一种环丙基甲基酮的制备方法与流程
1.本发明属于有机合成技术领域,具体地说,涉及一种环丙基甲基酮的制备方法。
背景技术:
2.环丙基甲基酮是具有刺激性气味的无色易挥发液体,部分溶于水,可与水形成共沸物,还可与醇类、醚类、酰胺类、烷烃类有机溶剂互溶。现有技术中,环丙基甲基酮主要作为一种重要的有机合成原料。例如,在农药方面主要用于合成抗真菌剂环唑醇与嘧菌环胺,在医药方面则用于制备抗艾滋病病毒药物依法韦伦。此外,环丙基甲基酮具有张力环结构,可用于制备一系列高能化合物,在含能材料领域也备受重视。
3.目前,国内环丙基甲基酮工业品的制备,主要采用糠醛为原料,经由催化氢化、酸性水解、氯化与成环4步制得。受限于生物质原料糠醛的价格经常大幅波动,导致环丙基甲基酮的售价一直不低于8万元/吨。另一方面,该工艺过程中使用盐酸酸化,产生大量焦油状副产物,直接排放会污染环境,且后处理较为繁琐,总体产率不高。
4.另一种现有的制备工艺是利用2-乙酰基-γ-丁内酯在碘化钠的催化下发生裂解,得到环丙基甲基酮。此工艺可实现一步法生产环丙基甲基酮,但由于2-乙酰基-γ-丁内酯的上游原料使用了高毒的环氧乙烷,对工艺安全性要求很高,不符合绿色化学理念。同时,催化剂碘化钠的价格高昂,约为22万元/吨,而反应过程中催化剂的损耗较大,进而导致成本偏高。而且,该反应多在170~200℃下进行,对设备耐高温、耐腐蚀的要求很高,不利于环丙基甲基酮的工业化生产。
5.现有的环丙基甲基酮工业制备方法,在成本或环境友好性上都存在局限性,或是使用了高毒、强腐蚀性的反应条件,或是会产生难处理的三废。另外,现有制备方法的总体产率不高,且有相当量的副产物生成,需要加以改进。
6.有鉴于此,特提出本发明。
技术实现要素:
7.本发明要解决的技术问题在于克服现有技术的不足,提供一种所用原料绿色低毒,产物收率高,且适合工业化放大的环丙基甲基酮的制备方法。
8.为解决上述技术问题,本发明采用技术方案的基本构思是:
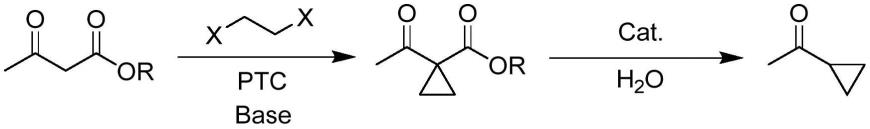
9.一种环丙基甲基酮的制备方法,包括如下步骤:
10.1)向乙酰乙酸酯、1,2-二卤代乙烷和相转移催化剂的混合物中投入固体碱,升温并搅拌进行反应;
11.2)反应结束后冷却,过滤,用去离子水对滤液进行洗涤,分离水相与有机相;
12.3)步骤2)得到的有机相减压蒸馏除去未反应的1,2-二卤代乙烷,得到中间产物1-乙酰基环丙烷-1-甲酸酯;
13.4)将步骤3)得到的中间产物与溶剂、水和酸催化剂混合,加热并保温进行反应;
14.5)继续升温后进行蒸馏,收集冷凝液,所得冷凝液即为目标产物环丙基甲基酮。
15.在上述方案中,采用乙酰乙酸酯和1,2-二卤代乙烷为原料,经由环丙烷化反应和脱羧反应两个阶段的反应,即可制得目标产物环丙基甲基酮。反应的原料简单廉价易得,且具有绿色低毒的特性,解决了现有制备环丙基甲基酮的工艺成本高,容易造成环境污染的问题。利用原料乙酰乙酸酯中亚甲基的高活性,以及1,2-二卤代乙烷中卤素离子良好的离去性,环丙烷化反应的产率可以达到较高水平。同时,中间产物中的酯基受到β-羰基吸电子作用,更易发生脱羧反应,且该反应不可逆,通过中间产物进一步发生脱羧反应以得到目标产物环丙基甲基酮,反应的选择性提升,进而相比于现有技术中的环丙基甲基酮制备工艺,产率得到了提高。本发明的制备方法符合绿色化学理念,产物收率高,同时可显著降低生产成本,适合进一步工业化放大。
16.各个步骤的反应条件简单,无需高温高压等复杂环境,进而对反应设备不存在严格要求。环丙烷化反应阶段结束后,仅需要水洗后蒸馏除去未反应完全的原料1,2-二卤代乙烷,无需进一步的提纯操作,所得中间产物即可直接用于后一阶段的脱羧反应,应用于工业化生产时可实现连续投料。
