一种吡唑取代的吡唑并嘧啶的TRK大环化合物和药物组合物及其应用
一种吡唑取代的吡唑并嘧啶的trk大环化合物和药物组合物及其应用
技术领域
1.本发明涉及生物医药领域,具体涉及一种吡唑取代的吡唑并嘧啶的trk大环化合物、一种含有吡唑取代的吡唑并嘧啶的trk大环化合物的药物组合物、前述含吡唑取代的吡唑并嘧啶的trk大环化合物及药物组合物的应用。
背景技术:
2.ntrk/trk(tropomyosin receptor kinase)为祌经营养因子酪氨酸受体,隶属于受体酪氨酸激酶家族。trk家族主要包括3个成员,ntrk1/trka,ntrk2/trkb和ntrk3/trkc。完整的trk激酶包括胞外区、跨膜区和胞内区三个部分。trk激酶的胞外区与相应的配体结合之后,能够引起激酶构型变化,形成二聚体。trk激酶的胞内区发生自体磷酸化从而激活自身的激酶活性,进而进一步激活下游的信号转导通路(如mapk,akt,pkc等),产生相应的生物学功能;其中ngf(神经生长因子)结合trka,bdnf(衍生的神经营养因子)结合trkb,以及nt3(神经营养因子3)结合trkc。
3.trk激酶在神经的发育过程中发挥重要的生理功能,包括神经元轴突的生长与功能维持、记忆的发生发展以及保护神经元免受伤害等等。同时,大量的研究表明trk信号转导通路的活化与肿瘤的发生发展密切相关,在神经细胞瘤、前列腺癌、乳腺癌等中都发现了活化的trk信号蛋白。
4.近几年来多种trk融合蛋白的发现,更显示了其促进肿瘤发生的生物学功能。最早的tpm3-trka融合蛋白是在结肠癌细胞中发现的,在检测的临床病人中约有1.5%的发生率。后来在不同类型的临床肿瘤病人样本如肺癌,头颈癌、乳腺癌、甲状腺癌、神经胶质瘤等中发现了不同类型的trk融合蛋白,如cd74-ntrk1,mprip-ntrk1,qki-ntrk2,etv6-ntrk3,btb1-ntrk3等。这些不同的ntrk融合蛋白在不需要配体结合的情况下,自身处于高度活化的激酶活性状态,因而能够持续性的磷酸化下游的信号途径,诱导细胞增殖,促进肿瘤的发生和发展。
5.因此,近几年来,trk融合蛋白己经成为一个有效的抗癌靶点和研究热点,例如wo2010048314、wo2012116217、wo2011146336、w02010033941、wo2018077246等均公开了具有不同结构类型的trk激酶抑制剂。
6.虽然第一代抑制剂拉罗替尼(larotrectinib)和恩曲替尼(entrectinib)在治疗ntrk融合阳性癌症方面取得了显著的临床疗效,但新出现的获得性耐药已成为一个未得到满足的临床需求。位于溶剂前沿突变(如trkag595r和trkcg623r)和激活环xdfg基序突变(如trkag667c/s和trkcg696a)是最常见的介导患者临床获得性耐药的trk突变(russo m等.cancer discovery,2016,6(1),36-44)、trkc g623r和g696a的突变(drilon a.等annals of oncology 2016,27(5),920-926),主要是突变后的氨基酸残基空间位阻增大,进而阻碍了抑制剂的结合。
7.目前,正在开发几种第二代抑制剂来解决这些突变,例如塞利替尼
(selitrectinib)和洛普替尼(repotrectinib),它们具有紧凑的大环结构可减轻突变氨基酸残基的影响(cancer discovery,2017,7(9),963-972)、(cancer discovery,2018,8(10),1227-1236)。这些第二代抑制剂可以有效规避溶剂前沿trka-g595r突变导致的临床耐药性。然而,临床病例结果表明,一些接受这些化合物治疗的患者最终由于xdfg区域的突变(trka-g667c)而变得无反应(cancer discovery,11,2021,126-141),这暗示着这些突变限制了对第二代抑制剂的敏感性。迄今为止,没有任何治疗药物被批准用于治疗经第一代trk抑制剂产生耐药性的患者。
