一种碳硼烷类化合物及其制备方法和应用
1.本发明涉及一种碳硼烷类化合物及其制备方法和应用,属于有机合成领域。
背景技术:
2.光引发剂(photoinitiator,pi)是一类能在紫外光区(250~420nm)或可见光区(400~800nm)吸收一定波长的能量,产生自由基、阳离子、阴离子等活性基团,从而引发单体聚合交联固化的化合物。光引发剂(photoinitiator,pi)是紫外光固化体系的重要组成部分,光引发剂决定了光固化反应的速度,影响光固化材料的性能。苯偶姻(俗称安息香,见式1)、氢化苯偶姻(见式2)及其衍生物是一种应用广泛的自由基引发剂,其量子产率和产生的自由基的反应活性都比较高,有着很好的引发效率,在工业上适用于引发苯乙烯单体的聚合。
[0003][0004]
但是由于分子中苯甲基醚碳上的氢原子比较活泼,容易生成苄基醚活性自由基,在外界没有提供光能的情况下也能引发聚合反应,因此这类引发剂的热稳定性较差,在室温的贮存时间较短。与不饱和聚醋及常用乙烯基单体交联剂配合时相溶性较差,易于引起热胶凝,引发光固化速度慢。虽然安息香类光引发剂有着很好的引发效率,但是为了进一步缩短光固化时间,提高生产效率,人们对光引发剂的引发效率提出了更高的要求。因此,对安息香衍生物进行改造以提升热稳定性和改善引发效果显得尤为重要。
技术实现要素:
[0005]
本发明的目的在于提供一种碳硼烷类化合物,该碳硼烷类化合物作为光引发剂时,具有较高的光引发效率。
[0006]
本发明的第二个目的在于提供一种碳硼烷类化合物的制备方法。
[0007]
本发明的第三个目的在于提供一种碳硼烷类化合物在光固化材料领域中的应用。
[0008]
本发明的碳硼烷类化合物的技术方案为:
[0009]
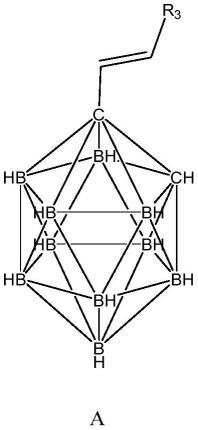
一种碳硼烷类化合物,所述碳硼烷类化合物具有式i所示的结构,
[0010][0011]
式i中,r为未取代的芳基、取代的芳基、未取代的芳杂环基或取代的芳杂环基。
[0012]
本发明的碳硼烷类化合物作为光引发剂时,具有较高的光引发效率,其具有较高的分子量,在引发光固化体系聚合的过程中,由于引发时间短,聚合反应后光引发剂残留的化合物和光解产物不易向涂层表面迁移,可以应用于光固化材料等领域。
[0013]
可以理解的是,芳杂环基是指构成芳香环的原子除碳原子外,还至少含有一个选自n、o或s的杂原子,包括单芳杂环基和由苯环和一个或两个以上单杂环稠合而成的稠芳杂环基。例如,单芳杂环基为呋喃基、吡咯基、噻吩基、噻唑基、咪唑基、吡啶基、吡喃基或嘧啶基,稠芳杂环基为吲哚基或喹啉基。芳杂环基的连接原子可以是芳香环上的碳原子也可以是芳香环上的氮原子。
[0014]
本发明的碳硼烷类化合物为外消旋体混合物。
[0015]
所述未取代的芳基优选为苯基、萘基或联苯基。
[0016]
进一步优选地,所述未取代的芳基为苯基或萘基。
[0017]
所述取代的芳基优选为取代的苯基、萘基或联苯基。
[0018]
进一步优选地,所述取代的芳基为取代的苯基或萘基。
[0019]
优选地,所述未取代的芳杂环基为呋喃基、吡咯基、噻吩基、噻唑基、咪唑基、吡啶基、吡喃基、嘧啶基、吲哚基或喹啉基。
[0020]
进一步优选地,所述未取代的芳杂环基为呋喃基、噻吩基或吡啶基。
[0021]
优选地,所述r为取代基r1取代的芳基或取代基r2取代的芳杂环基;所述取代基r1、r2各自独立地选自-f、-cl、-br、-i、c
1-c
10
烷基、c
1-c
10
烷氧基或c
1-c
10
氟代烷基中的至少一种。
[0022]
优选地,所述c
1-c
10
烷基为c
1-c5烷基。
[0023]
优选地,所述c
1-c5烷基为甲基或乙基。
[0024]
进一步优选地,所述c
