一种丙戊酸钠、丙戊酸共晶的制备方法及其产品与流程
1.本发明涉及丙戊酸及其盐类的制备领域,特别涉及一种丙戊酸钠、丙戊酸共晶的制备方法及其产品。
背景技术:
2.丙戊酸(2-丙基戊酸)及其盐类是脂肪酸衍生物药物,适应症主要为:癫痫、躁狂症,美国fda批准的适应症还包括双相性情感障碍和偏头痛,为窄治疗指数(nti)药物,丙戊酸类药物报道的有效治疗血药浓度为40-100μg/ml,且成人需以达1200mg的较高日剂量服药。而且由于其较短的生物半衰期,该药物更适宜制成口服缓释制剂。
3.由于丙戊酸在常温下为油状液体,在水中溶解度不高,除了制成软胶囊外,难以单独使用制成其他口服制剂产品,因此通常被制成钠盐或镁盐在口服制剂中应用。但丙戊酸钠具有极强的引湿性,且水溶性极好,若在相对湿度53%的环境中,放置过夜,该样品就会完全溶解水化在自身吸收的水分中,吸收水分高达43%。丙戊酸钠被报道有多种晶型,但无一例外,所有丙戊酸钠晶型均有很强的引湿性,导致其原料药生产和固体制剂开发难度均较大,原料药生产非常容易吸湿结块,而用于制剂采用常规制粒也非常难以干燥,且压片时操作环境若不能维持在低湿度(例如:相对湿度低于30%rh)则极易粘冲。
4.丙戊酸的不同盐在引湿性上具有非常大的差异,公知丙戊酸镁和双丙戊酸钠(又被称为:二丙戊酸钠、丙戊酸半钠、二丙戊酸氢钠等)的引湿性则相对较低,但其在制备过程中依然存在以下的问题:
5.1、目前,诸如专利文献us4988731、us5212326、ca1144558a、cn102942467a和 cn103183600b以及其他国内文献中公开的制备方法均为使用丙酮、乙腈、乙醇、乙二醇、异丙醇、正丙醇、丙三醇、正丁醇、戊醇、乙酸乙酯等有机溶剂进行结晶,而通过有机溶剂进行结晶的方法,一方面会引入新的杂质,另一方面会增大安全风险和产生潜在环境污染;
6.2、高温环境进行丙戊酸钠的溶解会导致物料变黄,影响后续的药品制剂用途,如美国专利us6077542公开了一种丙戊酸和丙戊酸钠固体混合物的制备方法:通过将丙戊酸钠溶解在热丙戊酸中得到溶液,混合物溶液凝固后熔点会显著升高,且引湿性显著减弱。其将丙戊酸钠80g溶解在热丙戊酸100g中得到溶液,凝固后熔点为73℃;将丙戊酸钠100g在165℃溶解于热丙戊酸100g中得到溶液,凝固后熔点≈100℃;然而其声称能在150℃时将丙戊酸钠115.2g完全溶解在165℃加热的100g丙戊酸中得到澄清溶液(此时丙戊酸钠和丙戊酸的摩尔比=1:1),但由于经历了较长时间的高温,此时物料已出现比较明显的变黄现象,影响后续应用;
技术实现要素:
7.本发明要解决的技术问题是提供一种丙戊酸钠、丙戊酸共晶的制备方法,该方法无需在有机溶剂中作进一步精制,符合药用标准,工序中物料暴露时间短,无需对环境温度进行严格控制,方法操作简单,安全环保,易于工业化生产,同时生产成本低廉;同时还提供
了由该方法所获得的产品,引湿性低,纯度高,无变黄现象,有利于后续的药品制剂应用。
8.为了解决上述技术问题,本发明的技术方案为一种丙戊酸钠、丙戊酸共晶的制备方法,包括如下步骤:
9.s1将氢氧化钠、碳酸钠、碳酸氢钠中至少一种与丙戊酸进行混合,在50~140℃条件下形成水溶液,其中氢氧化钠、碳酸钠、碳酸氢钠或其混合物总的摩尔量为丙戊酸摩尔量的0.4-0.8,当向丙戊酸中加入氢氧化钠、碳酸钠或碳酸氢钠其中之一或混合物时,既可以将干物质和水分别加入,也可以加入它们的水溶液,以最终达成在加热状态下部分中和以及充分溶解的目的;
10.s2将s1步骤中形成的水溶液蒸去水分,然后降温冷凝结晶;
11.s3将s2步骤中结晶体粉碎成颗粒或粉末形式,得到丙戊酸钠、丙戊酸共晶;
12.优选地,氢氧化钠、碳酸钠、碳酸氢钠或其混合物总的摩尔量为丙戊酸摩尔量的2/3或 1/2。