技术实现要素:
8.本发明的目的是为了提供一类新型的高抑制活性的吡唑取代的吡唑并嘧啶的trk大环化合物,以解决获得性耐药(尤其是位于激活环xdfg基序突变),满足临床需求。
9.现有技术中提供的塞利替尼(selitrectinib)虽然对溶剂前沿突变具有良好的抑制活性,但是,接受这些化合物治疗的患者最终由于xdfg区域的突变(trka-g667c)而变得无反应。本发明的发明人通过大量的科学研究发现,具有本发明式(i)所示结构的一种吡唑取代的吡唑并嘧啶的trk大环化合物对trk激酶表现出较高的抑制活性,且其抑制活性优于现有技术的塞利替尼(selitrectinib)。
10.为了实现上述目的,本发明的第一方面提供一种具有式(i)所示结构的含吡唑取代的吡唑并嘧啶的trk大环化合物或其药学上可接受的盐,或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物,或其前药,
[0011][0012]
其中,在式(i)中,
[0013]
r1和r2各自独立地选自h、甲基、三氟甲基;
[0014]
n为2-6。
[0015]
本发明的第二方面提供前述第一方面中所述的具有式(i)所示结构的含吡唑取代的吡唑并嘧啶的trk大环化合物或其药学上可接受的盐,或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物,或其前药在制备用于预防和/或治疗trk激酶受体介导的疾病的药物中的应用。
[0016]
本发明的第三方面提供一种药物组合物,其包含药学上可接受的载体、赋形剂或稀释剂,以及作为活性成分的本发明第一方面中所述的具有式(i)所示结构的含吡唑取代的吡唑并嘧啶的trk大环化合物或其药学上可接受的盐,或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物,或其前药。
[0017]
本发明的第四方面提供本发明第三方面所述的药物组合物在制备用于预防和/或治疗trk激酶受体介导的疾病的药物中的应用。
[0018]
本发明的第五方面提供本发明第一方面中所述的具有式(i)所示结构的含吡唑取代的吡唑并嘧啶的trk大环化合物或其药学上可接受的盐,或其立体异构体、几何异构体、
互变异构体、氮氧化物、水合物、溶剂化物、代谢产物,或其前药,或者本发明第三方面中所述的药物组合物在制备用于预防和/或治疗肿瘤的药物中的应用。
[0019]
本发明提供的具有式(i)所示结构的含吡唑取代的吡唑并嘧啶的trk大环化合物或其药学上可接受的盐,或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物,或其前药对trk激酶表现出优异的抑制活性。
具体实施方式
[0020]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0021]
如前所述,本发明的第一方面提供一种含吡唑取代的吡唑并嘧啶的trk大环化合物或其药学上可接受的盐,或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物,或其前药,
[0022][0023]
其中,在式(i)中,
[0024]
r1和r2各自独立地选自h、甲基、三氟甲基;
[0025]
n为2-6。
[0026]
以下提供本发明的所述式(i)所示结构的含吡唑取代的吡唑并嘧啶的trk大环化合物的几种优选的具体实施方式:
[0027]
具体实施方式1:
[0028]
在式(i)中,
[0029]
