去除细胞器基因组污染序列的染色体组装方法及装置
1.本发明涉及一种染色体组装方法,具体涉及一种构建本地的细胞器数据库并利用该数据库对样品进行检测并去细胞器基因组污染序列的染色体组装方法及装置。
背景技术:
2.染色体组装目前主要依赖于三代测序、二代测序以及hi-c测序混合组装。三代测序的序列长度非常长,通常单个分子被测序的长度可以达到100kb个碱基,因此,通常被用来组装连续交叠群(contig)水平的基因组。但是由于三代测序的错误率高达10%,因此,三代序列组装的contig水平基因组需要使用测序准确率更高的二代测序序列对其进行纠错。纠错后的基因组仍然属于contig水平基因组,组成基因组的序列片段数量仍然远远多于真实染色体数量,因此,需要利用hi-c测序序列将不同contig按照相应染色体进行聚类,并最终组装成相应的染色体。
3.细胞内除了细胞核中包含染色体dna序列,细胞器,如线粒体和叶绿体内同样包含相应的细胞器基因组dna序列。细胞器基因组通常呈环状存在,每个细胞内包含多个细胞器,而每个细胞器内通常包含多个细胞器基因组,因此,细胞器基因组在细胞内拷贝数远远多于染色体拷贝数。在进行基因组测序之前,需要先提取总的dna,然后构建测序文库并进行测序。由于提取的总dna中同时包含了细胞器基因组和染色体序列,因此,最终的测序结果中同时包含细胞器基因组序列和染色体序列。在进行染色体组装过程中,经过分析发现,当未提前将细胞器基因组从基因组序列中去除时,可能会最终导致细胞器基因组被错误地组装进染色体,从而导致染色体组装错误。发明人针对上述问题开创性地提出了新的染色体组装策略,并进行了进一步的研究,从而完成了本发明。
技术实现要素:
4.本发明的目的在于,提供一种去除细胞器基因组污染序列的染色体组装方法,该方法利用本地构建的细胞器数据库,在组装染色体之前,通过比较基因组方法,去除细胞器基因组序列,以减少细胞器基因组序列对于染色体组装的准确性的影响,提高染色体组装的准确性。
5.本发明的另目的在于,提供一种去除细胞器基因组污染序列的染色体组装装置,该装置利用本地构建的细胞器数据库,在组装染色体之前,通过比较基因组方法,去除细胞器基因组序列,以减少细胞器基因组序列对于染色体组装的准确性的影响,提高染色体组装的准确性。
6.根据本发明的一个实施方式,提供一种去除细胞器基因组污染序列的染色体组装方法,其可包括如下步骤:步骤s1,对样品进行二代测序、三代测序以及hi-c测序;步骤s2,对三代测序数据进行基因组组装;步骤s3,分别利用二代测序序列和三代测序序列对在所述步骤s2中得到的基因组进行纠错,并去除经纠错的基因组中的单倍体序列;步骤s4,利用细胞器数据库对在所述步骤s3中得到的基因组进行比对,鉴定并去除细胞器基因组序列;
步骤s5,将hi-c测序序列比对到在所述步骤s4中得到的基因组;以及步骤s6,利用在所述步骤s5中得到的比对结果,将在所述步骤s4中得到的基因组最终组装成染色体。
7.作为一个实施方式,所述细胞器数据库可以是通过如下步骤构建的:步骤s7,获取现有的线粒体和叶绿体的基因信息;步骤s8,根据在所述步骤s7中得到的信息,从不同物种的线粒体和叶绿体的蛋白编码基因中筛选出50%以上的物种中都存在的编码基因作为核心编码基因,再对每个核心编码基因分别按照95%的相似度进行聚类从包含两条以上的序列的类群中选择一条序列作为代表序列,组成最终的核心编码基因数据库;步骤s9,根据在所述步骤s7中得到的信息,对线粒体与叶绿体的基因组进行比对,去除错误组装的线粒体和叶绿体的基因组,剩下的序列组成细胞器基因组数据库;以及步骤s10,利用所述核心编码基因数据库和所述细胞器基因组数据库组成所述细胞器数据库。
8.作为一个实施方式,所述核心编码基因数据库可包括线粒体核心编码基因数据库和叶绿体核心编码基因数据库。
