一种苏沃雷生中间体的制备方法与流程
1.本发明涉及药物合成领域,尤其涉及一种苏沃雷生中间体的制备方法。
背景技术:
2.苏沃雷生,化学名为5-氯-2-[(5r)-5-甲基-4-[5-甲基-2-(2h-1,2,3-三唑-2-基)苯甲酰基]-1,4-二氮杂环庚烷-1-基]-1,3-苯并恶唑,是一种高度选择性食欲素受体拮抗剂,通过结合促觉醒神经肽食欲素a和食欲素b,选择性作用于介导唤醒和睡眠间转换的神经元,以此增加总睡眠时间及缩短入睡时间,改善过度觉醒,且不影响睡眠结构,从而达到治疗失眠和睡眠紊乱的目的。
[0003]
式i化合物能够作为一种有机合成中间体,用于合成苏沃雷生等高附加值化合物,填补目前失眠治疗市场未能满足的重要需求。式i化合物的结构式如下:但目前,关于式i化合物的合成方法报道很少,缺乏一种工艺简单、适合大规模工业化生产的合成方法。
技术实现要素:
[0004]
为了解决现有技术中缺乏工艺简单、适合大规模工业化生产的式i化合物合成方法的技术问题,本发明提供了一种苏沃雷生中间体的制备方法。该方法具有制备流程短、工艺步骤简单、适合大规模生产的优点,且能获得高纯度的苏沃雷生中间体。
[0005]
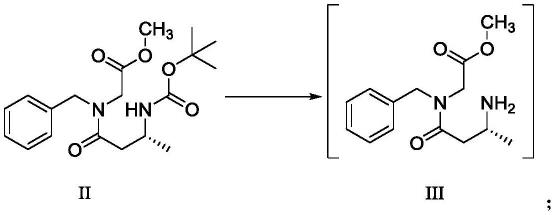
本发明的具体技术方案为:一种苏沃雷生中间体的制备方法,包括以下步骤:(1)将式ii化合物、有机溶剂和酸混合后,进行脱叔丁氧羰基反应,获得含有式iii化合物的混合液;反应路线如下:(2)将含有式iii化合物的混合液与碱混合,进行关环反应后,进行初步纯化去除碱,而后加入乙腈进行热打浆,趁热过滤后浓缩,再加入乙酸乙酯进行热打浆,过滤,获得式i化合物即苏沃雷生中间体;反应路线如下:
[0006]
本发明以式ii化合物(可采用常规方法,由2-(苄基氨基)乙酸甲酯和3-((叔丁氧羰基)氨基)丁酸通过缩合反应制得)为原料,经由“一锅法”连续反应制备苏沃雷生中间体(式i化合物)。在上述“一锅法”连续反应的过程中,式ii化合物经脱叔丁氧羰基反应生成式iii化合物(步骤(1))后,不进行中间产物(式iii化合物)的分离和提纯,直接向混合液中添加碱进行关环反应(步骤(2)),能够缩短制备流程,简化工艺步骤,从而大大提高生产效率,适合大规模工业化生产,此外还能避免中间产物在分离和提纯过程中损失,从而提高收率。
[0007]
本发明人团队关注到,当采用“一锅法”连续反应时,在步骤(1)脱叔丁氧羰基反应后,会存在少量原料(式ii化合物)未完全反应的情况,在步骤(2)中与碱接触后,在碱的作用下会发生酯水解,形成副产物,这些副产物难以通过常规的纯化方法去除,会造成最终制得的式i化合物品质较差,纯度较低。为此,本发明基于理论研究和试验,对纯化工艺进行了特殊设计,采用乙腈热打浆的纯化方法,能够有效去除由式ii化合物酯水解产生的副产物,使最终制得的式i化合物具有较高的纯度;同时,采用乙腈热打浆的纯化方法,还能够减少纯化过程中式i化合物的损失,从而实现较高的收率;并且,完成乙腈热打浆后,乙腈能够回收,因而生产过程中产生的三废较少。
[0008]
作为优选,步骤(2)中,所述碱为碳酸钾粉末。
[0009]
采用碳酸钾粉末作为催化剂,其在有机溶剂中的溶解度较小,在完成反应后,通过过滤即可去除大部分碳酸钾,因而能够简化产物的分离提纯步骤,提高生产效率。
