使用人多潜能干细胞诱导获得脊髓谷氨酸能中间神经元的方法及其应用
1.本发明涉及多潜能干细胞分化诱导技术领域,特别涉及使用人多潜能干细胞诱导获得脊髓谷氨酸能中间神经元的方法和应用。
背景技术:
2.脊髓损伤(spinal cord injury,sci)层被描述为一种“无法医治”的疾病。随着当代神经生物学研究的不断进步,有望将这种模式从缓解转变为更具治疗性的干预措施。科研工作者们利用动物中的sci模型研究中枢神经系统(central nervous system,cns)的功能,了解到脊髓中具有特征明确的上、下行束和各种形式的已经被明确定义的感觉输入,以及能够产生一系列可量化运动输出的自主环路。sci后的中枢脊髓和周围神经病变已成为基本神经生物学研究的关键,在为sci患者设计更有效、更具针对性的治疗策略中具有重要意义。
3.脊髓在受到损伤后,可一定程度上恢复各种感觉运动功能。完全性sci后的运动恢复取决于内在固有的脊髓环路(cpg)能够按照复杂的顺序激活各种腿部肌肉以产生动作,这个过程是通过cpg与腿部的感觉反馈协同实现。sci后,细胞和环路特性会发生不同变化,可以通过药理、电(生理)或康复策略促进恢复。在部分(或不完全)sci后,后肢运动恢复可能是由其余备用路径(下行束/纤维)的再生或(重新)长出引起的,也可能是由在完全性sci后观察到的内在机制引起的,即脊髓固有环路(cpg)和感觉输入的变化。
4.目前,大量研究工作集中在如何通过促进受损路径的再生,或通过未受损路径的侧支(重新)生长,促进功能性的接收,以重新连接病变的脊髓水平及其上段。在各种临床前研究和临床试验中已经评估了再生性干细胞疗法在靶向sci治疗中的应用。这些研究表明,干细胞具有自我更新和分化为具有不同功能的细胞的能力,如神经细胞能够建立新的突触连接,释放各种神经营养因子,并提供适当的导电微环境,促进受损脊髓的某些修复并改善运动功能。尽管许多实验研究已经评估了干细胞在sci治疗中的应用,但是干细胞治疗仍然远未达到有效的成果,仍然面临着几个重大挑战。例如,脊髓中移植的细胞的存活率非常低,目前的研究报道在脊髓内移植神经前体细胞可以达到相对其他干细胞类型较高的存活率,但尚不清楚这些细胞在sci后的哪个阶段可以达到最佳效果;此外,在针对不同的损伤类型及对应症状时,如何选择确定最适合的干细胞类型,仍然存在争议。
5.在对脊髓运动控制的稳健性和节律性方面,两类兴奋性谷氨酸能spins(脊髓中间神经元,包括v2a spins和v3 spins)起着关键性的作用。关于v2a spins在改善动物sci后运动功能方面的作用已经发表了相关文章,证明sci后v2a spins前体细胞的移植可以提供一种恢复脊髓内在环路神经元之间的功能连接的新疗法,以显著改善运动功能。而作为另一个重要的兴奋性谷氨酸能spins群体,sim1阳性的v3 spins在v1 ia上形成24%的谷氨酸能连接,在renshaw亚类上形成27%,在侧脑运动柱运动神经元上形成22%的谷氨酸能突触,在lhx3阳性的v2 spins及第
ⅷ
板层连合性spins上形成连接。研究表明,在行为学上,通
过破伤风毒素或咽侧体抑制素信号传导导致v3 spins活性丧失后,会导致cpg稳健性的丧失,并显示间歇或永久性跳跃步态。v3 spins还可以平衡脊髓两半之间的运动输出,确保步行过程中运动活动的对称模式。v3 spins通过在脊髓的两半之间分配兴奋性驱动力,建立规则而平衡的运动节律。由此可见,这类神经元在脊髓运动输出中的重要作用。
6.目前,中国授权专利cn109554342b提供了一种诱导性多功能干细胞诱导获得脊髓gaba能中间神经元的方法,采用诱导性多功能干细胞在chir99021、sb431542和dmh1,以及环巴胺和视黄酸等的先后诱导作用下,获得脊髓gaba能中间神经元。但是,尚未发现有人利用多潜能干细胞诱导分化获得v3 spins,而现代医学伦理亦难以从原代的胚胎脊髓中提取p3前体细胞(即v3 spins的前体细胞),这些都使得缺乏直接的来源以研究这类细胞的作用;同时,也尚未有人研究如何使用多能干细胞诱导获得脊髓p3前体细胞,并在体外成熟为v3谷氨酸能中间神经元(sim1
