喹喔啉类化合物马来酸盐的新晶型及其制备方法与流程
1.本发明涉及药物合成技术领域,具体涉及喹喔啉类化合物马来酸盐的新晶型及其制备方法。
背景技术:
2.受体酪氨酸激酶(receptor tyrosine kinase,rtk)是作用于细胞因子、生长因子、激素和其他信号分子受体的一种多基团跨膜蛋白。受体酪氨酸激酶是蛋白酪氨酸激酶家族分支的较大一部分。研究表明,受体酪氨酸激酶在多种细胞过程中起重要作用,包括生长、分化、血管生成以及多种癌症的发展进程。抑制受体酪氨酸激酶似乎是癌症治疗中的有效措施。已有超过20多种被fda批准的用于各种癌症治疗的小分子激酶抑制剂,目前已有大量的激酶抑制剂处于临床试验的各个阶段。wo2018071348a1公开了一种作为ⅲ型受体酪氨酸激酶抑制剂的喹喔啉化合物及其制备方法,然而,该专利文献并未公开6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈马来酸盐的晶型及制备方法。
3.在药物研发中晶体的研发非常重要,不同晶体形态的化合物具有不同的生物利用度和溶解度,晶型对化合物的稳定性、可加工性能、生物利用度、溶解度、制剂和工业生产运输等有较大的影响,因此,开发6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈(式i化合物)合适的盐的稳定晶体形态对于其药物研究具有重大的意义。
技术实现要素:
4.本发明要解决的技术问题是提供一种喹喔啉类化合物的马来酸盐新晶型及其制备方法。具体地,本发明提供了6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈的马来酸盐晶型a、晶型b及其制备方法。
5.为了解决上述技术问题,本发明提供了如下的技术方案:第一方面,本发明提供了6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈的马来酸盐的一种新晶型,定义为晶型a,所述晶型a的粉末x射线衍射图中,在7.7、12.5、13.6、14.5、16.8、26.3的2θ值处存在特征峰。
6.进一步地,所述晶型a的粉末x射线衍射图中,在7.7、12.5、13.6、14.5、16.8、21.0、
23.1、24.8、26.3的2θ值处存在特征峰。
7.进一步地,所述晶型a的粉末x射线衍射图如附图4所示。
8.进一步地,本发明提供了所述的6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈的马来酸盐晶型a的制备方法,包括以下步骤:将6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈游离碱溶于溶剂中,0℃下加入马来酸配体,在搅拌的条件下于25℃~50℃下反应,反应结束后降温至室温,过滤,滤液继续降温到0℃后再过滤,得到所述的晶型a。
9.本发明中,“室温”定义为5~35℃的温度范围,例如5℃、10℃、15℃、20℃、25℃、30℃、35℃等。
10.进一步地,所述游离碱与马来酸配体的摩尔比为1:1.2,所述溶剂选自四氢呋喃、二氯甲烷、乙醇、丙酮中的一种或多种的组合。
11.第二方面,本发明提供了6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈的马来酸盐的另一种新晶型,定义为晶型b,所述晶型b的粉末x射线衍射图中,在7.8、11.8、13.5、15.7、21.4、24.9的2θ值处存在特征峰。
12.进一步地,所述晶型b的粉末x射线衍射图中,在7.8、11.8、13.5、15.7、17.2、21.4、23.7、24.1、24.9的2θ值处存在特征峰。
13.进一步地,所述晶型b的粉末x射线衍射图如附图6所示。
14.进一步地,本发明提供了所述的6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈的马来酸盐晶型b的制备方法,包括以下步骤:将6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈游离碱溶于四氢呋喃中,冰浴下滴加马来酸的四氢呋喃溶液,搅拌,于50℃下反应;反应结束后降温到室温,过滤,再于50℃下真空干燥,得到所述晶型b。
