍生物或咔唑基甾体衍生物;
8.所述β-咔啉基甾体衍生物具有式i所示结构:
[0009][0010]
式i中,所述r1包括氢、c1~c6直链酰基、苯基酰基、取代苯基酰基;所述r2为氢、c1~c6烷基、苯基、取代芳香基;
[0011]
所述咔唑基甾体衍生物具有式ii所示结构:
[0012][0013]
式ii中,所述r3包括甲醛基或甲羟基。
[0014]
优选的,包括
[0015][0016][0017]
本发明还提供了上述技术方案所述甾体衍生物的制备方法,,其特征在于:
[0018]
式i所示结构的β-咔啉基甾体衍生物的制备方法,包括以下步骤:
[0019]
当r1为甲酰基时:
[0020]
将去氢表雄酮和甲酸混合,进行第一酯化反应,得到第一酯类化合物;
[0021]
将所述第一酯类化合物、第一有机溶剂和第一维尔斯迈尔试剂混合,进行第一氯代醛基化反应,得到第一氯代醛中间体;
[0022]
将所述第一氯代醛中间体、第二有机溶剂、第一碱性催化剂和第一β-咔啉类化合物混合,进行第一亲核加成反应,得到r1为甲酰基的β-咔啉基甾体衍生物;
[0023]
当r1为氢时:
[0024]
将所述r1为甲酰基的β-咔啉基甾体衍生物、第三有机溶剂和酸化剂混合,进行水解反应,得到r1为氢的β-咔啉基甾体衍生物;
[0025]
当r1为c2~c6直链酰基、苯基酰基、取代苯基酰基时:
[0026]
将去氢表雄酮、第四有机溶剂、酰化合物和第一缚酸剂混合,进行第二酯化反应,得到第二酯类化合物;所述酰化合物包括c2~c5直链酰酐、已酰氯、苯基酰氯、单取代苯基
酰氯或多取代苯基酰氯;
[0027]
将所述第二酯类化合物、第五有机溶剂和第二维尔斯迈尔试剂混合,进行第二氯代醛基化反应,得到第二氯代醛中间体;
[0028]
将所述第二氯代醛中间体、第六有机溶剂、第二碱性催化剂和第二β-咔啉类化合物混合,进行第二亲核加成反应,得到β-咔啉基甾体衍生物;
[0029]
所述第一β-咔啉类化合物和第二β-咔啉类化合物的结构式独立为所述r2包括氢、c1~c6烷基、苯基、取代芳香基;
[0030]
式ii所示结构的咔唑基甾体衍生物的制备方法,包括以下步骤:
[0031]
当r3为甲醛基时,
[0032]
将去氢表雄酮、第七有机溶剂、乙酸酐和第二缚酸剂混合,进行第三酯化反应,得到第三酯类化合物;
[0033]
将所述第三酯类化合物、第八有机溶剂和第三维尔斯迈尔试剂混合,进行第三氯代醛基化反应,得到第三氯代醛中间体;
[0034]
将所述第三氯代醛中间体、第九有机溶剂、第三碱性催化剂和咔唑混合,进行第三亲核加成反应,得到r3为甲醛基的咔唑基甾体衍生物;
[0035]
当r3为甲羟基时,
[0036]
将所述r3为甲醛基的咔唑基甾体衍生物、第十有机溶剂和还原剂混合,进行还原反应,得到咔唑基甾体衍生物。
[0037]
优选的,所述第一有机溶剂包括n,n-二甲基甲酰胺;所述第二有机溶剂包括二氧六环;所述第三有机溶剂包括乙醇;所述第四有机溶剂包括二氯甲烷;所述第五有机溶剂包括n,n-二甲基甲酰胺,所述第六有机溶剂包括二氧六环,所述第七有机溶剂包括二氯甲烷,所述第八有机溶剂包括n,n-二甲基甲酰胺,所述第九有机溶剂包括二氧六环,所述第十有机溶剂包括甲醇。
[0038]
优选的,所述第一氯代醛基化反应、第二氯代醛基化反应和第三氯代醛基化反应的温度为100℃,时间为5~10h。
[0039]
优选的,所述第一亲核加成反应、第二亲核加成反应和第三亲核加成反应的温度为60℃,时间为2~4h。
[0040]
优选的,所述水解反应在室温下进行;所述水解反应的时间为8~16h。
[0041]
优选的,所述还原反应在室温下进行,所述还原反应的时间为1~2h。
[0042]
本发明还提供了上述技术方案所述甾体衍生物或上述技术方案所述制备方法制备得到的甾体衍生物在制备抗肿瘤药物中的应用。
[0043]
本发明还提供了一种抗肿瘤药物组合物,所述抗肿瘤药物组合物包括甾体衍生物和药学上可接受的辅料;所述甾体衍生物为权利要求1或2所述甾体衍生物或权利要求3~8所述制备方法制备得到的甾体衍生物。
[0044]
本发明提供了一种甾体衍生物,包括β-咔啉基甾体衍生物或咔唑基甾体衍生物;
[0045]
所述β-咔啉基甾体衍生物具有式i所示结构:式 i,式i中,所述r1包括氢、c1~c6直链酰基、苯基酰基、取代苯基酰基;所述r2为氢、c1~c6烷基、苯基、取代芳香基;
[0046]
所述咔唑基甾体衍生物具有式ii所示结构:式ii,式ii中,所述r3包括甲醛基或甲羟基。
[0047]
本发明提供的甾体衍生物具有良好的抗肿瘤细胞增殖活性。本发明的实施例结果显示,在5μm浓度下对癌细胞的抑制率在85%以上,对a549(人肺腺癌细胞)、hela(子宫颈癌细胞系)、hepg2(人肝癌细胞)三种肿瘤细胞均有很好的细胞毒性,对三种癌细胞的毒性优于顺铂。
具体实施方式
[0048]
本发明提供了一种甾体衍生物,包括β-咔啉基甾体衍生物或咔唑基甾体衍生物;
[0049]
所述β-咔啉基甾体衍生物具有式i所示结构:式i,式i中,所述r1包括氢、c1~c6直链酰基、苯基酰基、取代苯基酰基;所述r2为氢、c1~c6烷基、苯基、取代芳香基;
