一种增强型吸附树脂材料的制备方法
1.本发明属于树脂材料领域,具体涉及一种增强型吸附树脂材料的制备方法。
背景技术:
2.近年来,日益严重的水生环境污染逐渐引起了全球范围内的关注。药品和个人护理品(ppcps)作为一类新兴污染物,种类繁多且具有环境持久性。因此,如何去除水体中的ppcps成为了环境领域的研究热点,树脂吸附法是去除水体中ppcps的一种高效且经济的方法。
3.waters公司的hlb固相萃取材料具有独特的亲疏水骨架,在对水体中微量有机物的富集分离中得到了广泛的应用。但是大量ppcps在环境水体中呈现电离状态,hlb对此类有机物萃取效率较差。为此,waters公司进一步推出了一系列离子交换萃取材料如wax、max、wcx、mcx等。这些基于亲水性骨架的离子交换树脂对水体中离子化的有机物实现了较为优异的萃取效果,其骨架为乙烯吡咯烷酮~二乙烯苯,但是制备工艺均未公开。申请人前期专利zl202111347253.2提出了一种改进的种子溶胀聚合工艺,制备了系列增强型萃取材料,对水体中带电ppcps的快速萃取效果较好。但是由于该材料的骨架结构为n-乙烯基吡咯烷酮-二乙烯苯,缺少胺化试剂的作用位点,所以在改性制备阴离子交换材料时,需要首先通过氯甲醚在树脂骨架上引入活性氯基再进行反应。但是,该反应所需的氯甲醚是一种剧毒物品,对人体有强致癌性,一旦泄露,对生命健康和环境都会造成不可逆的危害。同时,树脂上的氯甲基化反应效率不可控,不同批次树脂的离子交换容量波动较大,重复性较差。除此以外,胺化试剂的“缩孔”效应还会导致树脂的孔径、比表面积变小,降低树脂的传质速率,吸附过程中容易堵塞孔道。
4.甲基丙烯酸缩水甘油酯(gma)是一种常见的工业原料,其分子内部的环氧基团性质活泼,常用做功能单体参与共聚反应。wan等的研究结果显示,gma的环氧基中氧原子周围的电子云密度大,当亲核试剂靠近环氧基时,会使环氧基开环,形成羟基和目标功能基团。wang等的研究结果显示,在gma-dvb树脂中,gma的含量越高,树脂的平均孔径越大,但比表面积越小。可见,树脂中的gma的含量,对于调控树脂的离子交换容量、比表面积与孔道结构至关重要。目前,使用gma、nvp(n-乙烯基吡咯烷酮)和dvb(二乙烯苯)进行三元共聚的例子未见报道,因为这些单体间的亲疏水差异性较大,很难共聚,更缺少一种合适的工艺可以有效调控单体在聚合过程中参与反应的比例。
5.因此,开发出一种绿色环保、可以使gma、nvp和dvb同时共聚且可以调控聚合比例的工艺,是提高此类树脂吸附容量,改善传质效率,降低孔道堵塞问题的重要技术难题。
技术实现要素:
6.1.发明目的
7.本发明针对现有制备工艺和吸附材料在实际应用中的不足等问题之一,提供了一种增强型吸附树脂材料的制备方法及其制备的增强型吸附树脂,该方法以gma、nvp、dvb为
功能单体,在可逆加成-断裂链转移聚合(raft)链转移试剂存在条件下配制成三种油相,通过改变油相中链转移剂、功能单体的比例使其形成寡聚物,再通过控制聚合温度等聚合条件,将上述三种油相进行聚合,通过控制寡聚物在各个聚合阶段以不同的单体比、聚合度参与聚合反应,可以使亲疏水差异性较大的gma、nvp、dvb同时参与聚合,使用该方法合成的树脂白球,即具有多层结构的n-乙烯基吡咯烷酮、甲基丙烯酸缩水甘油酯和二乙烯苯的三元共聚物,无需使用氯甲醚即可实现树脂的胺化,得到增强型吸附树脂材料。
8.2.技术方案
9.为了解决上述问题,本发明所采用的技术方案如下:
10.本发明提供了一种增强型吸附树脂材料的制备方法,该方法将功能单体n-乙烯基吡咯烷酮(nvp)、甲基丙烯酸缩水甘油酯(gma)、二乙烯苯(dvb)和链转移剂、引发剂和致孔剂配制成三份油相,各油相的功能单体会在聚合初期形成的寡聚物,通过改变油相中各功能单体、链转移剂的比例与聚合温度,控制寡聚物在各个聚合阶段以不同的单体比、聚合度参与聚合反应,得到具有多层结构的n-乙烯基吡咯烷酮、甲基丙烯酸缩水甘油酯和二乙烯苯的三元共聚物,即树脂白球;再经胺化试剂胺化,得到了具有离子交换功能的树脂材料,使用该工艺合成的树脂白球,无需使用氯甲醚即可实现树脂的胺化。
11.优选地,上述一种增强型吸附树脂材料的制备方法包括如下步骤:
12.s1:配制分散相,向超纯水中加入分散剂、乳化剂并混合均匀,其中分散剂用量为超纯水质量的0.1%~5%,乳化剂用量为超纯水质量的0.01%~2%;
13.s2:配制第一油相,将功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮、二乙烯苯、链转移剂、引发剂、致孔剂混合均匀,其中所述功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为1:1:(0.1~10),链转移剂质量占上述功能单体总质量的0~10%,引发剂占上述功能单体总质量的1~20%,致孔剂占上述功能单体总质量1~30%;