17.进一步地,步骤4)中,将反应混合物加热至90~100℃进行反应;
18.步骤5)中,继续升温至114~117℃并保温,将目标产物环丙基甲基酮蒸出。
19.在上述方案中,步骤4)的温度相对较低,该温度范围下可进行反应生成目标产物环丙基甲基酮,同时,步骤4)中还保温一段时间,同时可以蒸出反应混合物中具有挥发性的酸催化剂。如果所采用的酸催化剂不具备挥发性,也可以将反应混合物中的水蒸出。步骤5)中进一步升温,从而通过蒸馏的方式将目标产物从反应混合物中提取出,余下的溶剂与不具备挥发性的酸催化剂可循环套用,节省成本。
20.进一步地,步骤4)中,所述溶剂与中间产物的质量比为(0.4~2):1,优选为(0.4~1.5):1,更优为(0.5~0.6):1。
21.优选地,所述溶剂包括乙酸、丙酸、正丁酸、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜中的一种或几种的组合。
22.进一步地,步骤4)中,水与中间产物的摩尔比为(2-6):1。
23.进一步地,步骤4)中,所述酸催化剂与中间产物的摩尔比为(1~10):1。
24.优选地,所述酸催化剂包括盐酸、氢溴酸、氢碘酸、硫酸、对甲苯磺酸、邻甲苯磺酸、甲磺酸、三氟甲磺酸、三氟乙酸和三氯乙酸中的一种或几种的组合。
25.进一步地,步骤1)中,步骤1)中,所述相转移催化剂的质量为乙酰乙酸酯质量的0.1%~2.0%,优选为0.5%~1.0%。
26.本发明中,所述相转移催化剂可以是以下所列出化合物中的任意一种:
27.(1)季铵盐型:四甲基卤化铵、乙基卤化铵、正丁基卤化铵、苄基三乙基卤化铵、十二烷基三甲基卤化铵、十四烷基三甲基卤化铵、十六烷基三甲基卤化铵、十八烷基三甲基卤化铵;
28.(2)季鏻盐型:四甲基卤化鏻、乙基卤化鏻、正丁基卤化鏻、苯基卤化鏻、十二烷基三甲基卤化鏻、十四烷基三甲基卤化鏻、十六烷基三甲基卤化鏻、十八烷基三甲基卤化鏻;
29.(3)非离子型:β-环糊精、聚乙二醇(平均分子量400-80000)、聚乙二醇二甲醚;
30.(4)两性离子型:甜菜碱。
31.本发明制备方法第一阶段的环丙烷化反应中,来自于乙酰乙酸酯的碳负离子与固
体碱中的阳离子结合生成固相的盐,通过相转移催化剂的添加,可以将碳负离子从固相中转移至溶液相中进行取代反应,实现乙酰乙酸酯和1,2-二卤代乙烷反应生成中间产物1-乙酰基环丙烷-1-甲酸酯的目的。
32.上述方案中,相转移催化剂优选季铵盐型相转移催化剂。相比于非离子型相转移催化剂,季铵盐型相转移催化剂对阴离子的转移能力较强,从而能够更充分地将碳负离子从固相转移到溶液相中进行取代反应,从而提高环丙烷化反应阶段中间产物的产率,进而提高制备目标产物环丙烷甲基酮的总体产率。本发明中利用很少量的相转移催化剂,就可以将目标产物的总体产率控制在不低于89%,可有效控制环丙基甲基酮的生产成本。
33.进一步地,步骤2)中,利用去离子水对滤液洗涤2~3次后,分离水相与有机相。
34.进一步地,步骤2)中,将分离出的水相加热蒸干,获取其中的相转移催化剂,用于循环套用。
35.在上述方案中,通过将步骤2)中所得水相加热蒸干的方式,还可以回收其中的相转移催化剂,进一步既降低成本。
36.进一步地,步骤1)中,所述固体碱与乙酰乙酸酯的摩尔比为(2-4):1;
37.优选地,所述固体碱包括氢氧化锂、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、磷酸钠、磷酸钾、甲醇钠、甲醇锂、乙醇钠、乙醇锂、叔丁醇钠或叔丁醇钾;
38.更优地,所述固体碱为氢氧化锂、氢氧化钠或氢氧化钾。