r1和r2各自独立地选自h、甲基、三氟甲基;
[0030]
n为2-6。
[0031]
具体实施方式2:
[0032]
在式(i)中,
[0033]
r1和r2各自独立地选自h、甲基、三氟甲基;
[0034]
n为3-6。
[0035]
具体实施方式3:
[0036]
在式(i)中,
[0037]
r1和r2各自独立地选自h、甲基、三氟甲基;
[0038]
n为4-6。
[0039]
具体实施方式4:
[0040]
所述含氟吡唑并嘧啶化合物为
[0041]
化合物1:
[0042]
化合物2:
[0043]
化合物3:
[0044]
化合物4:
[0045]
化合物5:
[0046]
化合物6:
[0047]
化合物7:
[0048]
化合物8:
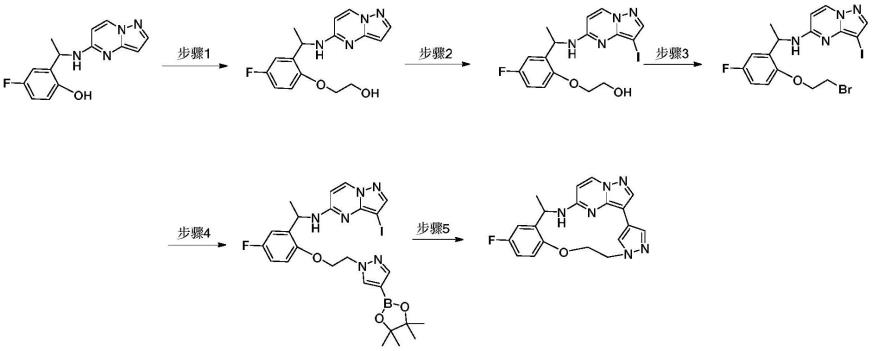
醇(9.8mmol),加入到200ml的梨形瓶中,向其中加入乙腈(50ml)。在室温,磁力搅拌条件下,加入n-碘代丁二酰亚胺(nis,14.85mmol)。室温反应1h,tlc监测反应完毕。减压尽量除去乙腈后用250ml乙酸乙酯稀释,并转移到分液漏斗中。用1mol/l得naoh洗3次,饱和食盐水洗两次,用无水硫酸钠干燥,浓缩得红色油状粗品,粗品经柱层析纯化得淡黄色固体,收率66%。
[0060]
步骤3):将2-(4-氟-2-(1-((3-碘吡唑[1,5-a]嘧啶-5-基)氨基)乙基)苯氧基)乙烷-1-醇(1.58mmol),加入到200ml的梨形瓶中,向其中加入二氯甲烷(30ml)。在0℃下,磁力搅拌条件下,依次加入三苯基膦(p(ph3)3,2.37mmol),n-碘代丁二酰亚胺(nis,2.37mmol)。冰浴下反应4h,tlc监测反应完毕。减压尽量除去二氯甲烷后用300ml乙酸乙酯稀释,并转移到分液漏斗中。饱和食盐水洗两次,用无水硫酸钠干燥,浓缩后,粗品经柱层析纯化得淡黄色固体,收率60%。
[0061]
步骤4):将n-(1-(2-(2-溴乙氧基)-5-氟苯基)乙基)-3-碘吡唑并[1,5-a]嘧啶-5-胺(0.20mmol),溶解在dmf(200ml)中,依次加入4-吡唑硼酸频哪醇酯(0.24mmol),无水碳酸铯(0.59mmol)。在120℃下反应过夜,tlc监测反应完毕。用300ml乙酸乙酯稀释,并转移到分液漏斗中。饱和食盐水洗两次,用无水硫酸钠干燥,浓缩后,粗品经柱层析纯化得淡黄色固体,收率65%。
[0062]
步骤5):n-(1-(5-氟-2-(2-(4-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1h-吡唑-1-基)乙氧基)苯基)乙基)-3-碘吡唑并[1,5-a]嘧啶-5-胺(0.16mmol),无水碳酸钾(0.64mmol),四(三苯基膦)钯(0.016mmol)加入到100ml反应管中,氩气置换3次,加入10ml无水dmf,2ml水。100℃,氩气氛围下反应3h,tlc监测反应完毕。冷却至50℃,硅藻土过滤,滤液加水并用乙酸乙酯萃取。有机相用饱和食盐水洗两次,用无水硫酸钠干燥,浓缩得黑色油状粗品,粗品经柱层析纯化得淡黄色固体,收率30%。
[0063]1h nmr(400mhz,dmso-d6)δ8.93(s,1h),8.48(d,j=7.5hz,1h),8.27(d,j=7.3hz,1h),7.92(s,1h),7.47(s,1h),7.16(dd,j=9.6,2.9hz,1h),6.99
–
6.82(m,2h),6.26(d,j=7.5hz,1h),5.88
–
5.77(m,1h),4.74
–
4.60(m,1h),4.59
–
4.46(m,1h),4.44
–
4.33(m,1h),3.89
–
3.75(m,1h),1.39(d,j=7.0hz,3h).
