化合物以及包含该化合物的有机电致发光器件的制作方法
1.本发明涉及有机电致发光技术领域,特别涉及一种新型有机化合物及其应用,包含该化合物的有机电致发光器件。
背景技术:
2.有机电致发光器件(oled:organic light emitting devices),是一种类似三明治结构的电流驱动式薄膜器件,在阳极和阴极之间夹杂单层或者多层有机功能材料层。oled在电场的作用下,阳极产生的空穴和阴极产生的电子就会发生移动,分别向空穴传输层和电子传输层注入,迁移到发光层,当二者在发光层相遇复合时,产生能量激子,从而激发发光分子最终产生可见光。oled具有自发光、视角广、色域广、响应时间短、发光效率高、工作电压低、成本低廉、生产工艺简单等特点,可以制作成大尺寸和/或柔性超薄面板,是一种发展迅速、工艺集成度较高的新型显示技术,目前已经广泛应用于电视、智能手机、平板电脑、车载显示、照明等显示产品中,还将进一步应用在大尺寸显示、柔性屏等创意显示产品中。
3.应用于oled器件的有机光电材料在用途上可以分为发光层材料和辅助功能层材料,其中,发光材层材料中包括客体材料(又称为发光材料、掺杂材料)和主体材料(又称为基质材料),发光材料根据不同的能量传递方式,分成荧光材料、磷光材料和热活化延迟荧光材料,辅助功能层材料按照电子或者空穴传输速度不同的性质,又分成电子注入材料、电子传输材料、空穴阻挡材料、电子阻挡材料、空穴传输材料、空穴注入材料。
4.显示产品中应用的oled器件主要由不同功能层构建的红、绿、蓝三原色器件,通过电路控制从而实现五彩缤纷的产品。而红、绿、蓝三原色器件中蓝光器件的最难开发,特别是市场对蓝光的性能要求越来越高。通常,器件有三个指标:低电压、高效率、长寿命,通过材料、器件优化可以实现三者中的两个指标,但另一个指标会降低。因此如何能够同时实现低电压、高效率、长寿命等指标是行业追求的目标。
5.因此,本领域亟需开发一类oled功能材料,获得低电压、高效率、长寿命的器件。基于此种考虑,本发明通过提供在芴结构中引入柔性烷基结构的化合物,同时可以调节材料的能级和空穴迁移率,使得器件可以在保持oled的驱动电压略有降低、寿命略有提高同时,提高效率。从而为获得高性能蓝光提供了一种可行性方案。
技术实现要素:
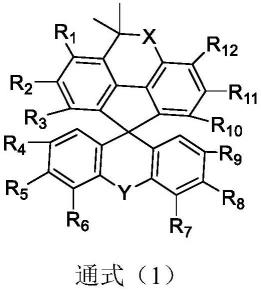
6.为解决上述技术问题,本发明提供了:1)一种用于有机电致发光器件的化合物,所述化合物具备下述通式(1)所表示的结构:
[0007][0008]
其中,r1~r
14
各自独立地选自氢原子或取代基,且其中至少一个具备下述通式(2)所表示的结构:
[0009][0010]
x选自cr
15r16
、o、s、se、nr
17
,sir
18r19
;y选自不存在、单键、cr
20r21
、o、s、se、nr
22
、sir
23r24
;r
15
~r
24
各自独立地选自氢原子或取代基;
[0011]
l选自单键、取代或未取代的亚芳基、取代或未取代的亚杂芳基;
[0012]
ar1,ar2各自独立地表示取代或未取代的芳基、取代或未取代的杂芳基。
[0013]
2)根据1)的用于有机电致发光器件的化合物,l选自单键、取代或未取代的亚芳基。
[0014]
3)根据1)的用于有机电致发光器件的化合物,l为单键。
[0015]
4)根据1)的用于有机电致发光器件的化合物,r1~r
24
各自独立地表示氢原子、取代或未取代的碳原子数1~50的烷基、取代或未取代的碳原子数1~20的烷氧基、取代或未取代的成环碳原子数6~50的芳基、或取代或未取代的成环碳原子数为2~30的杂芳基。
[0016]
5)根据1)的用于有机电致发光器件的化合物,所述化合物中“取代或未取代”中的“取代”是指取代基独立地选自氘原子、氚原子、羟基、碳原子数1~10的1价烷基或者环烷基、碳原子数6~30的1价单环芳基或者稠环芳基、碳原子数2~50的1价杂环基或者稠环杂芳基。
[0017]
6)根据1)的用于有机电致发光器件的化合物,其特征在于,所述化合物选自如下结构:
[0018]
[0019]
[0020]
[0021]
[0022]
[0023]
[0024]
[0025][0026]
7)一种有机电致发光器件,所述有机电致发光器件包括阳极、阴极以及位于所述阳极和阴极之间的至少一层的有机薄膜,所述有机薄膜中含有1)~6)中任一项所述的用于有机电致发光器件的化合物。
[0027]
8)根据7)所述的有机电致发光器件,所述有机薄膜包括空穴注入层、空穴传输层、电子阻挡层、发光层、激子阻挡层、空穴阻挡层、电子传输层、电子注入层中任意一种或者至少两种组合,且所述空穴传输层、电子阻挡层、发光层中至少一层含有1)~6)中任一项所述的用于有机电致发光器件的化合物。
[0028]
9)根据8)所述的有机电致发光器件,所述用于有机电致发光器件的化合物用作空穴传输层和/或电子阻挡层。
附图说明
[0029]
此处所说明的附图用来提供对本技术的进一步理解,构成本技术的一部分,本技术的示意性实施例及其说明用于解释本技术,并不构成对本技术的不当限定。在附图中:
[0030]
图1为本发明化合物应用的有机电致发光器件结构示意图,其中,器件各层结构代表含义如下:
[0031]
1、透明基板层,2、ito阳极层,3、空穴注入层,4、空穴传输层a,5、空穴传输层b(或者电子阻挡层),6、发光层,7、电子传输层b(或者空穴阻挡层),8、电子传输层a,9、电子注入层,10、阴极反射电极层。
具体实施方式
[0032]
下面将以多个合成实施例为例来进一步说明本发明的原理和特征,所举的实施例只用于解释本发明,但并非用于限定本发明的范围。
[0033]
以下所列举的式(1)的具体化合物的合成方法,除非另有说明,都在保护性气体气氛下在无水溶剂中进行。
[0034]
合成实施例1:化4的合成
[0035][0036]
将a4(5.76g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。4-氯-二苯甲酮b4(4.32g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0037]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.20g目标产物m4,产率76%,白色固体,质谱测得分子量为m/z=408.2(m+h)