[0010]
作为优选,步骤(2)中,所述初步纯化的过程包括以下步骤:过滤,对滤液进行中和,再次过滤后,浓缩。
[0011]
进一步地,步骤(2)中,所述初步纯化的过程包括以下步骤:过滤,将滤液进行中和至ph为7~8,再次过滤后,浓缩。
[0012]
作为优选,步骤(2)中,所述碳酸钾粉末的粒径小于100μm;步骤(1)中所述式ii化合物与步骤(2)中所述碳酸钾粉末的摩尔比为1:1.5~4。
[0013]
当碳酸钾粉末的用量过大时,会导致完成反应后碳酸钾粉末的去除难度增大(如过滤时易造成堵孔),三废增加,生产成本升高。本发明通过将碳酸钾粉末的粒径控制在小于100μm,能够使其具有较大的比表面积,提高催化效率,从而在确保收率的情况下,减少碳酸钾粉末的用量。
[0014]
作为进一步优选,步骤(2)中,所述碳酸钾粉末的粒径小于100μm;步骤(1)中所述式ii化合物与步骤(2)中所述碳酸钾粉末的摩尔比为1:1.5~2。
[0015]
作为优选,步骤(1)中所述式ii化合物与步骤(2)中所述乙腈的质量体积比为1kg:6~10l。
[0016]
作为进一步优选,步骤(1)中所述式ii化合物与步骤(2)中所述乙腈的质量体积比为1kg:6~8l。
[0017]
作为优选,步骤(2)中,所述加入乙腈进行热打浆的过程中,热打浆的温度为70~75℃。
[0018]
作为优选,步骤(1)中,所述有机溶剂为甲醇,所述酸为氯化氢甲醇溶液。
[0019]
进一步地,所述氯化氢甲醇溶液中,氯化氢的含量为3~5mol/l;所述式ii化合物与氯化氢甲醇溶液的质量体积比为1kg:1~2l。
[0020]
作为优选,步骤(2)中,所述关环反应的温度为0~35℃。
[0021]
作为进一步优选,步骤(2)中,所述关环反应的温度为20~30℃。
[0022]
作为优选,步骤(1)中,所述脱叔丁氧羰基反应的温度为20~30℃,时间为10~18h。
[0023]
与现有技术相比,本发明具有以下优点:(1)本发明采用“一锅法”连续反应,并配合特殊的产物纯化方法,能够在缩短制备流程、简化工艺步骤、提高生产效率的同时,使制得的苏沃雷生具有较高的纯度;(2)本发明采用碳酸钾粉末作为关环反应的催化剂,能够简化产物的分离提纯步骤;并且,通过控制碳酸钾粉末的粒径,能够减少初步纯化的过程中氯化钾的形成。
具体实施方式
[0024]
下面结合实施例对本发明作进一步的描述。
[0025]
总实施例一种苏沃雷生中间体的制备方法,包括以下步骤:(1)将式ii化合物、有机溶剂和酸混合后,在20~30℃下进行脱叔丁氧羰基反应10~18h,获得含有式iii化合物的混合液;反应路线如下:(2)将含有式iii化合物的混合液与粒径小于100μm的碳酸钾粉末混合,步骤(1)中所述式ii化合物与步骤(2)中所述碳酸钾粉末的摩尔比为1:1.5~4,在0~35℃下进行关环反应;充分反应后,过滤,对滤液进行中和,再次过滤后,浓缩,而后加入乙腈,在70~75℃下进行热打浆,步骤(1)中所述式ii化合物与步骤(2)中所述乙腈的质量体积比为1kg:6~10l,趁热过滤后浓缩,再加入乙酸乙酯进行热打浆,过滤,获得式i化合物即苏沃雷生中间体;反应路线如下:
[0026]