[0050]
所述咔唑基甾体衍生物具有式ii所示结构:式ii,式ii中,所述r3包括甲醛基或甲羟基。
[0051]
在本发明中,r1优选为氢、甲酰基或乙酰基;所述取代芳香基优选包括卤素、甲基、甲氧基、羟基、胺基、羧基或醛基;所述卤素优选包括f、cl 或br;所述取代芳香基的取代位置优选为邻位、间位或对位;所述r2优选为氢、c1~c3直链烷基、苯基、单取代苯基或具有两
个取代基的苯基,更优选为氢、苯基、4-氯苯基或4-甲基苯基。
[0052]
在本发明中,所述甾体衍生物优选为:
[0053][0054]
为了详实的描述式i和式ii的结构,本发明定义上下文中的术语。
[0055]
术语“烷基”应指从烷烃的任一碳原子上除去氢原子而衍生的一价基团,“烷基”的碳原子形成直链或支链的骨架,因此,“烷基”可分为“直链烷基”和“支链烷基”。该术语包括伯、仲、叔烷基子类,如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、正己基、异己基。特别地,术语“烷烃”是指仅仅含有碳、氢的饱和烃化合物。
[0056]
术语“芳香基”又可以称为“芳基”,应包括碳环芳环基团,碳原子数为 c6-c10芳环基团,比如苯基(c6)、萘基(c10)和c8芳环基团。
[0057]
术语“单取代芳香基”应理解为芳香基其中一个碳原子的氢被其它的基团取代。例如,芳香基若为苯基,这个氢可以被卤素(氟、氯、溴或碘)取代,形成邻、间、对卤代苯基;或被甲基取代,形成邻甲基苯基、间甲基苯基、对甲基苯基;或被甲氧基取代,形成邻甲氧基苯基、间甲氧基苯基、对甲氧基苯基。芳香基若为萘基,与此类似,在此不作详述。
[0058]
术语“多取代芳香基”应指芳香基其中两个或两个以上碳原子的氢被其它的基团取代。例如,芳香基若为苯基,两个氯原子,或两个氟原子,或一个氯原子一个氟原子可以取代不同碳原子上的氢;苯环上的氢被甲基和氟取代,可以形成2-氟对甲基苯基;或苯环上的氢被甲基和氯取代,可以形成 3-氯-2-甲基苯基;或苯环上的氢被两个三氟甲基取代,可以形成3,5-三氟甲基苯基。芳香基若为萘基,与此类似,在此不作详述。
[0059]
术语“酰基”应指有机酸去掉一个羟基后剩下的原子团其通式为 r-c=o-。
[0060]
本发明还提供了上述技术方案所述甾体衍生物的制备方法,式i所示结构的β-咔啉基甾体衍生物的制备方法,包括以下步骤:
[0061]
当r1为甲酰基时:
[0062]
将去氢表雄酮和甲酸混合,进行第一酯化反应,得到第一酯类化合物;
[0063]
将所述第一酯类化合物、第一有机溶剂和第一维尔斯迈尔试剂混合,进行第一氯代醛基化反应,得到第一氯代醛中间体;
[0064]
将所述第一氯代醛中间体、第二有机溶剂、第一碱性催化剂和第一β-咔啉类化合物混合,进行第一亲核加成反应,得到r1为甲酰基的β-咔啉基甾体衍生物;
[0065]
当r1为氢时:
[0066]
将所述r1为甲酰基的β-咔啉基甾体衍生物、第三有机溶剂和酸化剂混合,进行水解反应,得到r1为氢的β-咔啉基甾体衍生物;
[0067]
当r1为c2~c6直链酰基、苯基酰基、取代苯基酰基时:
[0068]
将去氢表雄酮、第四有机溶剂、酰化合物和第一缚酸剂混合,进行第二酯化反应,得到第二酯类化合物;所述酰化合物包括c2~c5直链酰酐、已酰氯、苯基酰氯、单取代苯基酰氯或多取代苯基酰氯;
[0069]
将所述第二酯类化合物、第五有机溶剂和第二维尔斯迈尔试剂混合,进行第二氯代醛基化反应,得到第二氯代醛中间体;
[0070]
将所述第二氯代醛中间体、第六有机溶剂、第二碱性催化剂和第二β-咔啉类化合物混合,进行第二亲核加成反应,得到β-咔啉基甾体衍生物;
[0071]
所述第一β-咔啉类化合物和第二β-咔啉类化合物的结构式独立为所述r2包括氢、c1~c6烷基、苯基、取代芳香基;
[0072]
式ii所示结构的咔唑基甾体衍生物的制备方法,包括以下步骤:
[0073]
当r3为甲醛基时,
[0074]
将去氢表雄酮、第七有机溶剂、乙酸酐和第二缚酸剂混合,进行第三酯化反应,得到第三酯类化合物;
[0075]
将所述第三酯类化合物、第八有机溶剂和第三维尔斯迈尔试剂混合,进行第三氯代醛基化反应,得到第三氯代醛中间体;
[0076]
将所述第三氯代醛中间体、第九有机溶剂、第三碱性催化剂和咔唑混合,进行第三亲核加成反应,得到r3为甲醛基的咔唑基甾体衍生物;
[0077]
当r3为甲羟基时,
[0078]
将所述r3为甲醛基的咔唑基甾体衍生物、第十有机溶剂和还原剂混合,进行还原反应,得到咔唑基甾体衍生物。
[0079]
如无特殊说明,本发明对所用制备原料的来源没有特殊要求,采用本领域技术人员所熟知的市售商品即可。
[0080]
式i所示结构的β-咔啉基甾体衍生物的制备方法包括以下步骤:
[0081]
当r1为甲酰基时,本发明将去氢表雄酮和甲酸混合,进行第一酯化反应,得到第一酯类化合物(结构式为)。
[0082]
在本发明中,所述去氢表雄酮和甲酸的摩尔比优选为1∶(10~20),更优选为1∶15;
[0083]