14.s3:配制第二油相,将功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮、交联剂二乙烯苯、链转移剂、引发剂、致孔剂混合均匀,其中所述功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为1:(0.1~10):3,链转移剂质量占上述功能单体总质量的0.1~10%,引发剂占上述功能单体总质量的1~20%,致孔剂占上述功能单体总质量1~30%;
15.s4:配制第三油相,将功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮、交联剂二乙烯苯、链转移剂、引发剂、致孔剂混合均匀,其中所述功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为(0.1~10):1:3,链转移剂质量占上述功能单体总质量的0.1~10%,引发剂占上述功能单体总质量的1~20%,致孔剂占上述功能单体总质量1~30%;
16.s5:制备白球,将分散相加热至60
±
2℃,在100~1000rpm的搅拌下加入第一油相,升温至80
±
2℃反应1h;降温至60
±
2℃后,在100~1000rpm的搅拌下加入第二油相,预聚20~120min后升温至80
±
2℃反应1h;降温至60
±
2℃后,在100~1000rpm的搅拌下加入第三油相,预聚20~120min后升温至80
±
2℃反应3h,得到白球,上述温度均为反应体系温度而非加热体系温度;
17.s6:制备功能树脂材料,s5中制备的白球加入到胺化试剂中,反应温度为55~100
℃,在400~1000rpm的搅拌下反应4~24h,得到了具有离子交换功能的增强型吸附树脂材料。
18.优选地,上述步骤s1中,使用的分散剂为聚乙二醇、六偏磷酸钠、液体石蜡、聚乙烯吡咯烷酮、羟乙基纤维素中的一种或几种;使用的乳化剂为阿拉伯胶、明胶、吐温80、十二烷基硫酸钠、十二烷基苯磺酸钠、脂肪酸皂中的一种或几种。
19.优选地,上述步骤s1中,分散剂用量为超纯水质量的0.1%~4%。
20.优选地,上述步骤s2-s4中,链转移剂为s,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯、甲基(苯基)氨基二硫代甲酸氰甲酯、2-(十二烷基硫基硫代羰基硫基)-2-甲基丙酸、4-氰基-4-[[(十二烷基硫基)硫代羰基]硫基]戊酸、双(硫代苯甲酰基)二硫醚中的一种或几种。
[0021]
优选地,上述步骤s2-s4中,引发剂为偶氮二异丁腈、偶氮二异庚腈、偶氮二异丁酸二甲酯、过氧化二苯甲酰、过氧化苯甲酰、过硫酸铵中的一种或几种。
[0022]
优选地,上述步骤s2-s4中,致孔剂为甲苯、二甲苯、乙酸乙酯、庚烷、正己烷中的一种或几种。
[0023]
优选地,上述步骤s2-s4中,引发剂占功能单体总质量的1~5%。
[0024]
优选地,上述步骤s2-s4中,致孔剂占功能单体总质量10~30%。
[0025]
优选地,上述步骤s2~s4中,第一油相、第二油相、第三油相中功能单体总质量的比为(0.1~10):(0.1~10):1。
[0026]
优选地,上述步骤s2~s4中,第一油相、第二油相、第三油相中功能单体总质量的比为3:2:2。
[0027]
优选地,上述步骤s2中,功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为1:1:(4~8),更优选为1:1:6。
[0028]
优选地,上述步骤s2中,链转移剂质量占功能单体总质量的0~5%。
[0029]
优选地,上述步骤s3中,功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为1:(1~4):3。
[0030]
优选地,上述步骤s3中,链转移剂质量占功能单体总质量的0.1~5%。
[0031]
优选地,上述步骤s4中,功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为(0.5~3):1:3。
[0032]
优选地,上述步骤s4中,链转移剂质量占功能单体总质量的0.1~5%;
[0033]
优选地,上述步骤s5中,在加入第一油相时的搅拌速度为800rpm,在加入第二油相时的搅拌速度为650rpm,在加入第三油相时的搅拌速度为600rpm。
[0034]
优选地,上述步骤s6中,所选的胺化试剂为二乙胺、三乙胺、二甲胺、三甲胺、二甲基丁胺中的一种或几种。
[0035]