作为一种具体实施方式,步骤(1)中,所述有机溶剂为甲醇,所述酸为氯化氢甲醇溶液(其中氯化氢的含量为3~5mol/l),所述式ii化合物与氯化氢甲醇溶液的质量体积比为1kg:1~2l;步骤(2)中,通过加入氯化氢甲醇溶液对滤液进行中和。
[0027]
实施例1通过以下步骤,制备苏沃雷生中间体:(1)式iii化合物的合成:向10l反应釜中加入1.8kg式ii化合物和6.2l甲醇,升温至30℃搅拌溶解,而后滴加2.5l4mol/l氯化氢甲醇溶液,30℃保温反应16h,获得含有式iii化合物的混合液;(2)式i化合物的合成:向含有式iii化合物的混合液中加入1.2kg粒径为d90=45μm的碳酸钾粉末,30℃保温反应8h,获得含有式i化合物的混合液;(3)式i化合物的分离纯化:对含有式i化合物的混合液进行过滤,用4mol/l氯化氢甲醇溶液调节滤液ph至7.5
±
0.5,再过滤一次,将滤液浓缩至油状物,加入12.6l乙腈,升温至70℃打浆1h后,过滤,再将滤液浓缩至油状物,加入3.6l乙酸乙酯,升温至50℃搅拌1h,过滤,获得897g(理论产量1.147kg)类白色固体式i化合物,即苏沃雷生中间体。
[0028]
经检测,本实施例中,制得的式i化合物纯度为98.05%,由式iii化合物制备式i化合物的收率为78.2%。
[0029]
实施例2通过以下步骤,制备苏沃雷生中间体:(1)式iii化合物的合成:向10l反应釜中加入1.8kg式ii化合物和6.2l甲醇,升温至30℃搅拌溶解,而后滴加2.5l4mol/l氯化氢甲醇溶液,30℃保温反应16h,获得含有式iii化合物的混合液;(2)式i化合物的合成:向含有式iii化合物的混合液中加入1.4kg粒径为d90=45μm的碳酸钾粉末,30℃保温反应8h,获得含有式i化合物的混合液;(3)式i化合物的分离纯化:对含有式i化合物的混合液进行过滤,用4mol/l氯化氢甲醇溶液调节滤液ph至7.5
±
0.5,再过滤一次,将滤液浓缩至油状物,加入15l乙腈,升温至70℃打浆1h后,过滤,再将滤液浓缩至油状物,加入3.6l乙酸乙酯,升温至50℃搅拌1h,过滤,获得845g(理论产量1.147kg)类白色固体式i化合物,即苏沃雷生中间体。
[0030]
经检测,本实施例中,制得的式i化合物纯度为98.55%,由式iii化合物制备式i化合物的收率为73.7%。
[0031]
对比例1通过以下步骤,制备苏沃雷生中间体:(1)式iii化合物的合成:向反应瓶中加入18g式ii化合物和62ml甲醇,升温至30℃搅拌溶解,而后滴加25ml 4mol/l氯化氢甲醇溶液,30℃保温反应16h,获得含有式iii化合物的混合液;(2)式iii化合物的分离纯化:将含有式iii化合物的混合液浓缩至干油状物,加入12ml饱和碳酸钠溶液,用体积比为2:1的二氯甲烷和乙醇的混合液(18ml
×
2)进行萃取,合并有机相后,用无水硫酸镁干燥,浓缩至油状物,加入乙酸乙酯60ml,20℃搅拌2h,过滤,干燥,获得9.8g(理论产量13.1g)式iii化合物,收率74.8%,纯度98.4%。
[0032]
(3)式i化合物的分离纯化:将式iii化合物加入60ml甲醇中,升温至30℃搅拌溶解后,向其中加入14g粒径为d90=45μm的碳酸钾粉末,30℃保温反应8h,获得含有式i化合物的混合液;(4)式i化合物的分离纯化:对含有式i化合物的混合液进行过滤,用4mol/l氯化氢甲醇溶液调节滤液ph至7.5