9.作为一个实施方式,所述线粒体核心编码基因数据库可包括动物线粒体核心编码基因数据库、植物线粒体核心编码基因数据库、真菌线粒体核心编码基因数据库以及原生生物线粒体核心编码基因数据库。
10.作为一个实施方式,所述步骤s4中可包括如下步骤:步骤s41,将在所述步骤s2中得到的基因与所述细胞器基因组进行比对,将比对长度大于1000bp的序列提取出来;步骤s42,将在所述步骤s41中提取出的序列与所述核心编码基因进行比对,将比对到核心编码基因的序列提取出来,根据与基因组序列比对到的长度以及比对到的核心编码基因数量,最终从基因组序列中鉴定出细胞器基因组序列;以及步骤s43,将在所述步骤s42中提取出的序列从在所述步骤s3中得到的基因组中去除。
11.根据本发明的另一实施方式,提供一种去除细胞器基因组污染序列的染色体组装装置,其可包括:细胞器数据库,包括核心编码基因数据库和细胞器基因组数据库;测序模块,对样品进行二代测序、三代测序以及hi-c测序;基因组组装模块,对三代测序数据进行基因组组装;纠错模块,分别利用二代测序序列和三代测序序列对由所述基因组组装模块组装的基因组进行纠错,并去除经纠错的基因组中的单倍体序列;细胞器基因组去除模块,利用所述细胞器数据库对经所述纠错模块处理的基因组进行比对,鉴定并去除细胞器基因组序列;比对模块,将hi-c测序序列比对到经所述细胞器基因组去除模块处理的基因组;以及染色体组装模块,利用通过所述比对模块得到的比对结果,将经所述细胞器基因组去除模块处理的基因组最终组装成染色体。
12.作为一个实施方式,所述核心编码基因数据库根据现有的线粒体和叶绿体的基因信息,从不同物种的线粒体和叶绿体的蛋白编码基因中筛选出50%以上的物种中都存在的编码基因作为核心编码基因,再对每个核心编码基因分别按照95%的相似度进行聚类并从包含两条以上的序列的类群中选择一条序列作为代表序列,从而组合而成,细胞器基因组数据库根据现有的线粒体和叶绿体的基因信息,对线粒体与叶绿体的基因组进行比对,去除错误组装的线粒体和叶绿体的基因组,并剩下的序列组合而成。
13.作为一个实施方式,所述核心编码基因数据库可包括线粒体核心编码基因数据库和叶绿体核心编码基因数据库。
14.作为一个实施方式,所述线粒体核心编码基因数据库可包括动物线粒体核心编码
基因数据库、植物线粒体核心编码基因数据库、真菌线粒体核心编码基因数据库以及原生生物线粒体核心编码基因数据库。
15.作为一个实施方式,所述细胞器基因组去除模块将由所述基因组组装模块组装的基因与所述细胞器基因组进行比对,将比对长度大于1000bp的序列提取出来,将所提取出的序列与所述核心编码基因进行比对,并将比对到核心编码基因的序列提取出来并从经纠错模块处理的基因组中去除。
16.本发明的去除细胞器基因组污染序列的染色体组装方法利用多种方法进行基因组测序,并且构建并利用细胞器数据库,在组装染色体之前,预先去除基因组序列中混杂的细胞器基因组序列,减少了细胞器基因组序列对于染色体组装的准确性的影响。
17.本发明的去除细胞器基因组污染序列的染色体组装装置利用多种方法进行基因组测序,并且构建并利用细胞器数据库,在组装染色体之前,预先去除基因组序列中混杂的细胞器基因组序列,减少了细胞器基因组序列对于染色体组装的准确性的影响。
附图说明
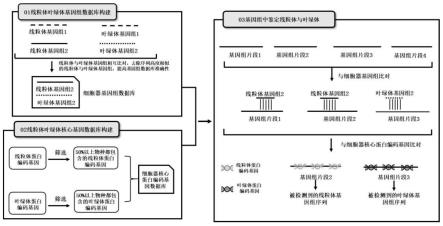
18.图1是本发明的一个实施方式的去除细胞器基因组污染序列的染色体组装方法的概略流程图。
19.图2是本发明的一个实施方式的构建本地的细胞器数据库以及鉴定并去除细胞器基因组的流程图。
20.图3是示出本发明的一实施例的鉴定到的河豚的基因组片段名称以及比对到细胞器基因组中的总长度的图表。
21.图4是示出本发明的一实施例的tig00001732_pilon_pilon片段与核心编码基因比对结果的图表。