优选地,上述步骤s6中,白球与胺化试剂的质量比为1:(2~4)。更进一步地,白球与胺化试剂的质量比为1:3。
[0036]
优选地,上述增强型吸附树脂材料平均粒径为30~100μm,比表面积为400~1200m2/g,平均孔径为1~100nm,接触角小于90
°
,交换容量为0.1~8mmol/g。
[0037]
本发明还提供了一种增强型吸附树脂材料,通过上述一种增强型吸附树脂的制备方法制备而成。
[0038]
优选地,上述的增强型吸附树脂材料平均粒径为30~100μm,比表面积为400~1200m2/g,平均孔径为1~100nm,接触角小于90
°
,离子交换容量为0.1~8mmol/g。
[0039]
本发明还提供了上述一种增强型吸附树脂材料的制备方法和/或一种增强型吸附树脂材料在去除水体中的ppcps中的应用。
[0040]
3.有益效果
[0041]
本发明与现有技术相比,其有益效果在于:
[0042]
(1)本发明提供的一种增强型吸附树脂材料的制备方法,以gma、nvp和dvb为功能单体,将包括上述功能单体的反应物组成的油相分为三份,每份油相根据聚合阶段和目的的不同调控链转移剂、功能单体、交联剂的比例,再通过改进的悬浮聚合工艺控制寡聚物在各个聚合阶段以不同的单体比、聚合度参与聚合反应,使得聚合反应开始后,聚合物链以寡聚物的形式存在,寡聚物由部分疏水性交联剂二乙烯苯和具有极性的单体gma、nvp组成,具有了一定的疏水性而在水体中形成胶束,疏水效应使得在聚合过程中小分子量的寡聚物不断被更大的油滴(疏水相)吸收,从而参与进一步反应,有效的避免了各功能单体较大的极性差异而导致的聚合反应不可控的问题。
[0043]
(2)本发明提供的一种增强型吸附树脂材料的制备方法,根据改进的悬浮聚合工艺控制寡聚物在各个聚合阶段以不同的单体比、聚合度,制备了具有多层结构的n~乙烯基吡咯烷酮、甲基丙烯酸缩水甘油酯和二乙烯苯的三元共聚物,利用gma结构中环氧基团的开环反应,无需使用氯甲醚,即可成功将离子交换基团枝接入树脂结构,通过胺化反应得到了阴离子强化的亲水性增强型吸附树脂材料,制备方法绿色环保。
[0044]
(3)本发明提供的一种增强型吸附树脂材料,具有多层结构特征,树脂外层的gma的含量较高,具有较大的孔径,有效的提高了树脂的传质效率,缓解了树脂平均孔径较小所带来的孔道堵塞问题;内层的dvb含量较高,具有较大比表面积和孔道结构,提高了树脂的吸附容量,即可以通过调节树脂中功能单体gma的含量,来调控离子交换容量,性能稳定,重复性好。
附图说明
[0045]
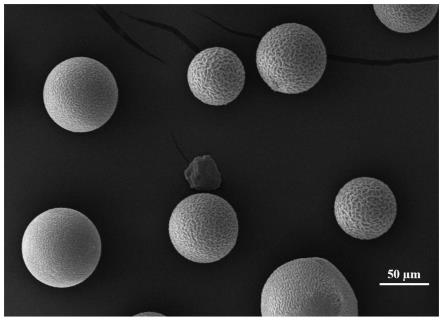
图1为本发明实施例1中制备的白球扫描电镜图(50μm);
[0046]
图2为本发明实施例1中制备的白球扫描电镜图(10μm);
[0047]
图3为本发明实施例1中制备的白球红外光谱图。
具体实施方式
[0048]
下面结合具体实施例对本发明进一步进行描述。
[0049]
需要说明的是,本说明书中所引用的如“上”、“下”、“左”、“右”、“中间”等用语,亦仅为便于叙述的明了,而非用以限定可实施的范围,其相对关系的改变或调整,在无实质变更技术内容下,当亦视为本发明可实施的范畴。
[0050]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同;本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0051]
实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂
或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0052]
如本文所使用,术语“约”用于提供与给定术语、度量或值相关联的灵活性和不精确性。本领域技术人员可以容易地确定具体变量的灵活性程度。
[0053]
如本文所使用,术语“......中的至少一个”旨在与“......中的一个或多个”同义。例如,“a、b和c中的至少一个”明确包括仅a、仅b、仅c以及它们各自的组合。
[0054]
浓度、温度、量和其他数值数据可以在本文中以范围格式呈现。应当理解,这样的范围格式仅是为了方便和简洁而使用,并且应当灵活地解释为不仅包括明确叙述为范围极限的数值,而且还包括涵盖在所述范围内的所有单独的数值或子范围,就如同每个数值和子范围都被明确叙述一样。例如,约1至约4.5的数值范围应当被解释为不仅包括明确叙述的1至约4.5的极限值,而且还包括单独的数字(诸如2、3、4)和子范围(诸如1至3、2至4等)。相同的原理适用于仅叙述一个数值的范围,诸如“小于约4.5”,应当将其解释为包括所有上述的值和范围。此外,无论所描述的范围或特征的广度如何,都应当适用这种解释。