[0064]
实施例2:(14z,23z,24e)-5
5-氟-4-甲基-11h-6-氧杂-3-氮杂-2(3,5)-吡唑并[1,5-a]嘧啶-1(4,1)-吡唑啉-5(1,2)-苯并环壬烷(化合物2)的制备
[0065]
制备方法同实施例1,不同之处在于将2-溴乙醇改为3-溴-1-丙醇。
[0066]1h nmr(600mhz,chloroform-d)δ8.52(d,j=7.5hz,1h),7.96(s,1h),7.83(d,j=9.0hz,1h),7.27
–
7.18(m,1h),7.08(s,1h),6.95(d,j=8.0hz,1h),6.91
–
6.81(m,2h),6.44(d,j=7.5hz,1h),4.82
–
4.66(m,2h),4.49(dt,j=12.4,3.1hz,1h),4.39
–
4.31(m,1h),4.10
–
3.97(m,1h),2.36
–
2.24(m,1h),2.17
–
2.06(m,1h),1.51(d,j=6.8hz,3h).
[0067]
实施例3:(14z,23z,24e)-5
5-氟-4-甲基-11h-6-氧杂-3-氮杂-2(3,5)-吡唑并[1,5-a]嘧啶-1(4,1)-吡唑啉-5(1,2)-苯并环十烷(化合物3)的制备
[0068]
制备方法同实施例1,不同之处在于将2-溴乙醇改为4-溴-1-丁醇。
[0069]1h nmr (600mhz,chloroform-d)δ8.57(d,j=7.5hz,1h),8.02(s,1h),7.90(d,j=8.9hz,1h),7.33
–
7.24(m,1h),7.00
–
6.86(m,3h),6.44(d,j=7.5hz,1h),6.33(s,1h),5.03
–
4.93(m,1h),4.67(td,j=12.3,2.0hz,1h),4.31(dt,j=12.5,2.8hz,1h),4.22
–
4.12(m,1h),4.01(td,j=11.5,3.1hz,1h),2.16
–
2.02(m,1h),1.90
–
1.70(m,2h),1.67
–
1.57(m,
f589l、trkc-g623r、trkc-g696a等突变的激酶,来源于carna biosciences 08-186,08-187,08-197;htrf kinease tk kit(cisbio公司62tk0pec);384孔板(greiner公司);atp(life technologies pv3227),mgcl2(sigma)公司;pherastar fs多功能酶标仪(bmg公司);低速离心机(staitexiangyi公司);恒温箱(binder公司)。
[0087]
化合物溶解及保存:根据溶解性,用二甲基亚砜(dmso)将受试化合物配置成10mmol/l的母液,分装后-20℃保存。
[0088]
化合物工作液的配制:测试前将分装的化合物从冰箱取出,用纯dmso稀释到100
×
所需浓度;然后用去离子水将化合物稀释至4
×
所需浓度。
[0089]
1.33
×
酶缓冲液(enzymatic buffer)的配制:将5
×
酶缓冲液(来源于htrf试剂盒)用去离子水稀释到1.33
×
,并且加入1.33
×
终浓度的相应成分:1.33mmol/l二硫苏糖醇(dtt)、1.33mmol/l mncl2、6.65mmol/l mgcl2和39.9nmol/l seb。
[0090]
激酶工作液的配制:用1.33
×
酶缓冲液将trka、trkb和trkc稀释到2
×
所需浓度分别为0.404ng/μl、0.304ng/μl和0.236ng/μl。
[0091]
底物工作液的配制:用1.33
×
酶缓冲液将tk substrate
–
biotin(来源于htrf试剂盒)和atp(10mm)稀释为4
×