13.优选地,s1步骤水溶液中水的重量为丙戊酸重量的8%~35%;进一步优选地,s1步骤水溶液中水的重量为丙戊酸重量的12-20%。
14.加水量不低于丙戊酸总重量的8%,才能充分将中和后的丙戊酸钠和丙戊酸充分溶解澄清,稍多加水(35%以内)则能使物料完全溶解时间加快,虽水量增加会延长后续水分蒸发时间但缩短了溶清时间,综合考虑生产效率和能耗是平衡的。但若超出上限加更多水则只能单纯延长蒸水时间并增加能耗,变得毫无意义。
15.优选地,s1步骤中采用氢氧化钠与丙戊酸进行混合。
16.优选地,s1步骤采用的温度优选80~120℃,进一步优选86~96℃;s2步骤中蒸发水分温度优选50~140℃,更优选80~120℃,最优选86~96℃;在该种温度条件下,维持搅拌干燥至水分≤1.5%时,可直接析出共晶粉末以及团块,并且析晶粉末和团块形成空隙更利于水分蒸发,使得最终干燥效率更高,而共晶产物再经过粉碎即可用于后续制剂用途。通常物料在105℃以上时呈澄清或微浊粘稠液体状态,但蒸发水分时的温度优选不超过100℃,可避免丙戊酸在高温下潜在的氧化分解和变色,以及高温蒸发损失丙戊酸等问题。更高的操作温度除了蒸发速度较快的优势以外,并不具有其他益处,反而增加能耗,也增加丙戊酸在高温下潜在的氧化分解、变色以及高温蒸发损失风险,并且超过120℃后,不可避免地会出现颜色加深情况,温度越高,颜色越深。当操作温度为140℃时,产物已呈类白色或微黄色粉末,触及可用于后续药品制剂的最低标准了。
17.优选地,s2步骤中水溶液蒸去水分后形成浓缩料液,可以将浓缩料液置于平板或托盘上自然冷却结晶,也可以将浓缩料液置于蜡质的商业化造粒机制粒,如回转带式冷凝造粒机或喷雾冷凝造粒机等制粒,并可以根据需要进一步粉碎成适宜药品制剂的粒度。
18.在浓缩干燥时,应采用密闭容器(微负压或减压)或局部单向流排风的设计,对蒸干过程产生的含有voc(挥发性有机碳)的废气进行冷凝捕集并单独处理,以预防蒸汽可能夹带的voc对操作人员的可能伤害或污染周界。
19.优选地,丙戊酸、氢氧化钠、碳酸钠或碳酸氢钠均符合各国药典标准或行业标准,适合药用的规格,投料时,对氢氧化钠、碳酸钠或碳酸氢钠带入的水分和可能存在的碳酸盐和结晶水进行折纯、折干计算。
20.优选地,水符合各国药典标准,适合药用的纯化水规格。
21.所制得的共晶产物中,丙戊酸钠与丙戊酸共晶的摩尔比为1.8~2.2:1或0.9~1.1:1。
22.进一步优选地,通过上述的方法制得的丙戊酸钠、丙戊酸共晶,具有以下两者之一的结构式:
[0023][0024]
优选地,丙戊酸钠、丙戊酸共晶中水分≤0.5%;进一步优选地,丙戊酸钠、丙戊酸共晶中水分≤0.2%。
[0025]
当工艺浓缩干燥的产物实现水分《0.5%后,可认为产物得到充分干燥,应通过对产物的工艺过程中进行在线或离线水分检测控制;检测方法可以采用业界公知的技术,如取样用红外快速水分测定仪测定,也可以采用卡氏水分测定仪测定,也可以安装在线的水分检测仪器(如近红外仪)测定。若未能充分干燥导致产物中水分》0.5%时,将导致产物不易析晶,以及制剂应用时稍有吸湿便易结块的问题。
[0026]
根据公知常识,碳酸盐被丙戊酸根取代后,二氧化碳将受热逸出,因此反应结束并浓缩干燥的产物脱水(水分≤0.5%)后也不会残留有碳酸根,自然也不会影响产物的纯度。
[0027]
所制得的丙戊酸钠、丙戊酸共晶还具有以下两者之一的特征:
[0028]
可用于生产赛诺菲(sanofi)上市的depakine500mg(丙戊酸钠缓释片)仿制药,每478mg所述丙戊酸钠、丙戊酸共晶中含丙戊酸钠333mg、丙戊酸145mg,换算成摩尔比,丙戊酸钠:丙戊酸=2:1。
[0029]