1-c5烷基为甲基。
[0025]
优选地,所述c
1-c
10
烷氧基为c
1-c5烷氧基。
[0026]
优选地,所述c
1-c
10
氟代烷基为c
1-c5氟代烷基。
[0027]
优选地,所述取代的芳基为间位和/或对位取代的芳基。
[0028]
优选地,所述取代的苯基为间位和/或对位取代的苯基。
[0029]
本发明的碳硼烷类化合物的制备方法的技术方案为:
[0030]
一种如上所述的碳硼烷类化合物的制备方法,包括以下步骤:将反应物m进行双羟化反应,即得;所述反应物m具有式a所示的结构,
[0031][0032]
式a中,r3为未取代的芳基、取代的芳基、未取代的芳杂环基或取代的芳杂环基。
[0033]
本发明利用双羟化反应制备上述碳硼烷类化合物,基于双羟化反应的优点,本发明的碳硼烷类化合物的制备方法操作简单,制备条件温和,制备时间较短,是一种高效的、环境友好的制备方法。
[0034]
本发明中反应物m中的烯烃为反式结构。
[0035]
所述未取代的芳基优选为苯基、萘基或联苯基。
[0036]
进一步优选地,所述未取代的芳基为苯基或萘基。
[0037]
所述取代的芳基优选为取代的苯基、萘基或联苯基。
[0038]
进一步优选地,所述取代的芳基为取代的苯基或萘基。
[0039]
优选地,所述未取代的芳杂环基为呋喃基、吡咯基、噻吩基、噻唑基、咪唑基、吡啶基、吡喃基、嘧啶基、吲哚基或喹啉基。
[0040]
进一步优选地,所述未取代的芳杂环基为呋喃基、噻吩基或吡啶基。
[0041]
优选地,所述r3为取代基r4取代的芳基或取代基r5的芳杂环基;所述取代基r4、r5各自独立地选自-f、-cl、-br、-i、c
1-c
10
烷基、c
1-c
10
烷氧基或c
1-c
10
氟代烷基中的至少一种。
[0042]
优选地,所述c
1-c
10
烷基为c
1-c5烷基。
[0043]
优选地,所述c
1-c5烷基为甲基或乙基。
[0044]
进一步优选地,所述c
1-c5烷基为甲基。
[0045]
优选地,所述c
1-c
10
烷氧基为c
1-c5烷氧基。
[0046]
优选地,所述c
1-c
10
氟代烷基为c
1-c5氟代烷基。
[0047]
优选地,所述取代的芳基为间位和/或对位取代的芳基。
[0048]
双羟化反应是将反应物m中的烯属碳碳双键转化为二元醇,双羟化反应的具体实施过程可以参考现有技术来完成。优选地,所述双羟化反应包括以下步骤:将反应物m加入到包含催化体系的溶液中,进行双羟化反应,反应完成后淬灭反应。所述包含催化体系的溶液由催化体系和溶剂组成。
[0049]
优选地,所述催化体系包括催化剂、助氧化剂、碱和水解促进剂。反应物首先在催化剂和助氧化剂的作用下被氧化成酯,然后生成的酯在水解促进剂的作用下通过水解即得碳硼烷类化合物。
[0050]
优选地,所述水解促进剂为甲磺酰胺。
[0051]
优选地,所述催化剂由三乙烯二胺和二水锇酸钾组成;所述三乙烯二胺和二水锇酸钾的摩尔比为(1~2):(0.1~0.5)。
[0052]
进一步优选地,所述三乙烯二胺和二水锇酸钾的摩尔比为1:0.2。
[0053]
优选地,所述反应物m的摩尔数、三乙烯二胺与二水锇酸钾的摩尔数之和、助氧化剂的摩尔数和水解促进剂的摩尔数之比为(1~2):(0.1~0.5):(1~3):(1~2)。
[0054]
进一步优选地,所述反应物m的摩尔数、三乙烯二胺和二水锇酸钾的摩尔数之和、助氧化剂的摩尔数和水解促进剂的摩尔数之比为1:0.12:(1~3):1。
[0055]
优选地,所述助氧化剂选自k3[fe(cn)6]和/或n-甲基吗啉-n-氧化物。
[0056]
优选地,所述溶液的溶剂由有机溶剂和水组成;所述有机溶剂选自叔丁醇、丙酮、二甲亚砜、乙腈中的一种或任意组合。
[0057]