[0055]
任何方法或过程权利要求中所述的任何步骤可以以任何顺序执行,并且不限于权利要求中提出的顺序。
[0056]
实施例1
[0057]
本实施例提供一种增强型吸附树脂的制备方法及其制备的吸附树脂材料,该方法包括如下步骤:
[0058]
s1:配置分散相,将0.8g聚乙烯吡咯烷酮,0.3g羟乙基纤维素,0.3g明胶和0.15g十二烷基硫酸钠加入500ml超纯水中作为分散相,在60℃下混合均匀;
[0059]
s2:配置第一油相,将3g甲基丙烯酸缩水甘油酯,3gn-乙烯基吡咯烷酮,18g二乙烯苯,0.96g偶氮二异丁腈(引发剂),4.8g甲苯(致孔剂)混合均匀组成第一油相;
[0060]
s3:配置第二油相,将3.2g甲基丙烯酸缩水甘油酯,3.2gn-乙烯基吡咯烷酮,9.6g二乙烯苯,0.64g偶氮二异丁腈(引发剂),0.64gs,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯(链转移剂),0.64g甲基(苯基)氨基二硫代甲酸氰甲酯(链转移剂),4.8g甲苯(致孔剂)混合均匀组成第二油相;
[0061]
s4:配置第三油相,将5.33g甲基丙烯酸缩水甘油酯,2.67gn-乙烯基吡咯烷酮,8g二乙烯苯,0.64g偶氮二异丁腈(引发剂),0.533gs,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯(链转移剂),1.066g甲基(苯基)氨基二硫代甲酸氰甲酯(链转移剂),4.8g甲苯(致孔剂)混合均匀组成第三油相;
[0062]
s5:制备白球,将分散相体系温度升至80℃后,将第一油相逐滴加入分散相中反应1h,维持搅拌速度为800rpm;将体系温度由80℃降至60℃后,将第二油相逐滴加入并预聚30min,之后再将体系升温至80℃反应1h,期间维持搅拌速度为650rpm;当体系温度降至60℃后,将第三油相逐滴加入并预聚30min,之后将体系升温至80℃反应3h得到成品白球,期间维持搅拌速度为600rpm;
[0063]
s6:制备功能树脂材料,取一定量的上述白球按照质量比1:3的比例加入三甲胺中,在60℃、400rpm的条件下反应24小时,得到增强型吸附树脂材料。
[0064]
本实施例制备的白球扫描电镜图如图1和图2所示,微球表面粗糙,成孔良好,有利于物质在微球表面进行传质;白球红外光谱图如图3所示,gma-nvp-dvb三元聚合物在1733.7和1687.4处均有明显的特征峰,系丙烯酸缩水甘油酯中c=o基在1770~1720之间的
伸缩振动和nvp中的c=o基在1700~1630之间的伸缩振动,表明三元聚合物形成;元素分析结果表明,白球的n元素含量为0.91%;进一步对得到的增强型吸附树脂材料进行表征:其静态接触角为74.54
°
,离子交换容量为2.26mmol/g,粒径分布为30~60μm,比表面积为854m2/g,孔径分布为1~100nm。
[0065]
实施例2
[0066]
本实施例提供一种增强型吸附树脂的制备方法及其制备的吸附树脂材料,其它条件与实施例1中的制备方法相同,不同之处在于,调整了第三油相中单体的比例,单体配比与树脂表征结果见表1。
[0067]
表1第三油相单体配比与树脂表征结果
[0068][0069]
实施例3
[0070]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第一油相中,链转移试剂s,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯的添加量为0.2g,甲基(苯基)氨基二硫代甲酸氰甲酯的添加量为0.2g。
[0071]
此方法制备的白球n元素含量为0.93%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为77.31
°
,离子交换容量为1.68mmol/g,粒径分布为30~100μm,比表面积为797m2/g,孔径分布为1~100nm。
[0072]
实施例4
[0073]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第二油相中,链转移试剂为s,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯和2-(十二烷基硫基硫代羰基硫基)-2-甲基丙酸,添加量分别为0.64g和0.64g。
[0074]
此方法制备的白球n元素含量为0.64%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为80.94
°
,离子交换容量为1.81mmol/g,粒径分布为30~100μm,比表面积为777m2/g,孔径分布为1~100nm。