39.进一步地,步骤1)中,分批次向混合物中投入固体碱;
40.优选地,固体碱分3~6批次投入,或者,每批次投入固体碱总质量的1/3~1/6;
41.优选地,对混合物进行搅拌,在搅拌过程中分批次投入固体碱;
42.更优地,固体碱全部投入后持续搅拌,待固体碱完全破碎后,开始升温。
43.在上述方案中,分批次投入固体碱,有利于固体碱在混合液体中快速破碎。固体碱完全破碎后再开始升温进行反应,保证反应在碱性环境下开始,避免了固体碱溶解不充分而不能充分发挥作用。
44.进一步地,步骤1)中,所述1,2-二卤代乙烷与乙酰乙酸酯的摩尔比为(1~12):1,优选为(6~10):1,更优为(7~9):1;
45.优选地,所述1,2-二卤代乙烷中的卤素为氯、溴或碘。
46.进一步地,步骤3)中,减压蒸馏的温度为80~90℃,优选为1,2-二卤代乙烷的沸点;
47.优选地,减压蒸馏的压强为0.1~0.6atm。
48.进一步地,收集减压蒸馏产生的冷凝液,得到1,2-二卤代乙烷用于循环套用。
49.在上述方案中,将减压蒸馏的温度控制在1,2-二卤代乙烷的沸点附近,从而将过量的反应原料1,2-二卤代乙烷从反应体系中除去,确保所得中间产物具有较高的纯度,进而可保证最终所得目标产物的纯度。减压蒸馏过程持续进行直到不再产生冷凝液为止,所收集的冷凝液即为过量的反应原料1,2-二卤代乙烷,可回收实现循环套用,进一步降低成本。
50.进一步地,步骤1)中,所述乙酰乙酸酯为c1-c4烷基乙酰乙酸酯。
51.进一步地,步骤1)中,升温至40~75℃进行反应,并保温1~12h,再进行步骤2);
52.优选地,步骤1)中升温至55~75℃进行反应,更优地,升温至60~70℃进行反应。
53.在上述方案中,将步骤1)中的反应温度控制在40~75℃,避免温度过高造成反应原料1,2-二卤代乙烷蒸发导致的原料损失,保温1~12h以确保第一阶段的环丙烷化反应充分进行,优选保温3~6h。
54.具体地,本发明中环丙基甲基酮的制备方法中,反应路线表示如下:
[0055][0056]
其中,r为c1~c4烷基,x为cl、br或i。base表示所使用的固体碱,ptc表示所使用的相转移催化剂,cat.表示所使用的酸催化剂。
[0057]
本发明中具体采用如下制备方法制备环丙基甲基酮:
[0058]
1)室温下,向反应瓶中投入乙酰乙酸酯、1,2-二卤代乙烷和相转移催化剂,搅拌进行混合,并在机械搅拌的状态下,向反应瓶中分批次混合物中投入固体碱,待固体碱完全破碎后,升温至40~75℃,保温搅拌反应1~12h;
[0059]
2)反应混合物冷却至室温,过滤除去不溶固体,将滤液用去离子水进行洗涤2~3次,分离水相与有机相,水相蒸干获取相转移催化剂循环套用;
[0060]
3)步骤2)得到的有机相在80~90℃下减压蒸馏,收集冷凝液得到未反应的1,2-二卤代乙烷用于循环套用,减压蒸馏直至不再产生冷凝液,反应瓶中即得到中间产物1-乙酰基环丙烷-1-甲酸酯;
[0061]
4)向步骤3)得到的中间产物中加入溶剂、水和酸催化剂,加热至90~100℃并保温1~2h进行反应,之后减压蒸馏除去低沸点杂质;
[0062]
5)继续升温至114~117℃并保温,继续减压蒸馏并收集产生的冷凝液,所得冷凝液即为目标产物环丙基甲基酮。
[0063]
采用上述技术方案后,本发明与现有技术相比具有以下有益效果。
[0064]
1、本发明采用乙酰乙酸酯和1,2-二卤代乙烷为原料制备目标产物环丙基甲基酮,所使用的原料简单廉价易得,且绿色无毒,反应过程中原子利用率较高,从而可实现产物的高产率,适合进一步工业化放大。
[0065]