优选地,所述碱选自碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、naoh、koh、lioh、nh4oh,tbuona、t-buok、t-buoli、碳酸铯、三乙胺、二异丙基乙基氨、dbu、吡啶和对二甲氨基吡啶中的一种或多种。
[0058]
进一步优选地,所述碱为碳酸钾。
[0059]
优选地,所述双羟化反应的时间为5~30h。
[0060]
进一步优选地,所述双羟化反应的时间为8~24h。
[0061]
优选地,所述双羟化反应的温度为0~25℃。
[0062]
优选地,所述淬灭反应包括以下步骤:将亚硫酸氢钠加入反应体系中,搅拌0.5h。
[0063]
优选地,所述双羟化反应还包括以下步骤:淬灭反应后依次进行萃取、干燥处理。
[0064]
优选地,所述萃取所用的有机溶剂选自二氯甲烷、乙酸乙酯、正丁醇中的一种或任意组合。
[0065]
本发明的碳硼烷类化合物在光固化材料领域中的应用的技术方案为:
[0066]
一种上述碳硼烷类化合物在光固化材料领域中的应用。
[0067]
将上述碳硼烷类化合物作为光引发剂应用于光固化材料领域中,可缩短光固化时间,减少残留的化合物和光解产物向涂层表面迁移,可以应用于光固化材料等领域。
[0068]
优选地,所述应用为碳硼烷类化合物作为光引发剂的应用。
附图说明
[0069]
图1为实施例11制得的碳硼烷类化合物(化合物2a)的核磁共振氢谱图;
[0070]
图2为实施例11制得的碳硼烷类化合物(化合物2a)的核磁共振碳谱图;
[0071]
图3为实施例11制得的碳硼烷类化合物(化合物2a)的核磁共振硼谱图。
具体实施方式
[0072]
下面结合具体实施方式对本发明的技术方案进一步说明。
[0073]
一、本发明的碳硼烷类化合物的具体实施例如下:
[0074]
实施例1
[0075]
本实施例的碳硼烷类化合物(化合物2a)的结构式如下:
[0076][0077]
实施例2
[0078]
本实施例的碳硼烷类化合物(化合物2b)的结构式如下:
[0079][0080]
实施例3
[0081]
本实施例的碳硼烷类化合物(化合物2c)的结构式如下:
[0082][0083]
实施例4
[0084]
本实施例的碳硼烷类化合物(化合物2d)的结构式如下:
[0085][0086]
实施例5
[0087]
本实施例的碳硼烷类化合物(化合物2e)的结构式如下:
[0088][0089]
实施例6
[0090]
本实施例的碳硼烷类化合物(化合物2f)的结构式如下:
[0091][0092]
实施例7
[0093]
本实施例的碳硼烷类化合物(化合物2g)的结构式如下:
[0094][0095]
实施例8
[0096]
本实施例的碳硼烷类化合物(化合物2h)的结构式如下:
[0097][0098]
实施例9
[0099]
本实施例的碳硼烷类化合物(化合物2i)的结构式如下:
[0100][0101]
实施例10
[0102]
本实施例的碳硼烷类化合物(化合物2j)的结构式如下:
[0103][0104]
二、本发明的碳硼烷类化合物的制备方法的具体实施例如下:
[0105]
以下碳硼烷类手性化合物的制备方法的实施例(实施例11-20)中的收率均为双羟化反应(步骤2)的收率。
[0106]
实施例11
[0107]
本实施例的碳硼烷类化合物的制备方法得到的是实施例1的碳硼烷类化合物,包括以下步骤:
[0108]
(1)在n2保护下,向反应瓶中加入38mg打磨过的碎镁屑和12ml无水乙醚,将反应瓶置于35℃油浴中搅拌,然后缓慢滴加1-溴亚甲基邻碳硼烷(295mg,1.2mmol)和90ml无水乙