+
,vglut2
+
);也未有人研究在选择合适的sci模型对实验动物造成了运动功能损伤后,移植外源性的脊髓p3前体细胞至损伤脊髓内,能否起到改善作用。
技术实现要素:
7.针对以上现有技术研究的空白,本发明提供了使用人多潜能干细胞诱导获得脊髓谷氨酸能中间神经元的方法及其应用,运用小分子组合法,利用人多潜能干细胞在体外获得富集的v3 spins的前体细胞,即脊髓p3前体细胞,并以其在动物sci模型中的功能验证作为基础,研究人类胚胎对脊髓p3前体细胞在人胚胎发育过程中的存在情况及其发生发育规律。具体通过以下技术实现。
8.使用人多潜能干细胞诱导获得脊髓谷氨酸能中间神经元的方法,包括以下步骤:
9.s1、使用人多潜能干细胞加至含有第一小分子组合物的诱导分化培养基中,诱导分化得到神经上皮细胞;所述第一小分子组合物包括gsk-3抑制剂、tgf-β/smad抑制剂和bmp抑制剂;
10.s2、取步骤s1所得神经上皮细胞加至含有第二小分子组合物的模式化培养基中(实际试验操作时,可以将步骤s1的所有产物直接加入维持模式化培养基中,也可以通过某种技术先分离纯化出神经上皮细胞,再将其加入维持模式化培养基中),诱导分化得到表达nkx2.2
+
/hoxb4
+
的脊髓p3前体细胞;所述第二小分子组合物包括bmp抑制剂、tgf-β/smad抑制剂、rar核受体激活剂、wnt通路抑制剂和sonic hedgehog信号通路激动剂;
11.s3、取步骤s2所得表达nkx2.2
+
/hoxb4
+
的脊髓p3前体细胞加至含有第三小分子组合物的维持模式化培养基中悬浮培养(实际试验操作时,可以将步骤s2的所有产物直接加入维持模式化培养基中,也可以通过某种技术先分离纯化出表达nkx2.2
+
/hoxb4
+
的脊髓p3前体细胞,再将其加入维持模式化培养基中),诱导分化得到表达nkx2.2
+
/neurog3
+
的脊髓p3前体细胞;所述第三小分子组合物包括rar核受体激活剂和sonic hedgehog信号通路激动剂;
12.s4、取步骤s3所得表达nkx2.2
+
/neurog3
+
的脊髓p3前体细胞贴壁培养,进行神经营养使其成熟得到脊髓v3谷氨酸能中间神经元(实际试验操作时,可以将步骤s3的所有产物直接加入神经营养培养基中,也可以通过某种技术先分离纯化出表达nkx2.2
+
/neurog3
+
的脊髓p3前体细胞,再将其加入神经营养培养基中)。
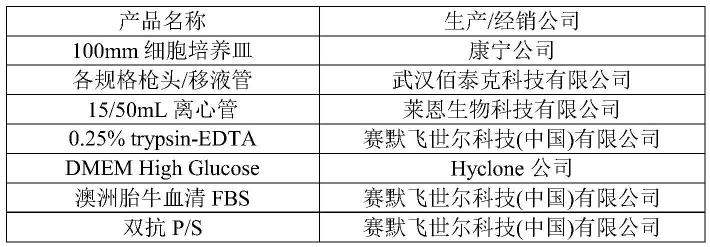
13.优选地,使用人多潜能干细胞诱导获得脊髓谷氨酸能中间神经元的方法中,步骤
s1的所述诱导分化培养基,包括体积比为1:1的dmem/f-12和neurobasal,还包括体积分数1%的neaa、体积分数1%的n-2、3μm的gsk-3抑制剂、2μm的tgf-β/smad抑制剂、2μm的bmp抑制剂混合配制而成。
14.进一步优选地,步骤s1的所述诱导分化培养基中,所述第一小分子组合物的浓度分别为chir99021(gsk-3抑制剂)3μm、sb431542(tgf-β/smad抑制剂)2μm和dmh1(bmp抑制剂)2μm。
15.优选地,步骤s2的所述模式化培养基,包括体积比为1:1的dmem/f-12和neurobasal,还包括体积分数1%的neaa、体积分数1%的n-2、体积分数2%的b27、2μm的bmp抑制剂、2μm的tgf-β/smad抑制剂、0.1μm的rar核受体激活剂、wnt通路抑制剂、1μm的sonic hedgehog信号通路激动剂混合配制而成,所述wnt通路抑制剂使用一种或两种,且每种所述wnt通路抑制剂的浓度为2-2.5μm。
16.更优选地,步骤s2中所述第二小分子组合物包括dmh1、sb431542、ra和sag,还包括iwr-1和/或iwp-2。
17.进一步优选地,步骤s2的所述模式化培养基中,所述第二小分子组合物的浓度分别为dmh1 2μm、sb431542 2μm、ra 0.1μm、sag 1μm,以及iwr-1 2μm和/或iwp-2 2.5μm。