2、本发明的制备方法中,各个步骤的反应条件简单,应用于工业生产的难度低,第一阶段的环丙烷化反应完成后,仅需水洗后蒸馏除去低沸点物质,即可得到可直接用于第二阶段脱羧反应的中间产物,无需通过精馏等进一步提纯操作以获得中间产物,适合应用于工业化生产中实现连续投料。
[0066]
3、本发明的制备方法中,环丙烷化反应阶段所用的相转移催化剂,脱羧反应阶段所用的溶剂和酸催化剂,以及可能投料过量的反应原料1,2-二卤代乙烷,均可以在制备过程中或制备完成后进行回收,进而实现循环套用,有利于应用于工业化生产时降低生产成本。
[0067]
下面结合附图对本发明的具体实施方式作进一步详细的描述。
附图说明
[0068]
附图作为本发明的一部分,用来提供对本发明的进一步的理解,本发明的示意性
实施例及其说明用于解释本发明,但不构成对本发明的不当限定。在附图中:
[0069]
图1是本发明实施例1中制备的中间产物1-乙酰基环丙烷-1-甲酸乙酯的核磁共振氢谱图;
[0070]
图2是本发明实施例1中制备的环丙基甲基酮的核磁共振氢谱图。
[0071]
需要说明的是,这些附图和文字描述并不旨在以任何方式限制本发明的构思范围,而是通过参考特定实施例为本领域技术人员说明本发明的概念。
具体实施方式
[0072]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对实施例中的技术方案进行清楚、完整地描述,以下实施例用于说明本发明,但不用来限制本发明的范围。
[0073]
实施例1
[0074]
本实施例采用如下步骤制备环丙基甲基酮:
[0075]
1)室温下,向2.5l圆底烧瓶中投入乙酰乙酸乙酯130g(1mol),1,2-二氯乙烷792g(8mol)和苄基三乙基氯化铵1.30g,搅拌进行混合,并在机械搅拌的状态下,向烧瓶中分批次混合物中投入氢氧化钾448g,待氢氧化钾完全破碎后,升温至65℃,保温搅拌反应12h;
[0076]
2)反应混合物冷却至室温,过滤除去不溶固体,将滤液用60ml去离子水洗涤2次,分离水相与有机相,水相蒸干获取苄基三乙基氯化铵循环套用;
[0077]
3)步骤2)得到的有机相在80℃下减压蒸馏,收集冷凝液得到未反应的1,2-二氯乙烷用于循环套用,减压蒸馏直至不再产生冷凝液,烧瓶中即得到中间产物1-乙酰基环丙烷-1-甲酸乙酯146g,产率为94%,气相色谱内标法测定其纯度为95%;
[0078]
4)向步骤3)得到的中间产物中加入乙酸67g,水36g和浓盐酸104g,加热至100℃并保温2h进行反应,之后减压蒸馏除去盐酸水溶液;
[0079]
5)继续升温至114℃并保温,继续减压蒸馏并收集产生的冷凝液,得到目标产物环丙基甲基酮75g,产率为95%,气相色谱内标法测定其纯度为97%。余下的盐酸溶液可循环套用。
[0080]
本实施例中,步骤3)所得中间产物1-乙酰基环丙烷-1-甲酸乙酯的核磁共振氢谱图如图1所示,目标产物环丙基甲基酮的核磁共振氢谱图如图2所示。以乙酰乙酸乙酯计,目标产物环丙基甲基酮的总体产率达到了89%。
[0081]
实施例2
[0082]
本实施例采用如下步骤制备环丙基甲基酮:
[0083]
1)室温下,向2.5l圆底烧瓶中投入乙酰乙酸乙酯130g(1mol),1,2-二溴乙烷188g(1mol)和四正丁基溴化铵1.30g,搅拌进行混合,并在机械搅拌的状态下,向烧瓶中分批次混合物中投入氢氧化钾336g,待氢氧化钾完全破碎后,升温50℃,保温搅拌反应12h;
[0084]
2)反应混合物冷却至室温,过滤除去不溶固体,将滤液用60ml去离子水洗涤2次,分离水相与有机相,水相蒸干获取四正丁基溴化铵循环套用;
[0085]