在本发明中,所述去氢表雄酮和甲酸的混合过程优选为将去氢表雄酮溶解于甲酸中,搅拌至去氢表雄酮完全溶解;所述第一酯化反应的过程优选为将所述去氢表雄酮和甲酸的混合所得溶液进行回流;所述回流的温度优选为甲酸的沸点;所述回流的时间优选为8h。
[0084]
第一酯化反应完成后,本发明优选对第一酯化反应所得产物依次进行洗涤、萃取、旋干和烘干,得到第一酯类化合物。在本发明中,所述洗涤所用溶液优选为水。本发明对所述洗涤、萃取、旋干和烘干的过程没有特殊限定,采用本领域熟知的洗涤、萃取、旋干和烘干即可。
[0085]
得到第一酯类化合物后,本发明将所述第一酯类化合物、第一有机溶剂和第一维尔斯迈尔试剂混合,进行第一氯代醛基化反应,得到第一氯代醛中间体(结构式为)。
[0086]
在本发明中,所述第一维尔斯迈尔试剂现用现制。在本发明中,所述第一维尔斯迈尔试剂的制备方法优选包括以下步骤:
[0087]
在0℃下,将三氯氧磷溶液滴加入至呈搅拌状态下的n,n-二甲基甲酰胺中,滴加完毕后,继续进行搅拌20min,待用。在本发明中,所述三氯氧磷溶液和n,n-二甲基甲酰胺的体积比优选为4.65∶20;所述三氯氧磷溶液的浓度优选为10.75mol/l;所述滴加的时间优选为30min。本发明对所述搅拌的过程没有特殊限定,采用本领域熟知的搅拌过程即可。
[0088]
在本发明中,所述第一有机溶剂优选包括n,n-二甲基甲酰胺;所述第一酯类化合物的摩尔和第一有机溶剂的体积之比优选为5mmol∶30ml。
[0089]
在本发明中,所述第一酯类化合物的摩尔和第一维尔斯迈尔试剂的体积之比优选为5mmol∶20ml;所述第一酯类化合物和第一维尔斯迈尔试剂的混合过程优选为将所述第一酯类化合物溶解于第一有机溶剂中,得到混合溶液,在0℃下,将所述混合溶液滴加入所述第一维尔斯迈尔试剂中。
[0090]
在本发明中,所述第一氯代醛基化反应的温度优选为100℃;所述第一氯代醛基化反应的时间优选为5~10h,更优选为5h。
[0091]
第一氯代醛基化反应完成后,本发明优选对所述第一氯代醛基化反应所得产物依次进行淬灭、抽滤、萃取、干燥、过滤和柱分,得到第一氯代醛中间体。
[0092]
在本发明中,所述淬灭的方式优选为检测所述氯代醛基化反应完全后,将所述氯代醛基化反应所得产物导入饱和的碳酸氢钠溶液中,搅拌至有固体析出。本发明对检测所述氯代醛基化反应的过程没有特殊限定,采用本领域熟知的检测过程即可。本发明对所述抽滤的过程没有特殊限定,采用本领域熟知的抽滤过程即可。在本发明中,所述萃取所用溶剂优选为二氯甲烷,所述萃取的次数为三次。本发明对所述干燥的过程没有特殊限定,采用本领域熟知的干燥过程即可。本发明对所述过滤的过程没有特殊限定,采用本领域熟知的过滤过程即可。在本发明中,所述柱分所用溶剂优选为石油醚和乙酸乙酯;所述石油醚和乙酸乙酯的体积比优选为10∶1。
[0093]
得到第一氯代醛中间体后,本发明将将所述第一氯代醛中间体、第二有机溶剂、第
一碱性催化剂和第一β-咔啉类化合物混合,进行第一亲核加成反应,得到r1为甲酰基的β-咔啉基甾体衍生物。
[0094]
在本发明中,所述第二有机溶剂优选包括二氧六环;所述第一氯代醛中间体的摩尔和第二有机溶剂的体积之比优选为2mmol∶20ml。
[0095]
在本发明中,所述第一碱性催化剂优选包括碳酸铯;所述第一氯代醛中间体和第一碱性催化剂的摩尔比优选为2∶5.5。
[0096]
在本发明中,所述第一β-咔啉类化合物的结构式为所述 r2包括氢、c1~c6烷基、苯基、取代芳香基,优选为氢、c1~c3直链烷基、苯基、单取代苯基或具有两个取代基的苯基,更优选为苯基、4-氟苯基或4
‑ꢀ
甲基苯基;所述第一氯代醛中间体和β-咔啉类化合物的摩尔比优选为2∶2.5。
[0097]
在本发明中,所述第一β-咔啉类化合物的制备方法优选为将色氨酸和醛类化合物在5%硫酸水溶液中反应24h,然后经重铬酸钾氧化后,利用乙酸乙酯萃取,得到第一β-咔啉类化合物。
[0098]
在本发明中,所述第一氯代醛中间体、第二有机溶剂、第一碱性催化剂和第一β-咔啉类化合物的混合过程优选为将第一氯代醛中间体溶解于第二有机溶剂中,然后加入第一碱性催化剂和第一β-咔啉类化合物。
[0099]
在本发明中,所述第一亲核加成反应的温度优选为60℃,时间优选为 2~4h,更优选为2h。
[0100]
第一亲核加成反应完成后,本发明优选对所述第一亲核加成反应依次进行检测,检测反应至完全后,依次进行稀释、萃取和柱分,得到r1为甲酰基的β-咔啉基甾体衍生物。
[0101]
本发明对所述检测、稀释、萃取和柱分的过程没有特殊限定,采用本领域熟知的检测、稀释、萃取和柱分的过程即可。在本发明中,所述稀释所用溶液优选为水;所述萃取所用溶剂优选为二氯甲烷。
[0102]
当r1为氢时,本发明将所述r1为甲酰基的β-咔啉基甾体衍生物、第三有机溶剂和酸化剂混合,进行水解反应,得到β-咔啉基甾体衍生物。
[0103]
在本发明中,所述第三有机溶剂优选包括为乙醇;所述r1为甲酰基的β-咔啉基甾体衍生物的摩尔和第三有机溶剂的体积之比为1mmol∶15ml;所述酸化剂优选包括盐酸或磷酸;所述酸化剂的浓度优选为2mol/l;本发明对所述酸化剂的用量没有特殊限定,根据实际需求进行调整即可。