或可用于生产雅培(abbott)上市的er(双丙戊酸钠缓释片)仿制药,每310.4mg所述丙戊酸钠、丙戊酸共晶(双丙戊酸钠)中含丙戊酸钠166.2mg、丙戊酸144.2mg,换算成摩尔比,丙戊酸钠:丙戊酸=1:1。
[0030]
采用上述技术方案,由于直接将氢氧化钠、碳酸钠、碳酸氢钠中至少一种与丙戊酸进行混合,并采用水形成水溶液,避免了现在技术中采用有机溶剂进行进一步精制,同时本发明的制备方法,物料在工序中暴露时间短,无需对环境湿度进行严格控制,方法操作简单,安全环保,易于工业化生产,同时生产成本低廉;
[0031]
通过本发明的制备方法制得的丙戊酸钠、丙戊酸共晶引湿性低,经测试,2小时引湿增重0-1.84%,6小时引湿增重0-3.58%,结晶粉末均为白色,无变黄现象,有利于后续的药品制剂应用。
附图说明
[0032]
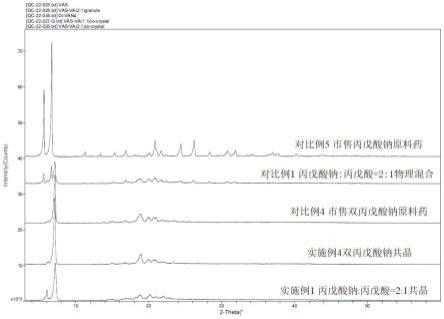
图1为实施例和对比例pxrd叠加对比图。
[0033]
图2为实施例和对比例dsc叠加对比图。
具体实施方式
[0034]
下面结合附图对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
[0035]
丙戊酸钠、丙戊酸共晶的制备方法:以下实施例均使用热夹套的w形捏合机进行加热混合搅拌并脱去水分,除部分实验直接取干燥后高温料液直接趁热浇筑冷凝形成2~5mm厚度的白色半透明均质圆饼外,其余工艺均采用蒸干水分后,逐渐降低夹套温度,料液随着温度下降会逐渐冷凝结晶,在维持搅拌情况下通常会得到白色粉末和团块的混合物,降温至室温后,使用锤式粉碎机将冷凝硬化过程中较大团块产物进一步粉碎,粉碎后颗粒或粉末的干燥失重(快速水分干燥仪)或水分(卡氏水分滴定仪)不大于0.2%,筛网可以是20~80目。
[0036]
上述统一制备方法只是保证所得样品为同一标准下制备得到的,但制备方法并不仅限于这一种,具体的操作可以根据实际情况进行调整,只要条件参数符合本发明的技术方案即可。
[0037]
本发明制得产物的质量(尤其是有关物质)评估,均采用业界公知的各国药典(中国药典、欧洲药典、美国药典)现行版丙戊酸、丙戊酸钠、双丙戊酸钠专论项下检验方法和标准,此处不再赘述。
[0038]
实施例1
[0039]
将丙戊酸866.6g加氢氧化钠160g,纯化水303g(相当于丙戊酸重量的35%),50℃搅拌 1小时完全溶清后,逐渐升温至94℃常压下蒸去水分至卡式水分滴定仪水分检测为0.15%,期间料液逐渐增稠并冷凝结晶,从无色澄清液体逐步转变为类白色半透明粘稠蜡质状,控制夹套降温,搅拌过程中物料逐渐冷凝结晶和硬化,形成白色粉末和团块的混合物;
[0040]
短时间暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,2小时引湿增重1.50%,6小时引湿增重2.96%。
[0041]
本实施例所得的共晶中,丙戊酸钠:丙戊酸=2:1(摩尔比),符合结构式1的结构,熔程为92.2~93.6℃(熔程相对较宽,说明可能同时存在两种或以上共晶混合物),和所用丙戊酸原料药比较,对有关物质进行检测,未发现单个杂质量和种类有变化。
[0042]
对比检测pxrd,从附图1可观察到市售丙戊酸钠原料药的pxrd图谱和本发明方法制得丙戊酸钠:丙戊酸=2:1共晶有显著差异。
[0043]
实施例2
[0044]
将丙戊酸439.86g加无水碳酸钠103.93g,纯化水130g(相当于丙戊酸重量的30%),80℃搅拌(期间释放大量二氧化碳气泡)1小时完全溶清后,升温至94℃常压下蒸去水分至卡式水分滴定仪水分检测为0.13%,期间料液逐渐增稠,从无色澄清液体逐步转变为类白色半透明粘稠蜡质状,控制夹套降温,搅拌过程中物料逐渐冷凝结晶和硬化,形成白