18.优选地,使用人多潜能干细胞诱导获得脊髓谷氨酸能中间神经元的方法中,步骤s3的维持模式化培养基,包括体积比为1:1的dmem/f-12和neurobasal,还包括体积分数1%的neaa、体积分数1%的n-2、体积分数2%的b27、0.1μm的rar核受体激活剂、0.5μm的sonic hedgehog信号通路激动剂混合配制而成。
19.更优选地,步骤s3的所述模式化培养基中,所述第三小分子组合物的浓度分别为ra(rar核受体激活剂)0.1μm,sag(sonic hedgehog信号通路激动剂)0.5μm。
20.优选地,步骤s4具体为:取步骤s3所得表达nkx2.2
+
/neurog3
+
的脊髓p3前体细胞,(经过/不经过accutase消化,以消化7-10min最佳,得到平均每个神经球约含50个细胞的神经球悬液),将神经球悬液接种在matrigel或plo/laminin处理的玻片上贴壁培养1-3周,进行神经营养使其成熟得到表达sim1
+
/vglut2
+
的脊髓v3谷氨酸能中间神经元;
21.神经营养所用的培养基的组分,包括neurobasal,还包括体积分数1%的neaa、体积分数1%的n-2、体积分数2%的b27、1μm aa、1μm camp、10ng/ml bdnf和10ng/ml gdnf。
22.本发明中,第一小分子组合物选择gsk-3抑制剂、tgf-β/smad抑制剂和bmp抑制剂,即分别抑制gsk-3信号通路、tgf-β/smad信号通路和bmp信号通路的物质,例如可以选择chir99021、sb431542、dmh1以及他们的衍生物。本发明中优选为chir99021、sb431542和dmh1,只要能够抑制上述三种通路,则三者的浓度就不用特别限定,例如三者的最佳浓度均可以选择3μm、2μm、2μm,但不限于此。
23.第二小分子组合物选择bmp抑制剂、tgf-β/smad抑制剂、rar核受体激活剂、wnt通路抑制剂和sonic hedgehog信号通路激动剂,例如可以选择dmh1、sb431542、ra和sag,还包括iwp-2和/或iwr-1及其衍生物,只要能够抑制tgf-β/smad信号通路、抑制bmp信号通路、激活rar核受体、抑制wnt通路和激活sonic hedgehog信号通路,其浓度就可以不用特别限定,例如可以选择bmp抑制剂2μm、tgf-β/smad抑制剂2μm、rar核受体激活剂0.1μm,wnt通路抑制剂(iwp-2 2.5μm、iwr-1 2μm),sonic hedgehog信号通路激动剂0.1μm,但不限于此。
24.第三小分子组合物可以选择rar核受体激活剂和sonic hedgehog信号通路激动
剂,例如可以选择ra、sag激起他们的衍生物,本发明优选地选择ra和sag,只要能够激活rar核受体和sonic hedgehog信号通路,ra和sag的浓度就不用特别限定,例如可以最佳选择ra 0.1μm、sag 0.5μm,但不限于此。
25.本发明还提供了一种基于上述方法制备获得的脊髓谷氨酸能中间神经元细胞群。
26.本发明还提供了基于上述方法的,用于诱导获得脊髓谷氨酸能中间神经元的组合物或试剂盒,这种组合物或试剂盒含有上述方法中提供的第一小分子组合物、第二小分子组合物和第三小分子组合物,或者含有上述方法中提到的诱导分化培养基、模式化培养基、维持模式化培养基和神经营养培养基。
27.本发明还提供了一种采用上述方法获得的脊髓谷氨酸能中间神经元细胞群的应用,主要可以用于制备治疗脊髓谷氨酸能中间神经元缺失或失衡,以及因神经系统疾病或损伤引起的运动障碍和/或感觉障碍的药物,或者用于以上疾病的病理学机制研究及药物筛选。
28.与现有技术相比,本发明的有益之处在于:本发明提供了一种利用人多潜能干细胞诱导分化,获得脊髓v3谷氨酸能中间神经元的方法。与以往的方法相比,本发明提供的诱导分化的方法获得的脊髓谷氨酸能中间神经元的纯度更高,对治疗脊髓谷氨酸能中间神经元缺失或失衡,以及因神经系统疾病或损伤引起的运动障碍和/或感觉障碍的药物,或者用于以上疾病的病理学机制研究及药物筛选。