+
。
[0038]
将中间体m4(4.08g,10.0mmol)、二苯胺c4(3.79g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.37g目标产物,产率87%。白色固体,质谱测得分子量为733.2。
[0039]
合成实施例2:化6的合成
[0040][0041]
将a6(6.44g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。二苯甲酮b6(3.62g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0042]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.04g目标产物m6,产率74%,白色固体,质谱测得分子量为m/z=408.2(m+h)
+
。
[0043]
将中间体m6(4.08g,10.0mmol)、c6(3.52g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到5.80g目标产物,产率82%。白色固体,质谱测得分子量为707.2。
[0044]
合成实施例3:化11的合成
[0045][0046]
将a11(6.28g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。3-氯-二苯甲酮b11(4.32g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0047]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.60g目标产物m11,产率76%,白色固体,质谱测得分子量为m/z=
434.2(m+h)
+
。
[0048]
将中间体m11(4.34g,10.0mmol)、c11(4.17g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.76g目标产物,产率85%。白色固体,质谱测得分子量为795.4。
[0049]
合成实施例4:化17的合成
[0050][0051]
将a17(7.64g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。二苯甲酮b6(3.62g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0052]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.75g目标产物m17,产率72%,白色固体,质谱测得分子量为m/z=469.2(m+h)
+
。
[0053]
将中间体m17(4.34g,10.0mmol)、c17(3.55g,21.0mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到5.95g目标产物,产率81%。白色固体,质谱测得分子量为734.4。
[0054]
合成实施例5:化22的合成
[0055][0056]
将a22(6.96g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。二苯甲酮b6(3.62g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过
夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0057]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.51g目标产物m22,产率75%,白色固体,质谱测得分子量为m/z=434.2(m+h)
+
。
[0058]
将中间体m22(4.34g,10.0mmol)、c22(3.79g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.31g目标产物,产率83%。白色固体,质谱测得分子量为760.1。
[0059]
合成实施例6:化32的合成
[0060][0061]
将a11(6.28g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。2-溴芴酮b32(5.18g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0062]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到7.24g目标产物m32,产率76%,白色固体,质谱测得分子量为m/z=476.2(m+h)
+
。
[0063]
将中间体m32(4.76g,10.0mmol)、c22(3.79g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.37g目标产物,产率84%。白色固体,质谱测得分子量为758.1。
[0064]
合成实施例7:化54的合成
[0065][0066]
将a11(6.28g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。4-溴芴酮b54(5.18g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0067]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到7.24g目标产物m32,产率76%,白色固体,质谱测得分子量为m/z=476.2(m+h)
+
。
[0068]
将中间体m54(4.76g,10.0mmol)、c54(4.21g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.78g目标产物,产率85%。白色固体,质谱测得分子量为798.1。
[0069]