醚形成的溶液,滴加完毕后,在室温下反应2.5h,待镁屑全部消失后,将反应体系降至0℃,继续搅拌20min,然后缓慢滴加苯甲醛(148.6mg,1.4mmol)和45ml无水乙醚形成的溶液,继续在0℃下反应3h,将反应产物进行减压蒸馏,得到固体产物,然后在n2保护下,将得到的固体产物、对甲苯磺酸(12mg,0.08mmol)和90ml甲苯加入单口瓶中,回流反应12h,反应结束后,将反应产物通过减压蒸馏和柱层析分离(以石油醚为展开剂)得到化合物1a。
[0109]
(2)在0℃下,将49.39g(0.15mol)铁氰化钾(k3[fe(cn)6])、20.73g(0.15mol)碳酸钾(k2co3)、0.368g(0.001mol)二水锇酸钾(k2oso4·
2h2o)、0.56g(0.005mol)三乙烯二胺(dabco)、50ml丙酮和50ml水置于反应瓶中,搅拌10min;然后将12.41g(0.05mol)化合物1a和4.75g(0.05mol)甲磺酰胺(ch3so2nh2)加入到反应瓶中,搅拌24h;再向反应瓶中加入12.5g亚硫酸氢钠,在25℃下进行搅拌30min,得到反应产物,然后将反应产物用300ml二氯甲烷萃取3次,然后将萃取液用干燥剂(无水硫酸钠)干燥后,再经减压蒸馏,除去溶剂,得到12.56g 2-邻碳硼烷-苯乙二醇,收率为89%。通过高效液相色谱检测可得,得到的产品中的2-邻碳硼烷-苯乙二醇外消旋体的含量(纯度)为90.92%。本实施例的碳硼烷类化合物(化合物2a)的合成路线如下:
[0110][0111]
对化合物2a进行核磁分析,结果如图1-3所示,所得化合物2a的表征数据如下:
[0112]
化合物2a:2-邻碳硼烷-苯乙二醇,白色固体,rf=0.23(己烷/乙酸乙酯=5/1);1h nmr(300mhz,cdcl3)δ=7.45-7.30(m,5h),4.99(d,j=3.7hz,1h),4.17(d,j=9.5hz,1h),4.09(s,1h),3.28(d,j=9.6hz,1h),2.34(d,j=4.3hz,1h);
13
c{1h}nmr(75mhz,cdcl3)δ=140.15,128.86,128.68,125.83,77.44,75.54,73.02,58.08;
11
b nmr(128mhz,cdcl3)δ=-3.33(t,j=160.4hz,3b),-11.31(dt,j=406.0,152.8hz,7b)。
[0113]
实施例12
[0114]
本实施例的碳硼烷类化合物的制备方法得到的是实施例2的碳硼烷类化合物,包括以下步骤:
[0115]
(1)在n2保护下,向反应瓶中加入1.5g打磨过的镁屑和100ml无水乙醚,将反应瓶置于35℃油浴中搅拌,然后缓慢滴加1-溴亚甲基邻碳硼烷(10g,0.04mol)和300ml无水乙醚
形成的溶液,滴加完毕后,在室温下反应4.5h,待镁屑全部消失后,将反应体系降至8℃,继续搅拌30min,然后缓慢滴加4-甲基苯甲醛(6g,0.05mol)和100ml无水乙醚形成的溶液,继续在8℃下反应3h,将反应产物进行减压蒸馏,得到固体产物,然后在n2保护下,将得到的固体产物、对甲苯磺酸(0.6g,3.5mmol)和500ml甲苯加入单口瓶中,回流反应14h,反应结束后,将反应产物通过减压蒸馏和柱层析分离(以石油醚为展开剂)得到化合物1b。
[0116]
(2)在25℃下,将127.1mg(0.5mmol)n-甲基吗啉-n-氧化物(nmo)、20.73g(0.15mol)碳酸钾(k2co3)、3.68mg(0.01mmol)二水锇酸钾(k2oso4·