22.图5是示出本发明的一实施例的河豚中鉴定到的线粒体基因组片段tig00001732_pilon_pilon与基因组平均测序深度比较结果的图表。
23.图6是示出本发明的一实施例的当未去除tig00001732_pilon_pilon片段时的染色体错误组装结果的图表。
24.图7是本发明的一个实施方式的去除细胞器基因组污染序列的染色体组装装置的示意图。
25.附图标记
26.1:去除细胞器基因组污染序列的染色体组装装置;11:细胞器数据库;12:测序模块;13:基因组组装模块;14:纠错模块;15:细胞器基因组去除模块;16:比对模块;17:染色体组装模块。
具体实施方式
27.以下,参照附图对本发明进行详细说明,但是本发明不限于附图和以下描述内容,本领域技术人员能够在本发明的技术思想的范围内进行多种修改和变更,并且这些修改和变更均属于本发明的权利范围。
28.另外,当对于本领域公知常识的描述可能会混淆本发明的技术要点时,省略对其说明。未记载于此的内容属于本领域技术人员能够推导出的内容。
29.图1是本发明的一个实施方式的去除细胞器基因组污染序列的染色体组装方法的概略流程图。图2是本发明的一个实施方式的构建本地的细胞器数据库的流程图。
30.如图1至图2所示,本发明提供一种去除细胞器基因组污染序列的染色体组装方法,其可包括如下步骤:步骤s1,对样品进行二代测序、三代测序以及hi-c测序;步骤s2,对三代测序数据进行基因组组装;步骤s3,分别利用二代测序序列和三代测序序列对在所述步骤s2中得到的基因组进行纠错,并去除经纠错的基因组中的单倍体序列;步骤s4,利用细胞器数据库对在所述步骤s3中得到的基因组进行比对,鉴定并去除细胞器基因组序列;步骤s5,将hi-c测序序列比对到在所述步骤s4中得到的基因组;以及步骤s6,利用在所述步骤s5中得到的比对结果,将在所述步骤s4中得到的基因组最终组装成染色体。
31.其中,作为一个实施方式,在所述步骤s2中,可使用canu软件对三代测序数据进行基因组组装;在所述步骤s3中,可使用pilon软件和racon软件分别利用二代测序序列和三代测序序列对在所述步骤s2中得到的基因组进行纠错,并利用purge_dups软件去除经纠错的基因组中的单倍体序列;在所述步骤s5中,可通过hic-pro软件将hi-c测序序列比对到在所述步骤s4中得到的基因组;在步骤s6中,可通过allhic软件利用在所述步骤s5中得到的比对结果,将在所述步骤s4中得到的基因组最终组装成染色体。上述步骤也可利用本领域常用的其他的软件进行,本发明不限于此。
32.具体地,作为一个实施方式,所述细胞器数据库可以是通过如下步骤构建的:步骤s7,获取现有的线粒体和叶绿体的基因信息;步骤s8,根据在所述步骤s7中得到的信息,从不同物种的线粒体和叶绿体的蛋白编码基因中筛选出50%以上的物种中都存在的编码基因作为核心编码基因,再对每个核心编码基因分别按照95%的相似度进行聚类并从包含两条以上的序列的类群中选择一条序列作为代表序列,组成最终的核心编码基因数据库;步骤s9,根据在所述步骤s7中得到的信息,对线粒体与叶绿体的基因组进行比对,去除错误组装的线粒体和叶绿体的基因组,剩下的序列组成细胞器基因组数据库;以及步骤s10,利用所述核心编码基因数据库和所述细胞器基因组数据库组成所述细胞器数据库。
33.其中,如表1所示,基于在所述步骤s7中得到的现有的线粒体和叶绿体的基因信息,对98%、90%、80%、70%、60%、50%、40%以及30%的物种中存在的细胞器基因数量进行了统计。经确认,从提取50%的物种中存在的细胞器基因作为核心基因开始,核心基因的数量趋于稳定,因此,在所述步骤s8中,将从不同物种的线粒体和叶绿体的蛋白编码基因中筛选出50%以上的物种中都存在的编码基因作为核心编码基因。然后,以本领域常规使用的cd-hit软件中的作为默认值的95%的相似度对每个核心编码基因进行聚类。为了提高核心基因序列的准确性,从包含两条以上的序列的类群中选择一条序列作为代表序列,从而组成了最终的核心编码基因数据库。