[0104]
在本发明中,所述r1为甲酰基的β-咔啉基甾体衍生物、第三有机溶剂和酸化剂的混合过程优选为将所述取代基r1为甲酰基的β-咔啉基甾体衍生物溶于第三有机溶剂中,在搅拌条件下加入酸化剂。本发明对所述搅拌的过程没有特殊限定,采用本领域熟知的搅拌过程即可。
[0105]
在本发明中,所述水解反应的温度优选为室温;所述水解反应的时间优选为8~16h,更优选为8h;所述水解反应优选在搅拌的条件下进行;本发明对所述搅拌的过程没有特殊限定,采用本领域熟知的搅拌过程即可。
[0106]
水解反应完成后,本发明优选对所述水解反应所得产物依次进行淬灭、萃取和柱分,得到取代基r1为氢的β-咔啉基甾体衍生物。
[0107]
在本发明中,所述淬灭的方式优选为检测所述水解反应完全后,将所述水解反应所得产物加入至饱和的碳酸氢钠溶液中进行淬灭;所述萃取所用溶剂优选为二氯甲烷,所述萃取的次数为三次;所述柱分所用溶剂优选为二氯甲烷和甲醇;所述二氯甲烷和甲醇的体积比优选为10∶1。
[0108]
当r1为c2~c6直链酰基、苯基酰基、取代苯基酰基时,
[0109]
本发明将去氢表雄酮、第四有机溶剂、酰化合物和第一缚酸剂混合,进行第二酯化反应,得到第二酯类化合物。
[0110]
在本发明中,所述第四有机溶剂优选为二氯甲烷;所述去氢表雄酮的摩尔和第四有机溶剂的体积之比优选为10mmol∶150ml。
[0111]
在本发明中,所述酰化合物包括c2~c5直链酰酐、已酰氯、苯基酰氯、单取代苯基酰氯或多取代苯基酰氯,更优选为乙酸酐;所述去氢表雄酮和酰化合物的摩尔比优选为10∶12。
[0112]
在本发明中,所述第一缚酸剂优选包括三乙胺;所述去氢表雄酮和第一缚酸剂的摩尔比优选为10∶25。
[0113]
在本发明中,所述去氢表雄酮、第四有机溶剂、酰化合物和第一缚酸剂的混合过程优选为将所述去氢表雄酮溶解于第四有机溶剂中,搅拌至完全溶解,然后加入酰化合物,再缓慢滴加入第一缚酸剂。本发明对所述搅拌的过程没有特殊限定,采用本领域熟知的搅拌过程即可。
[0114]
在本发明中,所述滴加的时间优选为30min;所述第二酯化反应的时间优选为5h;所述第二酯化反应的温度优选为室温;所述第二酯化反应优选在搅拌的条件下进行;所述搅拌的速率优选为700r/min。
[0115]
第二酯化反应完成后,本发明优选将所述第二酯化反应所得产物进行洗涤、萃取、旋干和烘干,得到第二酯类化合物。在本发明中,所述洗涤所用溶液优选为水;本发明对所述萃取、旋干和烘干的过程没有特殊限定,采用本领域熟知的萃取、旋干和烘干的过程即可。
[0116]
得到第二酯类化合物后,本发明将所述第二酯类化合物、第五有机溶剂和第二维尔斯迈尔试剂混合,进行第二氯代醛基化反应,得到第二氯代醛中间体。
[0117]
在本发明中,所述第二维尔斯迈尔试剂的制备方法与所述第一维尔斯迈尔试剂的制备方法一致,在此不再赘述。
[0118]
在本发明中,所述第五有机溶剂优选包括n,n-二甲基甲酰胺;所述第二酯类化合物的摩尔和第二维尔斯迈尔试剂的体积之比优选为 5mmol∶30ml;所述第二酯类化合物和第二维尔斯迈尔试剂的混合过程优选为将所述第二酯类化合物溶解于第五有机溶剂中,得到混合溶液,在0℃下,将所述混合溶液滴加入所述维尔斯迈尔试剂中;所述第二酯类化合物的摩尔和第五有机溶剂的体积之比优选为5mmol∶30ml;所述滴加的时间优选为 30min;所述第二氯代醛基化反应的温度优选为100℃;所述第二氯代醛基化反应的时间优选为5~10h,更优选为5h。
[0119]
第二氯代醛基化反应完成后,本发明优选对所述第二氯代醛基化反应所得产物依
次进行淬灭、抽滤、萃取、干燥、过滤和柱分,得到第二氯代醛中间体。
[0120]
在本发明中,所述淬灭的方式优选为检测所述第二氯代醛基化反应完全后,将所述第二氯代醛基化反应所得产物导入饱和的碳酸氢钠溶液中,搅拌至有固体析出。本发明对检测所述第二氯代醛基化反应的过程没有特殊限定,采用本领域熟知的检测过程即可。本发明对所述抽滤的过程没有特殊限定,采用本领域熟知的抽滤过程即可。在本发明中,所述萃取所用溶剂优选为二氯甲烷,所述萃取的次数为三次。本发明对所述干燥和过滤的过程没有特殊限定,采用本领域熟知的干燥和过滤过程即可。在本发明中,所述柱分所用溶剂优选为石油醚和乙酸乙酯;所述石油醚和乙酸乙酯的体积比优选为 10∶1。
[0121]
得到第二氯代醛中间体后,本发明将所述第二氯代醛中间体、第六有机溶剂、第二碱性催化剂和第二β-咔啉类化合物混合,进行第二亲核加成反应,得到β-咔啉基甾体衍生物。
[0122]
在本发明中,所述第六有机溶剂优选包括二氧六环;所述第二氯代醛中间体的摩尔和第六有机溶剂的体积之比优选为2mmol∶20ml。
[0123]
在本发明中,所述第二β-咔啉类化合物的结构式为所述 r2包括氢、c1~c6烷基、苯基、取代芳香基,优选为氢、c1~c3直链烷基、苯基、单取代苯基或具有两个取代基的苯基,更优选为苯基、4-氟苯基或4
‑ꢀ
甲基苯基;所述第二碱性催化剂优选包括碳酸铯;所述第二氯代醛中间体和第二碱性催化剂的摩尔比优选为2∶5.5;所述第二氯代醛中间体和第二β-咔啉类化合物的摩尔比优选为2∶2.5。