2h2o)、5.61mg(0.05mmol)三乙烯二胺(dabco)、5ml叔丁醇和5ml水置于反应瓶中,搅拌10min;然后将131.04g(0.5mmol)化合物1b和47.5g(0.5mmol)甲磺酰胺加入到反应瓶中,搅拌8h;再向反应瓶中加入125.0mg亚硫酸氢钠,在25℃下进行搅拌30min,得到反应产物,然后将反应产物用50ml乙酸乙酯萃取3次,然后将萃取液用干燥剂(无水硫酸钠)干燥后,再经减压蒸馏,除去溶剂,得到125.9mg 2-邻碳硼烷-对甲苯乙二醇,收率为85%。本实施例的碳硼烷类化合物(化合物2b)的合成路线如下:
[0117][0118]
对化合物2b进行核磁分析,所得化合物2b的表征数据如下:
[0119]
化合物2b:2-邻碳硼烷-对甲苯乙二醇,白色固体,rf=0.31(己烷/乙酸乙酯=5/1);1h nmr(300mhz,cdcl3)δ=7.20(s,4h),4.92(d,j=3.0hz,1h),4.14(d,j=9.2hz,1h),4.06(s,1h),3.31(d,j=9.3hz,1h),2.35(d,j=9.1hz,4h);
13
c{1h}nmr(75mhz,cdcl3)δ=138.62,137.14,129.51,125.78,77.62,72.92,58.05,21.12;
11
b nmr(128mhz,cdcl3)δ=-0.18
‑‑
5.63(m,3b),-7.20
‑‑
16.64(m,7b)。
[0120]
实施例13
[0121]
本实施例的碳硼烷类化合物的制备方法得到的是实施例3的碳硼烷类化合物,包括以下步骤:
[0122]
(1)在n2保护下,向反应瓶中加入66mg打磨后的碎镁屑和25ml无水乙醚,将反应瓶
置于35℃水浴中搅拌,然后缓慢滴加1-溴亚甲基邻碳硼烷(500mg,2.1mmol)和120ml无水乙醚形成的溶液,滴加完毕后,在室温下继续反应3h,待镁屑全部消失后,将反应体系降至3℃,继续搅拌20min,然后缓慢滴加3-甲基苯甲醛(300mg,2.5mmol)和90ml无水乙醚形成的溶液,继续在3℃下反应3h,将反应产物进行减压蒸馏,得到固体产物,然后在n2保护下,将得到的固体产物、对甲苯磺酸(15mg,0.1mmol)和500ml甲苯加入单口瓶中,回流反应14h,反应结束后,将反应产物通过减压蒸馏和柱层析分离(以石油醚为展开剂)得到化合物1c。
[0123]
(2)在25℃下,将98.77g(0.3mol)铁氰化钾(k3[fe(cn)6])、41.46g(0.3mol)碳酸钾(k2co3)、0.74g(0.002mol)二水锇酸钾(k2oso4·
2h2o)、1.12g(0.01mol)三乙烯二胺(dabco)、125ml乙腈(ch3cn)和125ml水置于反应瓶中,搅拌10min;然后将26.21g(0.1mol)化合物1c和9.5g(0.1mol)甲磺酰胺(ch3so2nh2)加入到反应瓶中,搅拌12h;再向反应瓶中加入25g亚硫酸氢钠,在25℃下进行搅拌30min,得到反应产物,然后将反应产物用100ml乙酸乙酯萃取3次,然后将萃取液用干燥剂(无水硫酸钠)干燥后,再经减压蒸馏,除去溶剂,得到2-邻碳硼烷-间甲苯乙二醇25.78g,收率为87%。本实施例的碳硼烷类化合物(化合物2c)的合成路线如下:
[0124][0125]
对化合物2c进行核磁分析,所得化合物2c的表征数据如下:
[0126]
化合物2c:2-邻碳硼烷-间甲苯乙二醇,白色固体,rf=0.29(己烷/乙酸乙酯=5/1);1h nmr(300mhz,cdcl3)δ=7.31-7.20(m,1h),7.12(q,j=7.7hz,3h),4.91(s,1h),4.13(d,j=9.1hz,1h),4.04(s,1h),3.28(d,j=9.3hz,1h),2.37(d,j=7.3hz,4h);
13
c{1h}nmr(75mhz,cdcl3)δ=140.08,138.69,129.41,128.75,126.44,122.82,77.42,75.51,73.01,58.05,21.43;