所需终浓度的混合液;trka、trkb和trkc的atp终浓度分别为:3.727μmol/l、2.56μmol/l和2.526μmol/l。tk substrate
–
biotin(来自htrf kinease tk kit)终浓度均为:0.2μmol/l。
[0092]
检测工作液的配制:用htrf测试缓冲液将16.67μmol/l的抗生蛋白链菌-xl665(streptavidin-xl665)稀释到4
×
所需终浓度,然后与等体积的抗体-铕隐酸盐(antibody-cryptate)混合(均来源于htrf试剂盒)。
[0093]
酶反应步骤:向低体积384微孔板的每个孔中加入4μl的激酶工作液,同时加入4μl的1.33
×
酶缓冲液作为阴性对照(negative);向孔加入2μl的化合物工作液,同时加入2μl的8%dmso水溶液作为零化合物浓度对照(即阳性对照,positive);于25℃,孵育5min;向孔中加入2μl底物工作液启动酶反应,于37℃,振荡反应30min。
[0094]
htrf试剂检测步骤:向孔加入8μl的检测工作液终止反应;25℃反应1h。
[0095]
htrf信号的读取:采用pherastar fs读数检测信号,仪器相应设置如下:
[0096]
optic module
[0097]
积分延迟(integration delay,lag time)50μs;
[0098]
积分时间(integration time)400μs;
[0099]
闪光次数(number of flashes)200;
[0100]
对于每孔读出的原始数据,比值=665nm/620nm。
[0101]
抑制率的计算:
[0102][0103]
ic
50
值的计算:以化合物浓度的对数为横坐标,抑制率为纵坐标,在graphpad prism 5中,拟合非线性曲线:log(inhibitor)vs.response
‑‑
variable slope,求出酶活抑制率为50%时的待测化合物浓度即ic
50
。
[0104]
实验结果:trka、trkb和trkc激酶活性半数抑制浓度(ic
50
,nm)。
[0105]
本发明提供结构如式(i)所示化合物以及对照化合物对trka、trkb和trkc的半数抑制浓度(ic
50
)见表1:
[0106]
表1:化合物的trka、trkb和trkc激酶抑制活性
[0107][0108]
如表1所示,本发明提供的化合物对野生的trka、trkb和trkc激酶均表现出优异的抑制活性。
[0109]
测试例2:大鼠体内药物代谢研究
[0110]
将实施例1、2和5提供的化合物以聚乙二醇400水溶液(70%)形式大鼠给药。对于口服给药,大鼠给予5mg/kg的剂量。口服组给药后15、30、45min、1、2、4、6、8、10、24h各采集血样约0.3ml至肝素化的eppendorf管中,于冰上暂存至离心。全血经8000rpm离心5min后收集血浆,转移血浆至96孔板中,于-20℃保存至lc-ms/ms检测。
[0111]
采用软件winnonlin软件的非室模型计算大鼠给药后的药代动力学参数。
[0112]
达峰浓度cmax:采用实测值。
[0113]
药时曲线下面积auc0-t值:采用梯形法计算;auc
0-∞
=auc
0-t
+ct/ke,ct为最后一个可测得时间点的血药浓度,ke为消除速率常数。
[0114]
消除半衰期t
1/2
=0.693/ke。
[0115]
绝度生物利用度f=dose iv
*auc
0-t,ig
/dose ig
*auc
0-t,iv
×
100%。
[0116]
表2列出了静注给药后,实施例1、2和5的化合物在大鼠体内的药代动力学参数。结果表明,实施例1、2和5的化合物具有良好的药代动力学性质,包括理想的清除率(cl)、半衰期(t