附图说明
29.图1为本发明提供的使用原代hescs细胞诱导分化获得脊髓v3谷氨酸能中间神经元的流程图;
30.图2为各阶段诱导分化获得的细胞的普通光学显微镜图;图2中,(a)原代hescs;(b)神经上皮诱导分化阶段细胞克隆呈现肠型管状上皮形态;(c)脊髓p3前体细胞模式化阶段示意图,细胞克隆呈现玫瑰花簇样;(d)脊髓p3前体维持模式化诱导阶段,呈现透亮的神经球状;(e)脊髓p3前体细胞神经球阶段贴壁生长图;(f)贴壁营养化促进成熟阶段;
31.图3中,(a)原代hescs-mcherry在共聚焦成像系统中自发红色荧光;神经上皮诱导分化阶段细胞表达标志物sox1;(b)两株hescs细胞系中sox1阳性表达率的差异无统计学意义(p>0.05);
32.图4中,(a)neps向脊髓p3前体细胞模式化阶段表达脊髓标志物hoxb4,不表达大脑标志物foxg1;(b)向脊髓p3前体细胞模式化阶段表达特异性标志物nkx2.2;(c)两株hescs细胞系中nkx2.2阳性表达率的差异无统计学意义(p>0.05);
33.图5为脊髓p0-p3前体细胞结构域模式化过程中的转录因子表达情况;
34.图6表示细胞向脊髓p3前体细胞结构域模式化过程中不表达p0-p2前体细胞标志物pax3/7,亦不表达p0-pmn前体细胞标志物neurog2;
35.图7中,(a)分化第14天和第21天单细胞测序热图,表示细胞向p3前体细胞结构域模式化过程中,逐渐从背侧推向腹侧,表达腹侧前体细胞结构域相关标志物;
36.图8表示两株hescs细胞系中nkx2.2或neurog3阳性表达率的差异无统计学意义(p>0.05);
37.图9表示细胞持续向脊髓p3前体细胞模式化过程中,在第21天表达两种特异性标
志物;
38.图10表示神经球贴壁培养成熟,获得脊髓v3谷氨酸能中间神经元。在第42天能够获得的nf200
+
/vglut2
+
(72%)、表达v3特异性标志物sim1(85%)的脊髓v3谷氨酸能中间神经元,不表达v2a的标志物chx10。
具体实施方式
39.下面将对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动条件下所获得的所有其它实施例,都属于本发明保护的范围。
40.以下具体实施方式中提供的使用人多潜能干细胞诱导获得脊髓谷氨酸能中间神经元的方法包括四个步骤:(1)利用原代hescs(人胚胎干细胞)诱导分化获得神经上皮细胞neps;(2)诱导分化获得到表达nkx2.2
+
/hoxb4
+
的脊髓p3前体细胞;(3)诱导分化获得表达nkx2.2
+
或neurog3
+
的脊髓p3前体细胞;(4)贴壁培养成熟,获得脊髓v3谷氨酸能中间神经元。
41.1、材料和装置
42.(1)人胚胎干细胞系来源
43.本发明研究使用的h9人胚胎干细胞系以及h9-mcherry人胚胎干细胞系皆由美国wicell公司(麦迪逊),杜克-新加坡国立大学张素春教授慷慨赠予。实验过程中将使用两株胚胎干细胞系进行对比验证。
44.h9-mcherry人胚胎干细胞系是由特定的遗传修饰技术经由化学修饰得来的,因其使用kord(k-opioid receptor dreadd)插入到在特定的基因位点从而靶标两种不同的受体并且带有一个荧光基团,得到的hm3dq-kord h9-mcherry细胞系可以在体外通过特定的化合物激动或抑制细胞活动,从而达到了双向调控的作用,化合物激动剂为氯氮平甲氧氮芥(clozapine n-oxide cno),以及化合物抑制剂为salvinorin b(salb)。
45.(2)试剂材料及耗材
46.采购的试剂材料和耗材如下表1所示。
47.表1采购的试剂材料和耗材
48.[0049][0050]
自配的试剂材料如下表2所示。
[0051]
表2自配的试剂材料
[0052]
[0053][0054]
所使用的仪器、设备和器械如下表3所示。
[0055]
表3使用的仪器、设备和器械
[0056]
[0057][0058]
所使用的抗体如下表4所示。
[0059]