[0124]
在本发明中,所述第二β-咔啉类化合物的制备方法优选为将色氨酸和醛类化合物在5%硫酸水溶液中反应24h,然后经重铬酸钾氧化后,利用乙酸乙酯萃取,得到第二β-咔啉类化合物。
[0125]
在本发明中,所述第二氯代醛中间体、第二有机溶剂、第二碱性催化剂和第二β-咔啉类化合物的混合过程优选为将第二氯代醛中间体溶解于第二有机溶剂中,然后加入第二碱性催化剂和第二β-咔啉类化合物。在本发明中,所述第二亲核加成反应的温度优选为60℃,时间优选为2~4h,更优选为2h。
[0126]
式ii所示结构的咔唑基甾体衍生物的制备方法包括以下步骤:
[0127]
当r3为醛基时,本发明将去氢表雄酮、第七有机溶剂、乙酸酐和第二缚酸剂混合,进行第三酯化反应,得到第三酯类化合物(结构式为)。
[0128]
在本发明中,所述第七有机溶剂优选包括二氯甲烷;所述去氢表雄酮的摩尔和第七有机溶剂的体积之比优选为10mmol∶150ml。
[0129]
在本发明中,所述去氢表雄酮和乙酸酐的摩尔比优选为10∶12。
[0130]
在本发明中,所述第二缚酸剂优选包括三乙胺;所述去氢表雄酮和第二缚酸剂的摩尔比优选为10∶25。
[0131]
在本发明中,所述去氢表雄酮、第七有机溶剂、乙酸酐和第二缚酸剂的混合过程优
选为将所述去氢表雄酮溶解于第七有机溶剂中,搅拌至完全溶解,然后加入乙酸酐,再缓慢滴加入第二缚酸剂。本发明对所述搅拌的过程没有特殊限定,采用本领域熟知的搅拌过程即可。
[0132]
在本发明中,所述滴加的时间优选为30min;所述第三酯化反应的时间优选为5h;所述第三酯化反应的温度优选为室温;所述第三酯化反应优选在搅拌的条件下进行;所述搅拌的速率优选为700r/min。
[0133]
第三酯化反应完成后,本发明优选将所述第三酯化反应所得产物依次进行洗涤、萃取、旋干和烘干,得到第三酯类化合物。在本发明中,所述洗涤所用溶液优选为水;本发明对所述萃取、旋干和烘干的过程没有特殊限定,采用本领域熟知的萃取、旋干和烘干的过程即可。
[0134]
得到第三酯类化合物后,本发明将所述第三酯类化合物、第八有机溶剂和第三维尔斯迈尔试剂混合,进行第三氯代醛基化反应,得到第三氯代醛中间体(结构式为)。
[0135]
在本发明中,所述第三维尔斯迈尔试剂的制备方法与所述第一维尔斯迈尔试剂的制备方法一致,在此不再赘述。
[0136]
在本发明中,所述第八有机溶剂优选包括n,n-二甲基甲酰胺;所述第三酯类化合物的摩尔和第三维尔斯迈尔试剂的体积之比优选为 5mmol∶30ml;所述第三酯类化合物和第三维尔斯迈尔试剂的混合过程优选为将所述第三酯类化合物溶解于第八有机溶剂中,得到混合溶液,在0℃下,将所述混合溶液滴加入所述第三维尔斯迈尔试剂中;所述第三酯类化合物的摩尔和第八有机溶剂的体积之比优选为5mmol∶30ml;所述滴加的时间优选为30min;所述第三氯代醛基化反应的温度优选为100℃;所述第三氯代醛基化反应的时间优选为5~10h,更优选为5h。
[0137]
第三氯代醛基化反应完成后,本发明优选对所述第三氯代醛基化反应所得产物依次进行淬灭、抽滤、萃取、干燥、过滤和柱分,得到第三氯代醛中间体。
[0138]
在本发明中,所述淬灭的方式优选为检测所述第三氯代醛基化反应完全后,将所述第三氯代醛基化反应所得产物导入饱和的碳酸氢钠溶液中,搅拌至有固体析出。本发明对检测所述第三氯代醛基化反应的过程没有特殊限定,采用本领域熟知的检测过程即可。本发明对所述抽滤的过程没有特殊限定,采用本领域熟知的抽滤过程即可。在本发明中,所述萃取所用溶剂优选为二氯甲烷,所述萃取的次数为三次。本发明对所述干燥和过滤的过程没有特殊限定,采用本领域熟知的干燥和过滤过程即可。在本发明中,所述柱分所用溶剂优选为石油醚和乙酸乙酯;所述石油醚和乙酸乙酯的体积比优选为 10∶1。
[0139]
得到第三氯代醛中间体后,本发明将所述第三氯代醛中间体、第九有机溶剂、第三碱性催化剂和咔唑混合,进行第三亲核加成反应,得到r3为甲醛基的咔唑基甾体衍生物。
[0140]
在本发明中,所述第九有机溶剂优选包括二氧六环;所述第三氯代醛中间体的摩尔和第九有机溶剂的体积之比优选为2mmol∶20ml。
[0141]
在本发明中,所述第三碱性催化剂优选包括碳酸铯;所述第三氯代醛中间体和第三碱性催化剂的摩尔比优选为2∶5.5;所述第三氯代醛中间体和咔唑的摩尔比优选为2∶
2.5。
[0142]
在本发明中,所述第三氯代醛中间体、第九有机溶剂、第三碱性催化剂和咔唑的混合过程优选为将第三氯代醛中间体溶解于第九有机溶剂中,然后加入第三碱性催化剂和咔唑。在本发明中,所述第三亲核加成反应的温度优选为60℃,时间优选为2~4h,更优选为2h。
[0143]
当所述r3为甲羟基时,本发明将所述r3为甲醛基的咔唑基甾体衍生物、第十有机溶剂和还原剂混合,进行还原反应,得到咔唑基甾体衍生物。
[0144]
在本发明中,所述第十有机溶剂优选包括甲醇;所述r3为甲醛基的咔唑基甾体衍生物的摩尔与第十有机溶剂的体积之比优选为0.5mmol∶10ml。
[0145]