34.表1:
35.[0036][0037]
另外,作为一个实施方式,所述现有的线粒体和叶绿体的基因信息可从美国国家生物技术信息中心(national center for biotechnology information;ncbi)获取,也可使用本领域常用的其他的数据库资源,本发明不限于此。然而,ncbi中的细胞器基因组序列缺少人工校验,可能存在错误组装的细胞器基因组,因此,所述步骤s9中的去除错误组装的线粒体和叶绿体的基因组的环节是必不可少的。
[0038]
具体地,作为一个实施方式,在所述步骤s8中,构建细胞器数据库代码create_organelle_db.py如表2所示。
[0039]
表2:
[0040]
[0041]
[0042]
[0043]
[0044]
[0045]
[0046]
[0047]
[0048]
[0049]
[0050][0051]
另一方面,作为一个实施方式,所述核心编码基因数据库可包括线粒体核心编码基因数据库和叶绿体核心编码基因数据库。
[0052]
其中,所述线粒体核心编码基因数据库可包括动物线粒体核心编码基因数据库、植物线粒体核心编码基因数据库、真菌线粒体核心编码基因数据库以及原生生物线粒体核心编码基因数据库。
[0053]
另外,作为一个实施方式,所述步骤s4中可包括如下步骤:步骤s41,将在所述步骤s2中得到的基因与所述细胞器基因组进行比对,将比对长度大于1000bp的序列提取出来;步骤s42,将在所述步骤s41中提取出的序列与所述核心编码基因进行比对,将比对到核心编码基因的序列提取出来,根据与基因组序列比对到的长度以及比对到的核心编码基因数量,最终从基因组序列中鉴定出细胞器基因组序列;以及步骤s43,将在所述步骤s42中提取出的序列从在所述步骤s3中得到的基因组中去除。
[0054]
其中,在基因组组装中,本领域通常将长度小于1000bp的contig视为组装不准确的序列,因此,在所述步骤s41中,将比对长度大于1000bp的序列提取出来,以作为潜在的细胞器基因组序列。
[0055]
具体地,作为一个实施方式,在所述步骤s4中,核心代码organelle_filter.py如表3所示。
[0056]
表3:
[0057]
[0058]
[0059]
[0060]
[0061]
[0062]
[0063][0064]
另一方面,本发明提供一种去除细胞器基因组污染序列的染色体组装装置1。如图7所示,去除细胞器基因组污染序列的染色体组装装置1可包括:细胞器数据库11,包括核心编码基因数据库和细胞器基因组数据库;测序模块12,对样品进行二代测序、三代测序以及hi-c测序;基因组组装模块13,对三代测序数据进行基因组组装;纠错模块14,分别利用二代测序序列和三代测序序列对由基因组组装模块13组装的基因组进行纠错,并去除经纠错的基因组中的单倍体序列;细胞器基因组去除模块15,利用细胞器数据库11对经纠错模块14处理的基因组进行比对,鉴定并去除细胞器基因组序列;比对模块16,将hi-c测序序列比对到经细胞器基因组去除模块15处理的基因组;以及染色体组装模块17,利用通过比对模块16得到的比对结果,将经细胞器基因组去除模块15处理的基因组最终组装成染色体。
[0065]
其中,作为一个实施方式,基因组组装模块13可使用canu软件对三代测序数据进行基因组组装;纠错模块14可使用pilon软件和racon软件分别利用二代测序序列和三代测序序列对由基因组组装模块13组装的基因组进行纠错,并利用purge_dups软件去除经纠错
的基因组中的单倍体序列;比对模块16可通过hic-pro软件将hi-c测序序列比对到经细胞器基因组去除模块15处理的基因组;染色体组装模块17可通过allhic软件利用通过比对模块16得到的比对结果,将经细胞器基因组去除模块15处理的基因组最终组装成染色体。上述操作也可利用本领域常用的其他的软件进行,本发明不限于此。