表4所使用的抗体
[0060]
抗体名称公司及货号种属及稀释比例alexa fluor 488invitrogen/a-21208小鼠/1:1000alexa fluor 594invitrogen/a-21209小鼠/1:1000cy5jackson/128457兔/1:400cy5jackson/129208小鼠/1:400nkx2.2dshb/74.5a5小鼠/1:50pax6biolegend/901301兔/1:500-1000irx3santa cruz/sc-166877小鼠/1:500olig2millipore/ab9610兔/1:500neurog3abcam/ab176124兔/1:500pax3r&d/mab1675鼠/1:500pax7r&d/mab1675鼠/1:500sox1r&d/af3369山羊/1:1000hoxb4millipore/abn174大鼠/1:500neurog2millipore/ab5682兔/1:500foxg1abcam/ab196868兔/1:500nf-200sigma,af5389小鼠/1:1000vglut2synaptic systems,135402兔/1:500map2sigma,m1406小鼠/1:1000sim1origene/ap53918pu-n兔/1:500chx10santa cruz,sc-374151小鼠/1:50
[0061]
2、试验方法
[0062]
图1为本发明提供的细胞分化过程的流程图。每一个阶段分别是7天,经过21天获得脊髓p3前体细胞。
[0063]
图2为各阶段诱导分化获得的细胞的普通光学显微镜图;其中图2-a为原代hescs;图2-b为神经上皮诱导分化阶段细胞克隆呈现肠型管状上皮形态;图2-c为脊髓p3前体细胞模式化阶段示意图,细胞克隆呈现玫瑰花簇样;图2-d为脊髓p3前体维持模式化诱导阶段,呈现透亮的神经球状;图2-e为脊髓p3前体细胞神经球阶段贴壁生长图;图2-f为贴壁营养化促进成熟阶段。
[0064]
(1)饲养层mef的制备
[0065]
①
胎鼠原代细胞提取及冻存
[0066]
将孕期为11.5天的cf-1雌鼠断颈处死后,放入75%酒精中浸泡1h,将其子宫内胚胎取出,在无菌条件下分离并去除器官后留取皮肤组织,每4个胚胎装入一个100mm培养皿中,捣碎并使用每孔3ml 0.05%含edta的胰酶进行消化,在显微镜下观察至80-90%的组织被消化为单个细胞并悬浮于培养基中后,加入每孔皿5ml的mef基础培养基终止消化,并且每个皿按1:3的比例接种于新皿中,待细胞长满底部的80-90%后,吸弃培养基,每个皿中加入2-3ml pbs缓冲液润洗,重复3次;即可加入0.25%的胰酶消化2-3min后,加入等量的培养基按1:4的比例进行传代,记为第一代,即p1。每只冻存管为原先一个大皿内的细胞,将其留取冻存于液氮罐中备用。
[0067]
②
原代mef的复苏及传代
[0068]
第一天,从液氮罐中取出p1的mef细胞,迅速放入37℃恒温水浴锅中轻微摇晃复温,待管内细胞悬液由固态完全转化为液态后,将融化后的液体加入事先在15ml离心管中准备好的mef培养基中(所有的细胞培养基需提前15-20min放入37℃恒温水浴锅中预热),离心后吸弃上清,按原先每只冻存管细胞1:2的比例接种至100mm细胞培养皿中,记为p2。
[0069]
每个培养皿中含7-8ml细胞悬液,十字摇匀后在培养皿表面标记后放入37℃恒温co2培养箱中培养;等待至第三天,在镜下观察到皿底细胞长至80-90%的融合度,即可进行传代,按每只皿1:4的比例传代至新皿中,记为p3,步骤同上,连续培养三天。
[0070]
③
细胞绝育处理及计数冻存
[0071]
第六天,将上一步中扩增传代的p3 mef收集并进行绝育照射,每个大皿中的培养基吸弃后,加入pbs缓冲液润洗3次;加入2-3ml胰酶进行消化,终止消化后将细胞悬液收集到50ml离心管中,并放入4℃环境中储存,将所有细胞收集完毕后,使用冰盒携带至放射中心,对mef细胞进行绝育照射以作为饲养层使用。
[0072]