[0075]
实施例5
[0076]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第二油相中,链转移试剂为s,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯和4-氰基-4-[[(十二烷基硫基)硫代羰基]硫基]戊酸,添加量分别为0.64g和0.64g。
[0077]
此方法制备的白球n元素含量为0.74%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为75.4
°
,离子交换容量为1.71mmol/g,粒径分布为30~100μm,比表面积
为711m2/g,孔径分布为1~100nm。
[0078]
实施例6
[0079]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第二油相中,链转移试剂为双(硫代苯甲酰基)二硫醚和甲基(苯基)氨基二硫代甲酸氰甲酯,添加量分别为0.56g和0.56g。
[0080]
此方法制备的白球n元素含量为0.45%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为76.81
°
,离子交换容量为1.8mmol/g,粒径分布为30~100μm,比表面积为871m2/g,孔径分布为1~100nm。
[0081]
实施例7
[0082]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第三油相中,链转移试剂为s,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯和2-(十二烷基硫基硫代羰基硫基)-2-甲基丙酸,添加量分别为0.533g和1.066g。
[0083]
此方法制备的白球n元素含量为0.69%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为86.18
°
,离子交换容量为1.12mmol/g,粒径分布为30~100μm,比表面积为814m2/g,孔径分布为1~100nm。
[0084]
实施例8
[0085]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第三油相中,链转移试剂为s,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯和4-氰基-4-[[(十二烷基硫基)硫代羰基]硫基]戊酸,添加量分别为0.533g和1.066g。
[0086]
此方法制备的白球n元素含量为0.82%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为77.24
°
,离子交换容量为1.49mmol/g,粒径分布为30~100μm,比表面积为864m2/g,孔径分布为1~100nm。
[0087]
实施例9
[0088]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第三油相中,链转移试剂为双(硫代苯甲酰基)二硫醚和甲基(苯基)氨基二硫代甲酸氰甲酯,添加量分别为0.533g和1.066g。
[0089]
此方法制备的白球n元素含量为0.76%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为86.18
°
,离子交换容量为1.74mmol/g,粒径分布为30~100μm,比表面积为752m2/g,孔径分布为1~100nm。
[0090]
实施例10
[0091]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,第三油相由2.67g甲基丙烯酸缩水甘油酯,1.33gn-乙烯基吡咯烷酮,4g二乙烯苯,0.32g偶氮二异丁腈(引发剂),0.267gs,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯(链转移剂),0.533g甲基(苯基)氨基二硫代甲酸氰甲酯(链转移剂),2.4g甲苯(致孔剂)组成。
[0092]
此方法制备的白球n元素含量为0.64%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为83.31
°
,离子交换容量为1.91mmol/g,粒径分布为30~100μm,比表面积为848m2/g,孔径分布为1~100nm。
[0093]
实施例11
[0094]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之