色粉末和团块的混合物。
[0045]
短时间暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,2小时引湿增重仅1.34%,6小时引湿增重仅2.84%。
[0046]
本实施例所得的共晶中,丙戊酸钠:丙戊酸=1.8:1(摩尔比),熔程为:92.1~93.3℃(熔程相对较宽,说明可能同时存在两种或以上共晶混合物),和所用丙戊酸原料药比较,对有关物质进行检测,未发现单个杂质量和种类有变化。
[0047]
实施例3
[0048]
将丙戊酸456.10g加氢氧化钠86.96g,纯化水160g(相当于丙戊酸重量的35%),80℃搅拌1小时完全溶清后,升温至94℃常压蒸去水分至卡式水分滴定仪水分检测为0.20%,物料此时呈类白色半透明粘稠蜡质状,夹套降温,搅拌过程中物料逐渐冷凝结晶和硬化,形成白色粉末和团块的混合物。
[0049]
短时间暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,2小时后引湿增重1.84%,6小时后引湿增重3.58%。
[0050]
本实施例所得的共晶中,丙戊酸钠:丙戊酸=2.2:1(摩尔比),熔程为:91.0~92.5℃(熔程相对较宽,说明可能同时存在两种或以上共晶混合物),和所用丙戊酸原料药比较,对有关物质进行检测,未发现单个杂质量和种类有变化。
[0051]
实施例4
[0052]
将丙戊酸865.8g加氢氧化钠120.58g,纯化水100g(相当于丙戊酸重量的11.6%),94℃搅拌1小时完全溶清后,维持94℃常压蒸去水分至卡氏水分滴定仪检测水分0.12%,搅拌过程中物料逐渐结晶,形成白色结晶针状松散粉末。
[0053]
暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,表面既未软化更不会水化,2~24小时均未发现引湿增重,引湿性极低。
[0054]
本实施例所得的共晶中,丙戊酸钠:丙戊酸=1:1(摩尔比),符合结构式2的结构,熔程为:98.0~98.4℃(狭窄的熔程说明单一共晶),本实施例制得的共晶,检验完全符合双丙戊酸钠的美国药典标准,对比所用丙戊酸原料药,对有关物质进行检验,未发现单个杂质量和种类有变化。
[0055]
对比检测pxrd和dsc,从附图1和附图2可观察到市售双丙戊酸钠原料药的pxrd和dsc 图谱和本发明方法制得丙戊酸钠:丙戊酸=1:1(摩尔比)共晶(双丙戊酸钠)没有显著差异。
[0056]
实施例5
[0057]
将丙戊酸432.57g加碳酸氢钠119.36g,纯化水70g(相当于丙戊酸重量的16.2%),80℃搅拌(期间释放大量二氧化碳气泡)1小时完全溶清后,升温至140℃常压蒸去水分至卡氏水分滴定仪检测水分0.09%,降温至85℃,搅拌过程中物料逐渐结晶,形成白色结晶针状油性粉末。
[0058]
短时间暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,2小时后引湿增重仅0.21%,6小时后引湿增重仅0.23%。
[0059]
本实施例所得的共晶中,丙戊酸钠:丙戊酸=0.9:1(摩尔比),熔程为:90.0℃~96.5℃(熔程相对最宽,说明可能同时存在两种或以上共晶),和所用丙戊酸原料药比较,对有关物质进行检测,未发现单个杂质量和种类有变化。
[0060]
实施例6
[0061]
将丙戊酸432.2g加氢氧化钠62.78g,纯化水34.6g(相当于丙戊酸重量的8%),80℃搅拌2小时完全溶清后,升温至94℃常压蒸去水分至卡氏水分滴定仪检测水分0.09%,物料此时呈微黄透明粘稠熔蜡状,夹套降温,搅拌过程中物料逐渐冷凝结晶和硬化,形成白色结晶针状松散粉末。