本实验过程中的细胞x射线辐照设备由华中科技大学同济医学院附属协和医院肿瘤中心提供。照射结束后尽快将悬液离心,并用冻存液重悬以供冻存,按之前每皿p3冻存一管,每管约含有1.5
×
106的细胞,供后续复苏使用。由于作为饲养层使用时,每个6孔板中按9.5
×
104个/ml,每孔2.5ml种入细胞悬液使用,故在冻存时可按不同比例进行冻存,方便取用。
[0073]
(2)原代hescs的复苏及维持培养
[0074]
提前在6孔板中加入每孔1ml 0.1%明胶,放入培养箱中待1h后吸弃,将饲养层mef按每孔2.25
×
106的细胞量加入孔板中,摇匀后放入培养箱,待8h后方可使用。将一支装有原代hescs的冻存管从液氮罐中取出后迅速复温,离心后加入5ml维持培养基后,并在每ml培养基中加入1μl的rock inhibitor,复苏至2个孔中,摇匀后放入培养箱,待第二天观察复苏的细胞是否贴在饲养层上生长,并将培养基吸弃换新。直至细胞克隆长出,并长至80-90%融合的程度,即可进行传代,注意须得在传代的前一天将饲养层mef进行铺板,以供隔天使用。传代时使用dispace ii胶原酶进行消化后,可在镜下观察至克隆边缘卷起,用dmem/f-12每孔2ml润洗两次后,加入每孔2ml培养基并使用200μl枪头沿克隆边缘吹打,将所需克隆都吹打至培养基后,吸取细胞悬液,按原先每孔1:6的传代比例种至预先铺好的饲养层mef上,以此方法对原代hescs维持培养。
[0075]
(2)neps的诱导分化培养
[0076]
将传代第2-3天的原代hescs吸弃培养基,换为每孔3ml的neps的诱导分化培养基,该阶段的细胞培养依旧在饲养层上进行,并且2天换一次细胞培养液即可。约在更换培养液后的第4-5天,可以在镜下观察到,细胞克隆的边缘出现肠型管状上皮样结构,细胞克隆增厚,透光性降低,约在第7天,可观察到孔板内90%以上的细胞克隆呈现管状上皮样结构,即得到表达sox1
+
的高纯度neps,可进入到下一阶段的模式化诱导阶段。与原代hescs的复苏及维持培养同样地,在传代前一天提前将mef铺板备用。
[0077]
(3)脊髓p3前体细胞的模式化诱导分化
[0078]
将上一阶段中诱导的neps以每孔加入2ml dmem/f-12润洗2次,加入每孔2ml的第二阶段推向脊髓p3前体细胞模式化阶段的模式化培养基,润洗和加液过程中可以发现,分化诱导7天后的neps呈现较为松散的状态,可以使用枪头吹打至培养基中,无需加入酶进行消化剥离,吹打后将细胞悬液收集至离心管后,进行离心,并按每孔1:8的比例接种于预先准备的饲养层细胞上。摇匀后放入细胞培养箱中,每两天进行换液,在换液后的第4-5天可以观察到细胞克隆呈现玫瑰花蔟样的形态特征,细胞边缘较为透亮,并且克隆中的单个细胞呈现梭形,在第6-7天,可以观察到约80%的细胞克隆呈现特异性的玫瑰花蔟样形态,且细胞克隆中有细胞脱落,呈现较为松散的状态,细胞梭形形态更加明显,直至换液后的第7天,脊髓p3前体细胞的模式化初始阶段已经完成,初步获得表达nkx2.2
+
/hoxb4
+
的脊髓p3前体细胞。下一步将维持其模式化的过程,以获得纯度更高的该类型细胞。
[0079]
(4)脊髓p3前体细胞的模式化维持诱导
[0080]
在这一阶段中,持续将上一阶段中得到的细胞维持诱导,推进其往脊髓p3前体细胞更多的分化。
[0081]
在每个t25细胞培养瓶中加入5ml维持模式化培养基,并平放入细胞培养箱中进行预热;将第二阶段的细胞在孔板中使用dmem/f-12润洗2次,步骤同前,加入第三阶段培养基并将孔板底部的细胞吹散至培养液中,放入离心机中离心后,以原先没个孔种入一个培养的密度进行接种,摇匀后放入细胞培养箱中维持培养,并且2天进行一次换液,每天进行吹打以防止细胞克隆过大影响生长。
[0082]
(5)脊髓v3谷氨酸能中间神经元的培养制备
[0083]