[0066]
另外,本发明的去除细胞器基因组污染序列的染色体组装方法中的关于细胞器数据库11的描述内容可以相同方式应用于本发明的去除细胞器基因组污染序列的染色体组装装置1中,因此,对该部分描述进行省略。
[0067]
另一方面,作为一个实施方式,细胞器基因组去除模块15将由基因组组装模块13组装的基因与细胞器基因组进行比对,将比对长度大于1000bp的序列提取出来,将所提取出的序列与核心编码基因进行比对,并将比对到核心编码基因的序列提取出来并从经纠错模块14处理的基因组中去除。
[0068]
实施例
[0069]
以下,参照图1至图7,通过一个实施例对本发明进行更加详细说明。
[0070]
如图1至图2所示,所述实施例可包括如下步骤:
[0071]
从ncbi下载河豚的二代测序、三代测序以及hi-c测序数据,其中,二代测序数据为56gb,三代测序数据为55gb,hi-c测序数据为55gb;
[0072]
通过canu软件对三代测序数据进行组装,得到初步组装好的基因组g1,其命令为:nohup canu-psample-dcanu_assemble_result genomesize=348m-pacbio tgs.fastq&,经过canu软件组装,得到445mb大小的基因组,共包含4957条基因组片段组成;
[0073]
使用racon软件利用tgs序列对基因组g1进行纠错,其命令为:nohup minimap2-ax map-pb-r2k genome1.fasta tgs.fastq|samtools sort|samtools view》dbg.srp.minimap2.sam&&racon tgs.fastq dbg.srp.minimap2.sam tgs_sequence_assembled_draft_genome.fasta》genome2.fasta&;
[0074]
使用pilon软件利用二代测序序列对基因组genome2.fasta进行纠错,最终获得二代测序序列纠错后的基因组genome3.fasta,其命令为:bwa index racon_correct_1.fasta&&bwa mem racon_correct_1.fasta ngs_1.fastq ngs_2.fastq|samtools view-bs-f 12|samtools sort》racon_bwa.sort.bam&&java-xmx250g-jar pilon-1.24.jar
‑‑
genome genome2.fasta
‑‑
frags racon_bwa.sort.bam
‑‑
output genome3.fasta
‑‑
changes;
[0075]
通过purge_dups软件根据测序深度在基因组中除去单体型序列,其命令为:minimap2-xmap-pb genome3.fasta tgs.fastq|gzip-c-》aligned.paf.gz&&pbcstat aligned.paf.gz&&calcuts pb.stat》cutoffs 2》calcults.log&&split_fa genome3.fasta》racon.pilon2.split&&minimap2-xasm5-dp racon.pilon2.split racon.pilon2.split|gzip-c-》racon.pilon2.split.self.paf.gz&&purge_dups-2-t cutoffs-c pb.base.covracon.pilon2.split.self.paf.gz》dups.bed 2》purge_dups.log&&get_seqs-edups.bed genome3.fasta&&cut-d""-f 1purged.fa》racon.pilon2.purged.fasta,终得到340m的基因组genome4.fasta,共包含1970条基因组片;