11
b nmr(128mhz,cdcl3)δ=-0.25
‑‑
6.27(m,3b),-11.29(dt,j=401.1,153.9hz,7b)。
[0127]
实施例14
[0128]
本实施例的碳硼烷类化合物的制备方法得到的是实施例4的碳硼烷类化合物,本实施例与实施例8的区别在于,将1.4mmol苯甲醛替换为1.4mmol对氟苯甲醛,本实施例制备得到的化合物2d的收率为93%,对化合物2d进行核磁分析,所得化合物2d的表征数据如下:
[0129]
化合物2d:rf=0.26(己烷/乙酸乙酯=5/1);1h nmr(300mhz,cdcl3)δ=7.34-7.29
(m,2h),7.10-7.04(m,2h),4.97(s,1h),4.13(s,1h),4.07(s,1h),3.33(s,1h),2.46(s,1h).
13
c{1h}nmr(75mhz,cdcl3)δ=164.35,161.07,135.90(d,j=3.2hz),127.71(d,j=8.3hz),115.77(d,j=21.7hz),75.52,72.40,57.97;
11
b nmr(128mhz,cdcl3)δ=-1.03
‑‑
5.95(m,3b),-7.04
‑‑
16.34(m,7b);
19
f nmr(376mhz,cdcl3)δ=-112.9。
[0130]
实施例15
[0131]
本实施例的碳硼烷类化合物的制备方法得到的是实施例5的碳硼烷类化合物,本实施例与实施例11的区别在于,将1.4mmol苯甲醛替换为1.4mmol对甲氧基苯甲醛,本实施例制备得到的化合物2e的收率为77%,对化合物2e进行核磁分析,所得化合物2e的表征数据如下:
[0132]
化合物2e:rf=0.19(己烷/乙酸乙酯=3/1);1h nmr(300mhz,cdcl3)δ=7.33
–
7.17(m,2h),6.89(d,j=8.6hz,2h),4.89(s,1h),4.12(d,j=8.3hz,1h),4.06(s,1h),3.80(s,3h),3.36(d,j=9.0hz,1h),2.34(s,1h);
13
c{1h}nmr(75mhz,cdcl3)δ=159.80,132.21,127.29,114.20,77.42,75.47,72.73,58.07,55.33;
11
b nmr(128mhz,cdcl3)δ=
‑‑
3.39(t,j=163.0hz,3b),-7.20
‑‑
16.89(m,7b)。
[0133]
实施例16
[0134]
本实施例的碳硼烷类化合物的制备方法得到的是实施例6的碳硼烷类化合物,本实施例与实施例11的区别在于,将1.4mmol苯甲醛替换为1.4mmol对三氟甲基苯甲醛,本实施例制备得到的化合物2f的收率为90%,对化合物2f进行核磁分析,所得化合物2f的表征数据如下:
[0135]
化合物2f:rf=0.22(己烷/乙酸乙酯=5/1);1h nmr(300mhz,cdcl3)δ=7.65(d,j=8.2hz,2h),7.46(d,j=8.2hz,2h),5.20
–
4.92(m,1h),4.15(d,j=9.8hz,1h),4.08(s,1h),3.29(d,j=10.0hz,1h),2.60(d,j=4.5hz,1h);
13
c{1h}nmr(75mhz,cdcl3)δ=143.96(d,j=1.2hz),130.81(q,j=32.4hz).126.23,125.76(q,j=3.7hz).123.82(q,j=270.9hz),77.20,75.50,72.43,57.88;
11
b nmr(128mhz,cdcl3)δ=-3.20(t,j=160.6hz,3b),-11.84(dt,j=252.8,208.9hz,7b);