±
0.5,再过滤一次,将滤液浓缩至油状物,加入150ml乙腈,升温至70℃打浆1h后,过滤,再将滤液浓缩至油状物,加入360ml乙酸乙酯,升温至50℃搅拌1h,过滤,获得7.3g(理论产量8.6g)类白色固体式i化合物,即苏沃雷生中间体,收率84.9%。
[0033]
经检测,本实施例中,制得的式i化合物纯度为98.32%,由式iii化合物制备式i化合物的总收率为63.6%。
[0034]
对比实施例1和对比例1,可以看出:实施例1获得的式i化合物纯度与对比例1相当,收率明显高于对比例1。说明相较于非“一锅法”(即在合成式iii化合物后进行分离纯化,再合成式i化合物)而言,采用“一锅法”连续反应,能够提高式i化合物的收率。这是由于:“一锅法”能够避免式iii化合物在分离纯化的过程中损失,因而能提高最终制得的式i化合物收率。
[0035]
对比例2通过以下步骤,制备苏沃雷生中间体:
(1)式iii化合物的合成:向反应瓶中加入18g式ii化合物和62ml甲醇,升温至30℃搅拌溶解,而后滴加25ml 4mol/l氯化氢甲醇溶液,30℃保温反应16h,获得含有式iii化合物的混合液;(2)式i化合物的合成:向含有式iii化合物的混合液中加入14g粒径为d90=45μm的碳酸钾粉末,继续30℃保温反应8h,获得含有式i化合物的混合液;(3)式i化合物的分离纯化:对含有式i化合物的混合液进行过滤,用4mol/l氯化氢甲醇溶液调节滤液ph至7.5
±
0.5,再过滤一次,将滤液浓缩至干,加入12ml饱和碳酸钠溶液,用体积比为2:1的二氯甲烷和乙醇的混合液(18ml
×
3)进行萃取,合并有机相后,用无水硫酸镁干燥,旋蒸去除溶剂,获得7.8g(理论产量11.47g)类白色固体式i化合物,即苏沃雷生中间体。
[0036]
经检测,本实施例中,制得的式i化合物纯度为97.89%,由式iii化合物制备式i化合物的收率为68%。
[0037]
对比实施例2、对比例1和对比例2,可以看出:实施例2获得的式i化合物纯度略高于对比例2,与对比例1相当;实施例2的收率明显高于对比例2。说明采用实施例2中的方法对式i化合物进行分离纯化,能有效去除“一锅法”连续反应所造成的副产物,使最终制得的式i化合物纯度达到与非“一锅法”(即在合成式iii化合物后进行分离纯化,再合成式i化合物)相当的水平,同时收率更高。此外,对比例2中的二氯甲烷和乙醇混合溶液几乎无法回收,三废过高。这是由于:当采用“一锅法”连续反应时,在步骤(1)脱叔丁氧羰基反应后,会存在少量原料(式ii化合物)未完全反应的情况,在步骤(2)中与碱接触后,在碱的作用下会发生酯水解,形成副产物,这些副产物难以通过二氯甲烷和乙醇的混合液萃取的方法去除,会造成最终制得的式i化合物纯度较低,并且,二氯甲烷和乙醇混合溶液萃取时分层较困难,会造成式i化合物损失较多,收率较低;而采用乙腈热打浆的纯化方法,能够有效去除由式ii化合物酯水解产生的副产物,且纯化过程中式i化合物的损失较少。
[0038]
本发明中所用原料、设备,若无特别说明,均为本领域的常用原料、设备;本发明中所用方法,若无特别说明,均为本领域的常规方法。
[0039]
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1