处在于,第二油相加入体系后预聚时间为60min;第三油相加入体系后预聚时间为60min。将分散相体系温度升至80℃后,将第一油相逐滴加入分散相中反应1h,维持搅拌速度为100~1000rpm;将体系温度由80℃降至60℃后,将第二油相逐滴加入并预聚60min,之后再将体系升温至80℃反应1h,期间维持搅拌速度为100~1000rpm;当体系温度降至60℃后,将第三油相逐滴加入并预聚60min,之后将体系升温至80℃反应3h得到成品白球,期间维持搅拌速度为100~1000rpm。
[0095]
此方法制备的白球n元素含量为0.55%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为77.1
°
,离子交换容量为1.88mmol/g,粒径分布为30~60μm,比表面积为603m2/g,孔径分布为1~100nm。
[0096]
实施例12
[0097]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,第二油相加入体系后预聚时间为120min;第三油相加入体系后预聚时间为120min。
[0098]
此方法制备的白球n元素含量为0.71%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为84.28
°
,离子交换容量为1.39mmol/g,粒径分布为30~60μm,比表面积为598m2/g,孔径分布为1~100nm。
[0099]
对比例1
[0100]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,仅添加了第一油相而未添加第二油相和第三油相。
[0101]
此方法制备的白球n元素含量为0.16%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为99.17
°
,离子交换容量为0.1mmol/g,粒径分布为1~30μm,比表面积为1183m2/g,孔径分布为1~10nm。树脂材料的接触角较大,亲水性不足,判断树脂主要由疏水单体dvb自聚而成。同时未获得目标粒径分布和孔径分布的树脂材料,聚合失败。
[0102]
对比例2
[0103]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,仅添加了第一油相和第二油相而未添加第三油相。
[0104]
此方法制备的白球n元素含量为0.63%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为83.61
°
,离子交换容量为0.31mmol/g,粒径分布为1~30μm,比表面积为891m2/g,孔径分布为1~10nm。树脂材料的粒径分布和孔径分布不合预期,聚合失败。
[0105]
对比例3
[0106]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第二油相中没有添加链转移剂。
[0107]
此方法制备的白球n元素含量为0.21%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为101.22
°
,离子交换容量为0.07mmol/g,粒径分布为1~30μm,比表面积为1100m2/g,孔径分布为1~10nm。树脂的接触角较大,亲水性不足,判断树脂主要由疏水单体dvb自聚而成,功能单体nvp、gma未充分参与反应,聚合失败。
[0108]
对比例4
[0109]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第三油相中没有添加链转移剂。
[0110]
此方法制备的白球n元素含量为0.68%;进一步对得到的增强型吸附树脂进行表
征:其静态接触角为93.71
°
,离子交换容量为0.55mmol/g,粒径分布为1~50μm,比表面积为900m2/g,孔径分布为1~10nm。判断树脂主要由dvb与部分亲水性单体nvp共聚,gma未充分参与反应,亲水性较差的同时粒径分布不符预期,聚合失败。
[0111]
对比例5
[0112]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第二油相和第三油相中均没有添加链转移剂。
[0113]
此方法制备的白球n元素含量为0.14%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为96.33
°