3)步骤2)得到的有机相在80℃下减压蒸馏,除去低沸点杂质,减压蒸馏直至不再产生冷凝液,烧瓶中即得到中间产物1-乙酰基环丙烷-1-甲酸乙酯154g,产率为99%,气相色谱内标法测定其纯度为99%;
[0086]
4)向步骤3)得到的中间产物中加入乙酸73g,水36g和浓盐酸104g,加热至100℃并
保温2h进行反应,之后减压蒸馏除去盐酸水溶液;
[0087]
5)继续升温至114℃并保温,继续减压蒸馏并收集产生的冷凝液,得到目标产物环丙基甲基酮82g,产率为95%,气相色谱内标法测定其纯度为97%。余下的盐酸溶液可循环套用。
[0088]
本实施例中,以乙酰乙酸乙酯计,目标产物环丙基甲基酮的总体产率达到了96%。
[0089]
实施例3
[0090]
本实施例采用如下步骤制备环丙基甲基酮:
[0091]
1)室温下,向2.5l圆底烧瓶中投入乙酰乙酸甲酯116g(1mol),1,2-二氯乙烷792g(8mol)和苄基三乙基氯化铵1.16g,搅拌进行混合,并在机械搅拌的状态下,向烧瓶中分批次混合物中投入氢氧化钾448g,待氢氧化钾完全破碎后,升温至65℃,保温搅拌反应12h;
[0092]
2)反应混合物冷却至室温,过滤除去不溶固体,将滤液用60ml去离子水洗涤2次,分离水相与有机相,水相蒸干获取苄基三乙基氯化铵循环套用;
[0093]
3)步骤2)得到的有机相在80℃下减压蒸馏,收集冷凝液得到未反应的1,2-二氯乙烷用于循环套用,减压蒸馏直至不再产生冷凝液,烧瓶中即得到中间产物1-乙酰基环丙烷-1-甲酸甲酯129g,产率为91%,气相色谱内标法测定其纯度为95%;
[0094]
4)向步骤3)得到的中间产物中加入乙酸66g,水36g和浓盐酸104g,加热至100℃并保温2h进行反应,之后减压蒸馏除去盐酸水溶液;
[0095]
5)继续升温至114℃并保温,继续减压蒸馏并收集产生的冷凝液,得到目标产物环丙基甲基酮71g,产率为93%,气相色谱内标法测定其纯度为97%。余下的盐酸溶液可循环套用。
[0096]
本实施例中,以乙酰乙酸甲酯计,目标产物环丙基甲基酮的总体产率达到了84%。
[0097]
实施例4
[0098]
本实施例采用如下步骤制备环丙基甲基酮:
[0099]
1)室温下,向2.5l圆底烧瓶中投入乙酰乙酸叔丁酯158g(1mol),1,2-二氯乙烷792g(8mol)和苄基三乙基氯化铵1.16g,搅拌进行混合,并在机械搅拌的状态下,向烧瓶中分批次混合物中投入氢氧化钾448g,待氢氧化钾完全破碎后,升温至65℃,保温搅拌反应12h;
[0100]
2)反应混合物冷却至室温,过滤除去不溶固体,将滤液用60ml去离子水洗涤2次,分离水相与有机相,水相蒸干获取苄基三乙基氯化铵循环套用;
[0101]
3)步骤2)得到的有机相在80℃下减压蒸馏,收集冷凝液得到未反应的1,2-二氯乙烷用于循环套用,减压蒸馏直至不再产生冷凝液,烧瓶中即得到中间产物1-乙酰基环丙烷-1-甲酸叔丁酯158g,产率为86%,气相色谱内标法测定其纯度为95%;
[0102]
4)向步骤3)得到的中间产物中加入乙酸82g,水36g和浓盐酸104g,加热至100℃并保温2h进行反应,之后减压蒸馏除去盐酸水溶液;
[0103]
5)继续升温至114℃并保温,继续减压蒸馏并收集产生的冷凝液,得到目标产物环丙基甲基酮69g,产率为95%,气相色谱内标法测定其纯度为97%。余下的盐酸溶液可循环套用。
[0104]
本实施例中,以乙酰乙酸甲酯计,目标产物环丙基甲基酮的总体产率达到了82%。
[0105]
对比例1
[0106]
本实施例采用如下步骤制备环丙基甲基酮:
[0107]