1/2
)、峰浓度(c
max
)和暴露量(auc
0-t
)。
[0117]
表3列出了口服给药后,实施例1、2和5的化合物在大鼠体内的药代动力学参数。结果表明,实施例1、2和5的化合物具有良好的药代动力学性质,包括理想的清除率(cl)、半衰期(t
1/2
)、峰浓度(c
max
)、暴露量(auc
0-t
)和口服生物利用度。
[0118]
表2:实施例1、2和5化合物的大鼠静注给药的主要药代动力学参数
[0119][0120][0121]
表3:实施例1、2和5化合物的大鼠口服给药的主要药代动力学参数
[0122][0123]
测试例3:本发明的化合物对五种trk激酶突变体的抑制活性
[0124]
本测试例采用与测试例1相同的测试方法进行。
[0125]
本测试例的结果列于表4中。
[0126]
表4
[0127][0128]
由表4可见,本发明的化合物对五种trk激酶突变体的抑制活性优于对野生的trk激酶的活性,有望有效克服临床报道的肿瘤耐药性。
[0129]
测试例4:本发明的化合物在裸鼠异体移植瘤模型上的抗肿瘤活性
[0130]
本发明化合物的药效通过移植肿瘤的标准鼠类模型来进行评价。
[0131]
人非小细胞肺癌h2228培养、收集后,于后腹侧皮下接种于5-6周龄的雌性裸小鼠体内(balb/c,上海灵畅生物科技有限公司)。当肿瘤体积达到100-150mm3时,动物随机地分为溶剂对照组(70%peg-400的水溶液)和化合物组(每组6只动物)。后续采用实施例的化合物对动物进行灌胃给药(相应剂量,溶解在70%peg-400的水溶液中),从肿瘤细胞接种后的0到7天中的任何地方开始,并且在试验中每天进行两次。
[0132]
实验指标为考察实施例化合物对肿瘤生长的影响,具体指标为t/c%或抑瘤率tgi(%)。
[0133]
每周二次用游标卡尺测量肿瘤直径,肿瘤体积(v)计算公式为:
[0134]
v=1/2
×a×
b2其中a、b分别表示长、宽。
[0135]
t/c(%)=(t-t0)/(c-c0)
×
100其中t、c为实验结束时的肿瘤体积;t0、c0为实验开始时的肿瘤体积。
[0136]
抑瘤率(tgi)(%)=100-t/c(%)。
[0137]
当肿瘤出现消退时,抑瘤率(tgi)(%)=100-(t-t0)/t0×
100。
[0138]
如果肿瘤比起始体积缩小,即t《t0或c《c0时,即定义为肿瘤部分消退(pr);如果肿瘤完全消失,即定义为肿瘤完全消退(cr)。
[0139]
二组肿瘤体积之间比较采用双尾student’s t检验,p《0.05定义为有统计学显著性差异。
[0140]
本测试例的结果列于表5中。
[0141]
以下bid表示一天两次给药。
[0142]
表5
[0143]
分组给药抑瘤率d14完全消退部分消退溶剂bid,d0-7
‑‑‑
化合物5,50mg/kgbid,d0-7155%3/62/6
[0144]
由表5可见,本发明的化合物在人非小细胞肺癌h2228裸鼠异体移植瘤模型上表现出优异的抗肿瘤活性。其中,化合物5(50mg/kg,bid
×
8)明显抑制人非小细胞肺癌h2228裸小鼠皮下移植瘤的生长,给药至d7时,抑瘤率为182%,4/6肿瘤完全消退;从d8开始停止给药,至实验结束(d14),抑瘤率为155%,3/6动物肿瘤完全消退,2/6动物肿瘤出现部分消退。
[0145]
上述结果表明,本发明提供的具有式(i)所示结构的含氟吡唑并嘧啶化合物或其药学上可接受的盐,或其立体异构体、几何异构体、互变异构体、氮氧化物、水合物、溶剂化物、代谢产物,或其前药对trk激酶表现出优异的抑制活性,同时,能够在动物水平上表现出良好的抗肿瘤活性。
[0146]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1