在本发明中,所述还原剂优选为硼氢化钠或四氢铝锂,更优选为硼氢化钠;所述r3为甲醛基的咔唑基甾体衍生物和还原剂的摩尔比优选为0.5∶1。
[0146]
在本发明中,所述r3为甲醛基的咔唑基甾体衍生物、第十有机溶剂和还原剂的混合过程优选为将所述r3为甲醛基的咔唑基甾体衍生物溶解于第十有机溶剂中,然后分批加入还原剂。在本发明中,所述还原反应优选在室温下进行;所述还原反应的时间优选为1~2h,更优选为1h。
[0147]
还原反应完成后,本发明优选对所述还原反应所得产物依次进行淬灭、萃取和柱分,得到咔唑基甾体衍生物。
[0148]
在本发明中,所述淬灭的方式优选为检测所述还原反应完全后,将所述还原反应所得产物加入至饱和的冰醋酸中进行淬灭;所述萃取所用溶剂优选为乙酸乙酯,所述萃取的次数为三次;所述柱分所用溶剂优选为石油醚和乙酸乙酯;所述石油醚和乙酸乙酯的体积比优选为2∶1。
[0149]
具体地,在本发明的实施例中,咔唑基甾体衍生物以及β-咔啉基甾体衍生物的合成路线包括下述步骤:
[0150]
(a)选用去氢表雄酮(化合物1)作为各个咔唑基甾体衍生物以及β
‑ꢀ
咔啉基甾体衍生物的合成中间体,分别与甲酸以及乙酸酐反应生成中间产物酯(化合物2和7);
[0151]
(b)化合物2和7在三氯氧磷的催化下发生反应,得到相应的氯代醛基化合物3和8;
[0152]
(c)将化合物3和8分别与咔唑以及β-咔啉发生取代反应生成化合物4、 5.1-5.3和9;
[0153]
(d)或以硼氢化钠为还原剂,将化合物4还原获得化合物6;或化合物9在稀盐酸的催化下水解得到化合物10。
[0154]
合成路线如下所示:
[0155][0156]
本发明还提供了上述技术方案所述甾体衍生物或上述技术方案所述制备方法制备得到的甾体衍生物在制备抗肿瘤药物中的应用。
[0157]
本发明还提供了一种抗肿瘤药物组合物,所述抗肿瘤药物组合物包括甾体衍生物和药学上可接受的辅料;所述甾体衍生物为上述技术方案所述甾体衍生物或上述技术方案所述制备方法制备得到的甾体衍生物。
[0158]
在本发明中,所述抗肿瘤药物组合物优选用于抑制人肺腺癌细胞、子宫颈癌细胞系或人肝癌细胞的活性。
[0159]
在本发明中,所述抗肿瘤药物组合物的供药剂型优选包括颗粒剂、片剂、丸剂、糖衣丸、栓剂、胶囊剂、胶囊缓释剂、缓释片剂、混悬剂或注射液;所述药学上可接受的辅料优选包括水、硬脂酸镁、滑石、淀粉、有机酸、葡聚糖或类脂质。
[0160]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
[0161]
实施例1
[0162]
将2.88g(10mmol)去氢表雄酮(化合物1)溶解于装有150ml二氯甲烷的250ml茄型烧瓶中,搅拌至完全溶解后,将乙酸酐(12mmol)缓慢加入反应体系中;室温下将3.46ml(25mmol)三乙胺在0.5h内缓慢滴入反应体系中,室温700r/min搅拌5h,用水洗涤并萃取,旋干,烘干,得到3.2g 化合物2,结构式为本步骤的产物无需分离可直接用于下一步反应;
[0163]
向充分干燥的圆底烧瓶内加入20ml干燥过的二甲基甲酰胺(dmf)溶液,在0℃的搅拌条件下0.5h内缓慢滴加4.65ml三氯氧磷溶液(50mmol) 后,继续搅拌反应20min,得到visemier(维尔斯迈尔)试剂;
[0164]
将所述5mmol化合物2溶解于30ml无水dmf溶液中,0℃下,在30min 内缓慢滴入所述vilsemier试剂中,滴毕后加热至100℃反应5h,检测反应至完全。此时将反应体系倒入饱和的碳酸氢钠溶液中,搅拌至有固体析出,抽滤得到目标产物的粗品然后溶解于二氯甲烷中萃取三次,所得有机相合并干燥,过滤、柱分(石油醚:乙酸乙酯=10:1),得到1.5g化合物3纯品,结构式为
[0165]
取2mmol化合物3溶于20ml二氧六环中,然后加入759mg碳酸铯 (5.5mmol)以及2.5mmol咔唑,加热至60℃反应2h,检测反应至完全,用水稀释,利用乙酸乙酯萃取三次,合并有机相,柱分得到1.5mmol化合物4,结构式为
[0166]
实施例2
[0167]
与实施例1的区别在于,将咔唑替换为β-咔啉,其与内容与实施例1一致,制备得到化合物5.1,结构式为
[0168]
实施例3
[0169]
与实施例1的区别在于,将咔唑替换为1-苯基-β-咔啉,其与内容与实施例1一致,制备得到化合物5.2,结构式为
[0170]
实施例4
[0171]
与实施例1的区别在于,将咔唑替换为1-(4-氟-苯基)-β-咔啉,其与内容与实施例1一致,制备得到化合物5.3,结构式为
[0172]
实施例5
[0173]
取0.5mmol化合物4溶于10ml甲醇溶液中,然后分批加入38mg硼氢化钠(1mmol),在室温下反应1h后,检测反应完毕用冰醋酸淬灭,然后用乙酸乙酯萃取三次,合并有机相,柱
分(石油醚:乙酸乙酯=2:1)得0.45mmol 化合物6,结构式为
[0174]
实施例6
[0175]
将2.88g(10mmol)去氢表雄酮(化合物1)溶解于装有150ml甲酸的 250ml茄型烧瓶中,搅拌至完全溶解,回流反应8h后,用水洗涤并萃取,旋干,烘干,得到3.0g化合物7,结构式为本步骤的产物无需分离可直接用于下一步反应;