在进行神经球贴壁之前,取若干24孔板,在24孔板的每孔铺一无菌玻片,在每一玻片上加80μl稀释好的matrigel,使matrigel刚好贴满玻片而不溢出,将24孔板放入培养箱中;配制第四阶段培养基,以50ml为例,在50ml离心管中加入50ml neurobasal,1ml b27,0.5ml neaa,0.5ml n-2,200μm aa,1μm camp,10ng/ml bdnf,10ng/ml gdnf。24孔板放入培养箱1小时后,每孔吸弃50μl matrigel,将t25瓶中获得的神经球,使用accutase消化7-10min,制备为神经球悬液接种到铺有matrigel的玻片上,均匀铺开,使每一玻片上约有20个神经球,一个神经球约含50个细胞;将24孔板放入37℃,5%co2的恒温细胞培养箱培养2h,待神经球贴附到玻片上,每孔添加神经营养培养基至500μl,放入培养箱,培养21天,隔天换液,用200μl枪头轻柔操作,每天观察神经元生长情况。
[0084]
以上所有的细胞培养皆在同济医院干细胞研究中心万级层流室中以严格无菌条件下操作,该过程中的操作步骤都在生物安全柜中完成。
[0085]
(6)单细胞测序
[0086]
实验中的单细胞测序技术由华中农业大学生物医学中心曹罡教授团队提供。在培养的第14天及21天,使用accutase将细胞解离为单个细胞。在chromium控制器中通过液滴封装,使用chromium single cell 3'v1文库和gel bead kit(10
×
genomics)进行文库制备,准备约8000个细胞用于单细胞分析;covaris s2超声仪剪切cdna,并在扩增过程中进行12个pcr循环;使用10
×
genomics cellranger v 1.2的默认设置,对序列进行多路分解并与人类参考基因组进行比对;经过cellranger过滤,剩余的>8500万条有效读数中有>70%映射到转录组;使用seurat进行下游分析并去除不表达500至5000个独特基因的细胞;使用seurat的“meanvarplot”功能(表达截止值≥0.25;分散截止值≥0.50)确定了高变异基因的子集,使用该参数将细胞分组为簇(簇分辨率参数=0.5);在热图中绘制每个簇的差异表达基因;最终使用panther和gorilla对具有统计学意义的差异表达基因(p≤0.05)进行基因本体分析。
[0087]
(7)细胞爬片处理及免疫荧光染色
[0088]
①
细胞爬片处理及固定
[0089]
将24孔板从培养箱中取出,在生物安全柜中,使用预热的pbs,以每孔300μl润洗3次,加入每孔200μl的通用型组织固定液固定30min后,再使用pbs润洗3次,之后可进行免疫荧光染色处理或将爬片暂时存放于4℃冰箱中,7天之内可用。
[0090]
②
免疫荧光染色
[0091]
细胞爬片使用的免疫荧光染色步骤,同第一部分中,贴片染色的步骤。这部分不再进行详细描述。注意最后不使用盖玻片而是将圆形玻片直接向下盖在滴加了抗荧光淬灭封片剂的载玻片上即可。
[0092]
③
荧光显微镜及共聚焦成像系统下的图像采集
[0093]
将片子置于普通免疫荧光显微镜下查看染色结果,在镜下挑选合适的片子,寻找并选取合适视野后,在共聚焦成像系统中进行后续图像采集工作,实验中出现的结果均已重复3次独立实验,并且每次采集须得有3张以上片子,取单张片子3个以上视野进行结果分析,出现可重复结果为宜,采集后使用image j/fiji进行图像处理,标尺的添加,统一格式设置等操作。
[0094]
每个结果均已重复3次独立实验,且每次采集图像时,至少每个染色结果是由3个以上样本,经由单个样本取3-5个克隆比对得出。
[0095]
3、结果分析
[0096]
如图3所示,为验证该过程中得到的细胞是否遵循向neps的神经诱导的规律,使用neps特异性标志物sox1进行免疫荧光染色标记(图3-a),与此同时使用h9 hescs细胞系与h9 mcherry(图3-a)细胞系进行比对。计算三次重复独立实验后得到的结果,在h9 hescs株系中sox1阳性率为93.4
±
4.1%,h9 mcherry hescs株系中sox1阳性表达率为91.7
±
3.7%,两株细胞系中sox1阳性表达率差异没有统计学意义(p>0.05)(图3-b)。
[0097]