,离子交换容量为0.13mmol/g,粒径分布为1~30μm,比表面积为1037m2/g,孔径分布为1~10nm。树脂的接触角较大,亲水性不足,判断树脂主要由疏水单体dvb自聚而成,功能单体nvp、gma未充分参与反应,聚合失败。
[0114]
对比例6
[0115]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在第一油相、第二油相和第三油相中均没有添加功能单体gma。
[0116]
此方法制备的白球n元素含量为0.74%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为78.49
°
,离子交换容量为0.06mmol/g,粒径分布为1~50μm,比表面积为810m2/g,孔径分布为1~10nm。树脂材料的离子容量较低且未能获得目标孔径分布和粒径分布,聚合失败。
[0117]
对比例7
[0118]
本实施例中制备增强型吸附树脂,其它条件与实施例1中的制备方法相同,不同之处在于,在分别加入第二油相和第三油相后,没有在60℃进行预聚,而是直接升温至80℃进行反应。
[0119]
此方法制备的白球n元素含量为0.24%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为97.59
°
,离子交换容量为0.18mmol/g,粒径分布为1~50μm,比表面积为920m2/g,孔径分布为1~10nm。树脂的接触角较大,亲水性不足,判断树脂主要由疏水单体dvb自聚而成,功能单体nvp、gma未充分参与反应,聚合失败。
[0120]
对比例8
[0121]
本实施例中制备增强型吸附树脂,其它条件与实施例1中白球的制备方法相同,不同之处在于,在分别加入第二油相和第三油相后,在70℃的温度下进行了预聚。
[0122]
此方法制备的白球n元素含量为0.34%;进一步对得到的增强型吸附树脂进行表征:其静态接触角为94.67
°
,离子交换容量为1.27mmol/g,粒径分布为1~100μm,比表面积为310m2/g,孔径分布为1~50nm。树脂的粒径分布过于分散,亲水性不足,比表面积较小,离子交换容量较少聚合失败。
[0123]
以上内容是对本发明及其实施方式进行了示意性的描述,该描述没有限制性,实施例中所示的也只是本发明的实施方式之一,实际的实施方式并不局限于此。按照技术方案中的试剂和比例,如分散相中分散剂用量为超纯水质量的0.1%~5%,乳化剂用量为超纯水质量的0.01%~2%;第一油相中功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为1:1:(0.1~10),链转移剂质量占上述功能单体总质量的0~10%,引发剂占上述功能单体总质量的1~20%,致孔剂占上述功能单体总质量1~30%;第二油相中功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为1:(0.1~
10):3,链转移剂质量占上述功能单体总质量的0.1~10%,引发剂占上述功能单体总质量的1~20%,致孔剂占上述功能单体总质量1~30%;第三油相中功能单体甲基丙烯酸缩水甘油酯、n-乙烯基吡咯烷酮和二乙烯苯的质量比为(0.1~10):1:3,链转移剂质量占上述功能单体总质量的0.1~10%,引发剂占上述功能单体总质量的1~20%,致孔剂占上述功能单体总质量1~30%;第一油相、第二油相、第三油相中功能单体总质量的比为(0.1~10):(0.1~10):1;分散剂为聚乙二醇、六偏磷酸钠、液体石蜡、聚乙烯吡咯烷酮、羟乙基纤维素中的一种或几种;乳化剂为阿拉伯胶、明胶、吐温80、十二烷基硫酸钠、十二烷基苯磺酸钠、脂肪酸皂中的一种或几种;链转移剂为s,s
’‑
二(α,α
’‑
二甲基-α
”‑
乙酸)三硫代碳酸酯、甲基(苯基)氨基二硫代甲酸氰甲酯、2-(十二烷基硫基硫代羰基硫基)-2-甲基丙酸、4-氰基-4-[[(十二烷基硫基)硫代羰基]硫基]戊酸、双(硫代苯甲酰基)二硫醚中的一种或几种;引发剂为偶氮二异丁腈、偶氮二异庚腈、偶氮二异丁酸二甲酯、过氧化二苯甲酰、过氧化苯甲酰、过硫酸铵中的一种或几种;致孔剂为甲苯、二甲苯、乙酸乙酯、庚烷、正己烷中的一种或几种;胺化试剂为二乙胺、三乙胺、二甲胺、三甲胺、二甲基丁胺中的一种或几种;白球与胺化试剂的质量比为1:(2~4)时,同样能够制备平均粒径为30~100μm,比表面积为400~1200m2/g,平均孔径为1~100nm,接触角小于90
°
,交换容量为0.1~8mmol/g的增强型吸附树脂。所以,如果本领域的普通技术人员受其启示,在不脱离本发明创造宗旨的情况下,不经创造性的设计出与该技术方案相似的实施方式及实施例,均应属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1