[0076]
使用organelle_filter.py脚本将基因组genome4.fasta与细胞器数据库进行比
对,并鉴定和去除细胞器基因组,得到去除细胞器基因组后的基因组genome5.fasta,其命令为:python organelle_filter.py organelles.csv 0.90true 1000 2genome4.fasta output,最终从基因组genome4.fasta中鉴定到一条线粒体基因组序列(tig00001732_pilon_pilon),该序列与线粒体基因组序列有32384个碱基相匹配,与13个线粒体核心编码基因相匹配,将tig00001732_pilon_pilon序列从基因组genome4.fasta中去除,得到新的基因组genome5.fasta;
[0077]
使用hic-pro软件将hi-c测序序列比对到基因组genome5.fasta,得到比对好的文件genome5.sam,其命令为:python hicpro_align.py hic_1.fastq hic_2.fastq mboi 60;
[0078]
使用allhic软件根据hic-pro比对结果genome5.sam将基因组genome5.fasta组装成染色体,其命令为:python allhic_assemble_tgs.py mboi 22 60。
[0079]
具体地,河豚基因组与细胞器基因组数据库中比对,共发现54条基因组片段与细胞器基因组中的序列比对长度超过1000bp。图3中示出了鉴定到的54条基因组片段名称以及比对到细胞器基因组中的总长度。
[0080]
将54条序列片段与细胞器核心基因数据库比对,将比对到核心编码基因的片段提取出来,最终发现tig00001732_pilon_pilon片段包含13个核心编码基因,因此tig00001732_pilon_pilon片段属于河豚的线粒体基因组。图4中示出了tig00001732_pilon_pilon片段比对到的13个核心编码基因。
[0081]
关于河豚中鉴定到的线粒体基因组片段tig00001732_pilon_pilon与基因组平均测序深度比较结果,如图5所示,细胞器基因组测序深度明显高于基因组平均测序深度,这个现象与细胞器基因组拷贝数多于染色体的事实相吻合,因此,可以进一步确认tig00001732_pilon_pilon片段就是河豚的线粒体基因组。
[0082]
如果不去除tig00001732_pilon_pilon片段,而是用所有的基因组片段进行染色体组装,如图6所示,tig00001732_pilon_pilon片段最终会被错误地组装到7号染色体上,因此,能够确认,在组装染色体之前,去除细胞器基因组污染序列是一个非常重要的步骤。
[0083]
经以上步骤,最终获得22条河豚染色体,总基因组大小为340mb,将22条河豚染色体与鉴定到的线粒体基因组(tig00001732_pilon_pilon)相比对可以确认,在22条河豚染色体已经没有了线粒体的序列。
[0084]
综上所述,本发明的去除细胞器基因组污染序列的染色体组装方法及装置利用多种方法进行基因组测序,并且构建并利用细胞器数据库,在组装染色体之前,预先去除基因组序列中混杂的细胞器基因组序列,减少了细胞器基因组序列对于染色体组装的准确性的影响。
[0085]
以上,对本发明的一个实施方式进行了详细说明,但本发明不仅限于此,本发明的范围仅由所附的权利要求定义,本发明的多种修改和变形均属于本发明的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1