19
f nmr(376mhz,cdcl3)δ=-62.6。
[0136]
实施例17
[0137]
本实施例的碳硼烷类化合物的制备方法得到的是实施例7的碳硼烷类化合物,本实施例与实施例11的区别在于,将1.4mmol苯甲醛替换为1.4mmol 2-萘甲醛,本实施例制备得到的化合物2g的收率为97%,对化合物2g进行核磁分析,所得化合物2g的表征数据如下:
[0138]
化合物2g:rf=0.21(己烷/乙酸乙酯=3/1);1h nmr(300mhz,cdcl3)δ=7.93-7.78(m,4h),7.58-7.45(m,2h),7.40(d,j=8.6hz,1h),5.16(s,1h),4.27(s,1h),4.12(s,1h),3.33(s,1h),2.48(s,1h);
13
c{1h}nmr(75mhz,cdcl3)δ=137.45,133.25,133.07,128.89,128.08,127.83,126.80,126.68,125.01,123.35,77.52,75.47,73.13,58.12;
11
b nmr(128mhz,cdcl3)δ=-0.25
‑‑
6.82(m,3b),-11.29(dt,j=404.2,153.4hz,7b)。
[0139]
实施例18
[0140]
本实施例的碳硼烷类化合物的制备方法得到的是实施例8的碳硼烷类化合物,本实施例与实施例11的区别在于,将1.4mmol苯甲醛替换为1.4mmol 2-呋喃甲醛,本实施例制备得到的化合物2h的收率为88%。
[0141]
实施例19
[0142]
本实施例的碳硼烷类化合物的制备方法得到的是实施例9的碳硼烷类化合物,本实施例与实施例11的区别在于,将1.4mmol苯甲醛替换为1.4mmol 2-噻吩甲醛,本实施例制备得到的化合物2i的收率为82%。
[0143]
实施例20
[0144]
本实施例的碳硼烷类化合物的制备方法得到的是实施例10的碳硼烷类化合物,本实施例与实施例11的区别在于,将1.4mmol苯甲醛替换为1.4mmol 2-吡啶甲醛,本实施例制备得到的化合物2j的收率为84%。
[0145]
三、本发明的碳硼烷类化合物在光固化材料领域中的应用的具体实施例如下:
[0146]
将实施例1-10中任一碳硼烷类化合物应用于光固化材料中即可。
[0147]
实验例
[0148]
将0.4g安息香及实施例11-13制备的碳硼烷类化合物分别加入到由5g交联剂(乙酸乙烯酯、甲基丙烯酸、苯乙烯的混合液)和5g不饱和聚酯(自制的聚乙二醇改性不饱和聚酯树脂)组成的混合物中,搅拌均匀,形成光固化组合物,然后将光固化组合物倒入预先贴好边框(厚0.1cm)的玻璃板上,使光固化组合物流平并填充边框与玻璃板形成的空间,待气泡消失后,在光固化组合物表面复盖一层涤纶薄膜。用黑光灯(20*20w,灯距9cm)进行单面曝光,当光固化组合物材料不沾涤纶薄膜时,记下相应的曝光时间,即为光固化时间;同时将上述配制的光固化组合物避光放置于室温环境中,通过光固化组合物发生胶凝所需的时间(室温储存时间)来评价光引发剂的热稳定性,实验结果如表1所示。
[0149]
表1光固化时间测定结果
[0150][0151][0152]
由表1可知,化合物2a、2b和2c对光固化过程都有较好的引发作用,其中化合物2a的引发作用较强,对苯乙烯的光固化过程的引发作用优于安息香,并且含有本发明的碳硼烷类化合物的光固化组合物发生胶凝所需的时间远大于含有安息香的光固化组合物,说明本发明的碳硼烷类化合物不仅能够缩短光固化过程的时间,而且热稳定性良好,在光固化材料领域具有潜在的用途。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1