合成实施例8:化117的合成
[0070][0071]
将a11(6.28g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。2-溴芴酮b32(5.18g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0072]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到7.24g目标产物m32,产率76%,白色固体,质谱测得分子量为m/z=476.2(m+h)
+
。
[0073]
将中间体m32(4.76g,10.0mmol)、c117(4.63g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(2.76g,20mmol)的碳酸钾、30ml的甲苯和10ml水,对反应体系抽换气三
次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.98g目标产物,产率88%。白色固体,质谱测得分子量为794.1。
[0074]
合成实施例9:化128的合成
[0075][0076]
将a128(6.44g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。芴酮b128(3.60g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0077]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.25g目标产物m128,产率77%,白色固体,质谱测得分子量为m/z=407.1(m+h)
+
。
[0078]
将中间体m128(4.07g,10.0mmol)、c128(5.05g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(2.76g,20mmol)的碳酸钾、30ml的甲苯和10ml水,对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到7.19g目标产物,产率89%。白色固体,质谱测得分子量为808.0。
[0079]
合成实施例10:化149的合成
[0080][0081]
将a22(7.00g,20mmol)与20ml四氢呋喃的混合物滴加到装有干燥镁屑、催化剂量碘和30ml四氢呋喃的100ml三口瓶中,滴加完毕后,回流反应2h。b149(3.92g,20mmol)与20ml四氢呋喃的混合物缓慢滴加到制备的格氏试剂中。滴加完毕,回流反应过夜。将反应液倒入饱和氯化胺溶液中淬灭反应,乙酸乙酯萃取三次,旋蒸除掉溶剂,干燥后,直接用于下一步反应。
[0082]
将上一步粗品、20ml盐酸和40ml醋酸加入到反应瓶中,80℃反应2h。反应完毕,倒入冰水中,乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,乙酸乙酯/石油醚(1:10)淋洗,
旋干溶剂,真空干燥得到6.56g目标产物m149,产率73%,白色固体,质谱测得分子量为m/z=448.2(m+h)
+
。
[0083]
将中间体m149(4.48g,10.0mmol)、c149(3.37g,10.5mmol)、三(二亚苄基丙酮)二钯(0.18g,0.2mmol)、(1.92g,20mmol)的叔丁醇钠。对反应体系抽换气三次,氮气保护。再用针管加入(0.8ml,0.8mmol)三叔丁基膦的甲苯溶液(1mol/l)和30ml的甲苯。氮气保护下,加热搅拌,120℃回流反应12h。停止反应,冷至室温,加入100ml去离子水,用乙酸乙酯萃取三次。旋蒸除掉溶剂,硅胶柱分离,二氯甲烷/石油醚(1:10)淋洗,旋干溶剂,真空干燥得到6.17g目标产物,产率84%。白色固体,质谱测得分子量为733.4。
[0084]
类似的反应,可以制备以下化合物:
[0085]
表1
[0086]
[0087]
[0088]
[0089]
[0090][0091]
表2用于oled的材料
[0092][0093]
器件实施例1:
[0094]
ito(130)/hatcn(15)/化4(60)/ebl(5)/bh:bd(20,重量2%)/hbl(5)/etl:liq(30,重量50%)/liq(1)/al(80)。
[0095]
器件实施例2-20与器件实施例1的区别仅在于将空穴传输层中所用的本发明化4替换为本发明的其它化合物,具体详见表3。
[0096]
比较例1~3:
[0097]
本比较例与器件实施例1相比,不同之处在于有机电致发光器件中的htl变更为业内公知且已商业化应用的,所得器件性能测试数据如表3所示。
[0098]
通过标准方法表征所述oled。为了这个目的,确定电致发光光谱、电流效率(以cd/a度量)、功率效率(以lm/w度量)和外量子效率(eqe,以%度量),其作为发光密度的函数从呈现朗伯发射特征的电流/电压/发光密度特征线(iul特征线)计算。在1000cd/m2的亮度下确定所需的电压v1000。ce1000表示在1000cd/m2下达到的电流效率。最后,eqe1000表示在1000cd/m2的工作亮度下的外量子效率,t90表示器件在1000cd/m2的起始亮度下器件亮度减弱到95%的工作时间。将本发明的实施例1~20和比较例1~3的器件性能总结如表3所示。
[0099]
表3
[0100][0101][0102]
从表3中可以看出,相对于现有技术,使用本发明的其中一类材料,器件可以在保持oled的驱动电压略有降低、寿命略有提高同时,提高效率。如化48所在器件实施例7相比于比较例2,ce1000提升了11.8%,同时寿命提高12%。
[0103]
通过表3可以看出,在芴结构中引入柔性烷基结构,同时可以调节材料的能级和空穴迁移率,可以更好地和电子阻挡层搭配,既保证空穴高效的注入传输、又避免空穴在电子阻挡层和发光层界面聚集导致的器件恶化;从而能够有效提升器件的效率和寿命。从而为
获得高性能蓝光提供了一种可行性方案。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1