1)室温下,向1l不锈钢反应釜中加入2-甲基呋喃200g,10%盐酸150g和5%钯炭催化剂25g,搅拌均匀,持续通入0.3mpa氢气,继续搅拌24h;
[0108]
2)将反应混合物转入中和釜,加入8%碳酸钠水溶液中和剩余盐酸,静置分液,弃去下层水相液体,有机相减压蒸馏得到5-羟基-2-戊酮236g,产率为95%,纯度为92%;
[0109]
3)向1l反应瓶中加入步骤2)所得的5-羟基-2-戊酮200g和20%盐酸560g,升温至60℃,保温并搅拌约1h,继续升温至90℃,保温,收集蒸出产物,得到5-氯2-戊酮145g,产率为61%,纯度为94%,盐酸回收套用;
[0110]
4)向1l反应瓶中加入步骤3)所得的5-氯2-戊酮110g,氢氧化钠43g和去离子水66g,搅拌,加热回流2h,冷却至室温,加入60ml叔丁基甲醚萃取,分液弃去下层水相,有机相减压蒸除溶剂,即得目标产物环丙基甲基酮74g,产率为97%,纯度为95%。
[0111]
本对比例中,以初始原料2-甲基呋喃计,最终所得目标产物环丙基甲基酮的总体产率为56%。
[0112]
由以上实施例1-4及对比例1可以看出,采用本发明的制备方法进行环丙基甲基酮的制备,总体产率可达到不低于89%,而对比例1采用现有技术中环丙基甲基酮工业品的制备方法,总体产率仅为56%。可见,本发明中环丙基甲基酮的制备方法原子利用率高,可实现更高的目标产物总体产率。
[0113]
试验例1
[0114]
本试验例用于考察反应原料中乙酰乙酸酯与1,2-二卤代乙烷的配比对目标产物的影响,实施方法与实施例1相同,仅改变步骤1)中1,2-二氯乙烷的投入量,结果如表1所示。
[0115]
表1乙酰乙酸酯与1,2-二卤代乙烷配比对目标产物影响的试验数据表
[0116][0117][0118]
在本发明制备方法的环丙烷化反应阶段,参与反应的1,2-二卤代乙烷与乙酰乙酸酯的摩尔比理论上应为1:1。由以上试验结果可以看出,当1,2-二卤代乙烷的投料量与理论用量相比过量时,例如1,2-二卤代乙烷与乙酰乙酸酯的摩尔比大于3:1时,目标产物纯度可达到70%以上。而将两者的摩尔比控制在(6~10):1时,中间产物,也即环丙烷化反应的产率达到了85%及以上,进而目标产物的纯度不低于86%,且产率不低于80%。进一步将两者
的摩尔比控制在(7~9):1时,中间产物的产率提升至不低于90%,进而目标产物的纯度提升至不低于95%,产率提升至不低于85%。
[0119]
因此,本发明中在步骤1)中投入过量的1,2-二卤代乙烷,1,2-二卤代乙烷与乙酰乙酸酯的摩尔比优选采用(6~10):1,更优采用(7~9):1。
[0120]
试验例2
[0121]
本试验例用于考察相转移催化剂的用量对目标产物的影响,实施方法与实施例1相同,仅改变步骤1)中苄基三乙基氯化铵的投入量,将苄基三乙基氯化铵与乙酰乙酸乙酯的质量比值记为x,结果如表2所示。
[0122]
表2相转移催化剂用量对目标产物影响的试验数据表
[0123]
苄基三乙基氯化铵(g)x(%)中间产物产率(%)目标产物纯度(%)总体产率(%)0.130.15395500.390.36995660.650.58297781.301.09497891.951.59497892.602.09497893.252.59497893.903.0949789
[0124]
由以上试验结果可见,随着相转移催化剂(即苄基三乙基氯化铵)投入质量的增加,中间产物的产率上升,进而目标产物的纯度和产率也相应提升。而当相转移催化剂的投入质量超过乙酰乙酸乙酯质量的1%后,中间产物的产率,以及相应的目标产物的纯度和产率不再出现明显增加。
[0125]