如图4所示,在脊髓p3前体细胞的模式化诱导分化这一阶段,是通过模拟体内neps的腹侧化过程来推向脊髓p3前体细胞的形成,将neps暴露于ra(0.1mm)和shh高效激动剂sag(1μm),wnt信号通路阻滞剂iwp-2(2.5μm)及iwr-1(2μm)培养7天后。可以初步获得表达脊髓特异性标志物hoxb4,但不表达大脑标志物foxg1(图4-a),表达脊髓p3前体特异性tf nkx2.2,且不表达pmns特异性标志物olig2的脊髓p3前体细胞(图4-b)。如图4-c所示,同样
使用两株细胞系进行比较,在h9 hescs株系中nkx2.2阳性率为48.7
±
5.2%,h9 mcherry hescs株系中sox1阳性表达率为44.6
±
7.3%,两株细胞系中该标志物的阳性表达率差异没有统计学意义(p>0.05)。
[0098]
为验证在这一阶段中,我们使用的小分子组合可以使得neps继续向p3前体细胞结构域模式化,如图5所示,p3前体细胞在其有丝分裂阶段表达特异性标志物nkx2.2,以及neurog3。我们此前已经观察到这一时期存在nkx2.2的表达情况,但未见neurog3表达,将在下一阶段中进行检测。初步获得的细胞不表达脊髓腹侧p0-p2前体细胞结构域的特异性标志物pax3,亦不表达p0-pmn的标志物neurog2(图6)。
[0099]
将模式化诱导分化后得到的脊髓p3前体细胞,以及模式化维持诱导分化后获得的脊髓p3前体细胞进行单细胞测序。收集第14天及第21天获得的细胞进行重复3次测序,结果如图7,第14天得到的细胞仍存在表达脊髓背侧的基因表达,而第21天获得的细胞表达腹侧基因,各阶段样本之间重复性较佳。利用如前所述的两株稳定细胞系,对这一时期获得的细胞进行免疫荧光染色,观察到在h9 hescs株系中nkx2.2及neurog3阳性率为64.5
±
3.1%,h9 mcherry hescs株系中阳性表达率为62.3
±
3.7%,两株细胞系中的阳性表达率差异没有统计学意义(p>0.05)(图8,9)。
[0100]
由此可见,在培养过程中,由于前期预实验中使用了不同浓度的sag进行诱导分化,可以推断sag能够提高诱导nkx2.2表达所需的shh信号阈值。然而,随着浓度的增加,细胞的透光性变差,且死亡率增高,可能是由于shh高效激动剂sag会在对neps诱导的过程中产生了细胞毒性。为降低sag对细胞的伤害性,且同时提高分化率,本发明通过抑制脊髓的背侧分子bmp/wnt信号通路,降低shh信号的阈值。双抑制剂sb和dmh1联合wnt通路抑制剂iwp-2及iwr-1的应用使得在第二阶段逐渐获得了表达阳性的nkx2.2细胞群体,且在第三阶段的维持诱导过程中,使用较低浓度的sag依旧可以将上一阶段细胞向腹侧化推进。因此,可以认为,协调具体小分子以形成不同组合对神经上皮的形成和模式化可以影响区域特异性脊髓p3前体细胞的生成。
[0101]
与以往的方法相比,我们获得了更高纯度的细胞,分化过程整体遵循神经发育过程中脊髓的形成及模式化特征。单细胞测序的分析方法使我们能够在分化的第14天及第21天对细胞群体进行全面的分析。k均值聚类显示nkx2.2及neurog2细胞主要包含在一个簇内,这表示富集的p3前体细胞特征。且在分化的过程中,逐渐向腹侧化推进,到分化的第21天,明显表达腹侧细胞群体身份的特异性标志物。
[0102]
随着第三阶段的神经球贴壁培养,并继续神经营养支持,脊髓v3中间神经元的继续成熟,可以表达谷氨酸能神经元标志物vglut2,在第42天能够获得的nf200
+
/vglut2
+
(72%)、表达v3特异性标志物sim1(85%)的脊髓v3谷氨酸能中间神经元,不表达v2a的标志物chx10(图10)。
[0103]
以上具体实施方式详细描述了本发明的实施,但是,本发明并不限于上述实施方式中的具体细节。在本发明的权利要求书和技术构思范围内,可以对本发明的技术方案进行多种简单改型和改变,这些简单变型均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1