[0062]
短时间暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,2小时后引湿增重0.91%,6小时后引湿增重1.68%。
[0063]
本实施例所得的共晶中,丙戊酸钠:丙戊酸=1.1:1(摩尔比),熔程为:93.5~97.0℃(熔程相对较宽,说明可能同时存在两种或以上共晶混合物),和所用丙戊酸原料药比较,对有关物质进行检测,未发现单个杂质量和种类有变化。
[0064]
对比例1
[0065]
按照us5017613方法:将油状的丙戊酸145g常温下喷雾加入丙戊酸钠333g中湿法制粒,能形成大小不一颗粒和粉末,但肉眼可见并非均质状态。
[0066]
将颗粒暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,2小时后引湿增重1.76%,6小时后引湿增重3.66%。
[0067]
对比检测pxrd和dsc,从附图1和附图2可观察到丙戊酸钠:丙戊酸=2:1(摩尔比)物理混合物的pxrd和dsc图谱和本发明方法制得丙戊酸钠:丙戊酸=2:1(摩尔比)共晶有显著差异。
[0068]
对比例2
[0069]
按照us6077542方法:将丙戊酸钠333g部分溶解在180℃加热145g丙戊酸中,搅拌2 小时,只能得到部分溶解的淡黄白色粘稠不透明混悬溶液,冷凝硬化的混溶凝固物呈现部分丙戊酸钠结晶析出的非均质固体分散体状态,且颜色因为高温氧化的原因泛微黄。得到的固体混合物,冷凝后进一步粉碎成20-60目微黄色粉末。
[0070]
暴露在常规环境条件(25
±
2℃/60
±
5%rh)中,2小时后引湿增重1.68%,6小时后引湿增重2.89%。
[0071]
对比例3
[0072]
按照us6077542方法:将丙戊酸钠115.2g溶解在165℃加热100g丙戊酸中,搅拌2小时,只能得到溶解的浅黄色粘稠溶液,颜色因为长时间高温的原因从无色变为泛黄。得到的固体混合物,冷凝后进一步粉碎成20-60目类白色粉末,接近双丙戊酸钠的性能,暴露在在常规环境条件(25
±
2℃/60
±
5%rh)中2-24小时后均未观察到引湿增重。
[0073]
对比例4
[0074]
取中国台湾旭富生产商业化上市销售的双丙戊酸钠作为对比例4,除检测引湿性外,对比检测pxrd和dsc,从附图1和附图2可观察到市售双丙戊酸钠原料药的pxrd和dsc图谱和本发明方法制得丙戊酸钠:丙戊酸=1:1(摩尔比)共晶(双丙戊酸钠)没有显著差异。
[0075]
对比例5
[0076]
取印度太阳药业生产商业化上市销售的丙戊酸钠原料药作为对比例5,除检测引湿性外,对比检测pxrd,从附图1可以看出市售丙戊酸钠原料药的pxrd和本发明方法制得共晶有显著差异。
[0077]
将不同的丙戊酸钠与丙戊酸共晶摩尔比的实施例和对比例进行归类对比,具体如
下:
[0078][0079][0080]
综上所述,丙戊酸钠:丙戊酸=1:1(摩尔比)共晶,暴露在常规环境条件(25
±
2℃/60
ꢀ±
5%rh)下吸湿性极低或不具有引湿性,和商业化上市销售的双丙戊酸钠原料药防潮性能基本等效,制备方法更简单,成本更低,也最适合后续制剂使用;而其他配比,随着共晶中丙戊酸钠比例的增加,引湿性也会逐渐增强,但相比采用us5017613方法简单物理混合制得混合物,引湿性被削弱。
[0081]
以上结合附图对本发明的实施方式作了详细说明,但本发明不限于所描述的实施方式。对于本领域的技术人员而言,在不脱离本发明原理和精神的情况下,对这些实施方式进行多种变化、修改、替换和变型,仍落入本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1