在考虑生产成本的情况下,本发明中优选将相转移催化剂的质量控制为乙酰乙酸酯质量的0.5%~1.0%,目标产物的纯度可达到97%,产率可达到不低于78%。
[0126]
试验例3
[0127]
本试验例用于考察第一阶段的环丙烷化反应中,反应温度对目标产物的影响,实施方法与实施例1相同,仅改变步骤1)中的保温温度,结果如表3所示。
[0128]
表3环丙烷化反应温度对目标产物影响的试验数据表
[0129]
步骤1)保温温度(℃)中间产物产率(%)目标产物纯度(%)总体产率(%)4073976945809576508496805587948360899586659197897095929075988493
[0130]
由以上表3可见,将步骤1)中的保温温度控制在45~75℃,可以控制中间产物的产率不低于80%,进而目标产物的纯度不低于84%,产率不低于76%。而当保温温度控制在55
~75℃时,中间产物的产率提升至87%及以上,同时目标产物的产率提升至不低于83%。当保温温度在60~70℃之间时,可实现目标产物的纯度达到92%~95%,产率达到86%~90%。
[0131]
因此,本发明在步骤1)中对反应混合物升温至45~75℃进行反应,优选升温至55~75℃,更优升温至60~70℃。
[0132]
试验例4
[0133]
本试验例用于考察第二阶段的脱羧反应中,溶剂用量对目标产物的影响,实施方法与实施例1相同,仅改变步骤4)中乙酸的加入量,将乙酸与中间产物的质量比即为y:1,结果如表4所示。
[0134]
表4脱羧反应溶剂用量对目标产物影响的试验数据表
[0135][0136][0137]
以上表4中的试验结果显示,随着脱羧反应阶段中溶剂(即乙酸)质量的增加,目标产物的纯度逐渐上升并最终稳定在97%左右,而目标产物的总体产率呈下降趋势。本发明中将脱羧反应阶段溶剂与中间产物的质量比控制在(0.4~1.5):1,目标产物的纯度可达到90%~97%,总体产率控制在72%~89%。而将溶剂与中间产物的质量比控制在(0.5~0.6):1时,目标产物的纯度可提升至96%~97%,总体产率提升至86%~89%。
[0138]
试验例5
[0139]
本试验例用于考察第一阶段的环丙烷化反应中,采用不同类型的相转移催化剂对目标产物的影响,实施方法与实施例1相同,仅改变步骤1)中相转移催化剂的类型,结果如表5所示。
[0140]
表5相转移催化剂类型对目标产物影响的试验数据表
[0141]
相转移催化剂中间产物产率(%)目标产物纯度(%)总体产率(%)苄基三乙基氯化铵949789四正丁基溴化铵939788四苯基氯化鏻749870四正丁基氯化鏻919986β-环糊精829477聚乙二醇二甲醚709267甜菜碱859581
[0142]
由以上试验结果可以看出,在相同用量的情况下,以苄基三乙基氯化铵和四正丁基溴化铵为代表的季铵盐型相转移催化剂与其他类型的相转移催化剂相比,在第一阶段的环丙烷化反应中,所得中间产物的产率更高,达到了93%~94%。而其他类型的相转移催化剂,相应的中间产物产率偏低,尤其是非离子型的相转移催化剂,如β-环糊精和聚乙二醇二甲醚,其所得中间产物的产率分别仅为82%和70%。
[0143]
对于目标产物环丙烷甲基酮来说,季铵盐型相转移催化剂达到了97%的目标产物纯度,与其他类型的相转移催化剂相比达到了较高水平,同时,其具有88%~89%的总体产率,高于其他类型的相转移催化剂。
[0144]
因此,本发明中优选季铵盐型相转移催化剂,可以在相转移催化剂用量较少的情况下同时实现目标产物环丙烷甲基酮的高纯度与高总体产率。
[0145]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1