[0176]
向充分干燥的圆底烧瓶向内加入20ml干燥过的dmf溶液,在0℃的搅拌下在0.5h内缓慢滴加4.65ml三氯氧磷溶液(50mmol),滴毕后搅拌反应20min,得到visemier试剂;
[0177]
将5mmol化合物7溶解于30ml无水dmf中,0℃下,在30min内缓慢滴入所述visemier试剂中,滴毕后加热至100℃反应5h,检测反应至完全,此时将反应体系倒入饱和的碳酸氢钠溶液中,搅拌至有固体析出,抽滤得到目标产物的粗品,然后溶解于二氯甲烷中萃取三次,所得有机相合并干燥,过滤、柱分(石油醚:乙酸乙酯=10:1),得到1.2g化合物8,结构式为
[0178]
取2mmol化合物8溶于20ml二氧六环中,然后加入759mg碳酸铯 (5.5mmol)以及2.5mmolβ-咔啉,加热至60℃反应2h,检测反应至完全,用水稀释,利用乙酸乙酯萃取三次,合并有机相,柱分,得到1.5mmol化合物9,结构式为
[0179]
实施例7
[0180]
将1mmol化合物9溶于装有15ml乙醇的25ml干燥圆底烧瓶中,在室温搅拌条件下加入2n盐酸溶液反应8h,检测反应,待反应完全后加入饱和的碳酸氢钠溶液淬灭,然后用二氯甲烷萃取三次,合并有机相,柱分(二氯甲烷:甲醇=10:1),得到0.71mmol化合物10,结构式为
[0181]
性能测试
207.5,170.6,145.2,141.0,140.9,140.1,138.3,133.6,132.8,132.0,129.7,129.6, 129.1,128.9,128.1,122.5,121.9,121.8,121.7,113.5,111.8,73.8,50.3,49.4, 47.9,38.1,37.0,36.9,31.5,31.0,30.9,28.7,27.8,21.5,20.4,19.5,14.2。
[0191]
经检测,化合物5.3的核磁数据为:
[0192]1h-nmr(cdcl3,500mhz):δ8.64(d,j=5.0hz,1h),8.19(d,j=7.8hz, 1h),7.98(d,j=5.0hz,1h),7.63(t,j=7.5hz,3h),7.59(s,1h),7.41(t,j= 7.5hz,1h),7.34(d,j=8.3hz,1h),7.10(t,j=8.4hz,2h),5.28(d,j=4.9hz, 1h),4.67
–
4.50(m,3h),2.02(s,3h),1.93
–
1.78(m,5h),1.68(d,j=10.8hz, 2h),1.63
–
1.53(m,2h),1.51
–
1.41(m,2h),1.33
–
1.27(m,2h),1.02(s,3h), 0.73(s,3h);
13
c-nmr(cdcl3,125mhz):δ(ppm)207.4,170.63,163.2(j
cf
= 247.5hz),143.8,141.1,140.9,140.1,134.2,133.5,133.0,132.3,131.5(j
cf
= 8.75hz),129.4,129.4,122.4,122.0,121.9,121.6,115.0(j
cf
=22.5hz),113.7, 111.8,73.8,50.3,49.5,47.9,38.1,36.9,31.5,31.0,30.9,29.8,28.7,27.8,21.5, 20.3,19.5,14.1。
[0193]
经检测,化合物6的核磁数据为:
[0194]1h-nmr(cdcl3,500mhz):δ8.10(d,j=7.7hz,2h),7.45(t,j=7.6hz, 2h),7.33
–
7.22(m,3h),6.97
–
6.85(m,1h),5.23(dd,j=5.2,1.9hz,1h), 4.64
–
4.54(m,1h),4.35(t,j=2.4hz,1h),3.10
–
3.01(m,1h),2.27(d,j=7.8 hz,2h),2.14(dd,j=16.9,6.5hz,1h),2.03(s,3h),1.98(d,j=12.3hz,1h), 1.90
–
1.83(m,2h),1.81
–
1.69(m,2h),1.61(d,j=42.4hz,2h),1.51
–
1.43 (m,2h),1.37(t,j=7.3hz,2h),1.33
–
1.26(m,1h),1.18
–
1.07(m,2h),1.00 (s,3h),0.88(s,3h);
13
c-nmr(cdcl3,125mhz):δ(ppm)170.7,147.6,139.8, 139.6,125.8,123.2,122.0,120.3,119.5,118.0,110.0,83.4,73.9,50.2,48.3,45.8, 43.5,38.1,36.9,36.7,36.1,31.3,31.3,29.0,27.7,21.5,20.7,19.4,11.4,8.6。
[0195]
经检测,化合物9的核磁数据为:
[0196]1h-nmr(cdcl3,400mhz):δ8.82(s,1h),8.57(d,j=4.6hz,1h),8.15 (d,j=7.7hz,2h),8.03(d,j=8.4hz,1h),7.98(d,j=5.2hz,1h),7.64(t,j= 8.3hz,1h),7.50(d,j=8.3hz,1h),7.41(t,j=7.5hz,1h),5.32(d,j=4.8hz, 1h),4.77
–
4.64(m,1h),2.49
–
2.39(m,2h),2.38
–
2.32(m,2h),2.09
–
2.01 (m,1h),1.95
–
1.86(m,2h),1.81
–
1.72(m,2h),1.63(d,j=28.3hz,2h), 1.55
–
1.46(m,2h),1.41
–
1.33(m,1h),1.13(s,3h),1.09(s,3h);
13
c-nmr (cdcl3,100mhz):δ(ppm)208.6,160.7,141.5,140.5,139.6,134.4,130.7, 129.3,
128.6,127.0,122.8,122.3,122.1,122.1,114.9,111.7,73.7,50.2,49.8, 48.0,38.1,36.9,36.9,31.5,31.1,30.9,30.3,27.8,20.5,19.5,14.6。
[0197]
经检测,化合物10的核磁数据为:
[0198]1h-nmr(cdcl3,400mhz):δ8.81(s,1h),8.56(s,1h),8.14(d,j=8.1 hz,2h),7.98(d,j=5.0hz,1h),7.64(t,j=8.3hz,1h),7.51(d,j=8.3hz, 1h),7.41(t,j=7.5hz,1h),5.25(d,j=5.2hz,1h),3.59
–
3.44(m,1h),2.48
ꢀ–
2.41(m,2h),2.30
–
2.21(m,1h),2.08
–
2.01(m,1h),1.99
–
1.91(m,1h), 1.89
–
1.72(m,5h),1.63
–
1.57(m,1h),1.53
–
1.44(m,3h),1.40
–
1.31(m, 1h),1.13(s,3h),1.07(s,3h);
13
c-nmr(cdcl3,100mhz):δ(ppm)208.7, 141.4,141.1,140.6,134.4,130.7,129.3,128.6,127.0,122.8,122.3,122.1,121.4, 120.8,113.6,111.7,71.6,50.4,49.9,48.0,42.4,37.3,36.8,31.6,31.2,31.1,30.9, 30.3,20.5,19.6,14.6。
[0199]
(2)采用rsb法测定实施例2~7制备的甾体衍生物(化合物5.1、5.2、 5.3、6、9和10)对人肺腺癌细胞(a549)、子宫颈癌细胞系(hela)、人肝癌细胞(hepg2)三种肿瘤细胞的增殖抑制活性,三种肿瘤细胞购置于誉驰(上海)生物科技有限公司。
[0200]
具体方法为准确称量待测化合物1~3mg,以二甲基亚砜(dmso)为溶剂,配制成浓度为10mmol/l的溶液,室温下静置半小时待样品完全溶解后保存待用;取处于对数生长期的a549、hela、hepg2细胞,清洗并用胰酶消化后制成细胞悬液,细胞计数并稀释至适当浓度,将细胞悬液均匀的加入 96孔板中,每孔100μl,每组设置三个重复,同时设置阴性对照和阳性对照孔。取一定量的事先配置好的待测化合物加入到96孔板中,保证化合物的终浓度为5μm,然后将处理好的细胞置于5%二氧化碳浓度,37℃培养箱中培养72h。培养时间过后,终止培养,移去上清液,每个孔加入冷的10%三氯乙酸(tca)100μl,4℃条件下固定2h,用二次蒸馏水反复冲洗5次, 50℃烘箱中烘干约半小时。待其干燥后,加入冰醋酸配制的磺酰罗丹明b (srb)溶液100μl,室温染色半小时后用1%冰醋酸冲洗5次以除去非特异性结合的染料,此步需要操作迅速以防止已经与蛋白特异性结合的染料分解。50℃烘箱烘干半小时,加150μl三羟甲基氨基甲烷(tris)溶液溶解拍打混匀,酶标仪a540nm测定吸光度,计算其抑制率,用excel计算其ic50,结果见表1。
[0201]
表1部分化合物抗细胞增殖活性ic
50
(μmol/l)
[0202][0203]
表1的结果说明,式i所示β-咔啉基甾体衍生物或式ii所示咔唑基甾体衍生物的抗肿瘤细胞增殖活性,在5μm浓度下对癌细胞的抑制率在85%以上。本实施例挑选化合物5.1、5.2、5.3、6、9和10进行生物活性评价,并以抗癌药物顺铂作为对照药品,咔唑基甾体衍生物以及β-咔啉基甾体衍生物 (化合物5.1、5.2、5.3、6、9和10)对a549(人肺腺癌细胞)、hela(子宫颈癌细胞系)、hepg2(人肝癌细胞)三种肿瘤细胞有很好的细胞毒性,对三种癌细胞的毒性优于顺铂。
[0204]
通过本实施例可以初步推测,本发明式i所示β-咔啉基甾体衍生物和式 ii所示咔唑基甾体衍生物的抗肿瘤细胞增殖活性优于顺铂,具有进一步研究开发的前景。
[0205]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1