15.进一步地,本发明还提供了另一种制备方法,包括以下步骤:将6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈游离碱溶于2-甲基四氢呋喃中,-15℃下滴加马来酸的2-甲基四氢呋喃溶液,搅拌,于50℃下反应;反应结束后降温至室温,过滤,接着采用冷的2-甲基四氢呋喃淋洗,滤饼于50℃下真空干燥,得到所述晶型b。
16.与现有技术相比,本发明的有益效果在于:本发明提供了6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈的两种马来酸盐新晶型——晶型a和晶型b,相比于游离碱、富马酸盐晶型a和乙醇酸盐晶型a,这两种马来酸盐新晶型在一定条件下具有更好的溶解性和更高的稳定性。
附图说明
17.图1为游离碱的xrpd图谱;图2为游离碱的tga/dsc叠图;图3为游离碱的plm图谱;图4为马来酸盐晶型a的xrpd图谱;图5为马来酸盐晶型a的tga/dsc叠图;
图6为马来酸盐晶型b的xprd图谱;图7为马来酸盐晶型b的tga/dsc叠图;图8为马来酸盐晶型b的dvs曲线;图9为马来酸盐晶型b样品dvs测试前后的xrpd叠图;图10为富马酸盐晶型a的xrpd谱图;图11为富马酸盐晶型a的tga/dsc谱图;图12为富马酸盐晶型a的dvs曲线;图13为乙醇酸盐晶型a的xrpd谱图;图14为乙醇酸盐晶型a的tga/dsc谱图;图15为乙醇酸盐晶型a的dvs曲线。
具体实施方式
18.下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
19.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“及/或”包括一个或多个相关的所列项目的任意的和所有的组合。
20.一、分析试验方法1. x射线粉末衍射仪(xrpd)利用x-粉末衍射仪对样品进行晶型分析。样品的2θ扫描角度为3
°
到42
°
,扫描步长为0.02
°
,每步的扫描时间为0.2 s。光管电压和电流分别为40 kv和40 ma。制样时将适量样品放到载样盘上,用勺子或玻璃片等工具压平,确保其表面光滑平整。
21.2. 差示扫描量热分析(dsc)采用ta instruments discovery dsc 25对样品进行分析。将称量过的样品放入载样盘中,在氮气(50 ml/min)的保护下将样品以10℃/min的速率升高到最终温度。
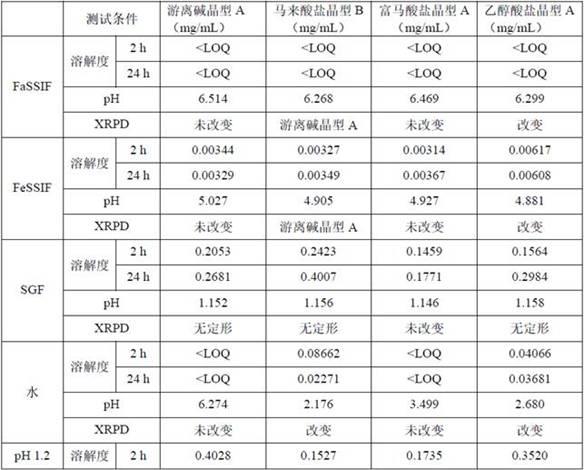
22.3. 热重分析仪 (tga)采用ta instruments tga discovery 550对样品进行分析。将样品放入去掉皮重的铝盘中,系统自动称重,然后在氮气的保护下将样品以10℃/min的速率升高到最终温度。
23.4. 高效液相色谱(hplc)试验中纯度和溶解度由安捷伦1260高效液相色谱仪测试,分析条件如下。
24.色谱条件:色谱柱:zorbax eclipse xdb-c18,4.6 mm *150 mm,5-micron流动相:a相:0.1 mol/l乙酸钠水溶液 (乙酸调ph= 5
±
0.5)
ꢀꢀꢀꢀꢀꢀꢀ
b相:乙腈洗脱程序:时间(min)0.09.015.020.025.026.030.0b(%)30.045.045.090.090.030.030.0 流速:1.0 ml/min
柱温:30℃样品盘温度:4℃进样体积:10 μl稀释剂:乙腈检测波长:254 nm二、制备例实施例1:游离碱晶型a的制备参照wo2018071348a1中公开的方法制备得到6-((5-甲氧基-6-((6-甲基吡啶-3-基)甲氧基)吡啶-3-基)氨基)-3-吗啉代喹喔啉-5-甲腈游离碱化合物(游离碱晶型a),其xrpd图谱如图1所示,tga/dsc图谱如图2所示,偏光显微镜(plm)图谱如图3所示。
25.实施例2:马来酸盐晶型a的制备称量约30 mg由实施例1制得的游离碱化合物(api),加入600 μl二氯甲烷(dcm),再称取适量的马来酸配体在0℃加入,api和配体的摩尔比为1:1.2。在磁力搅拌的作用下于50℃反应16 h,然后降到室温后过滤,滤液继续降温到0℃后再过滤。然后对固体样品进行分析,得到马来酸盐晶型a。
26.制备得到的马来酸盐晶型a的 xrpd图谱如图4所示, tga/dsc叠图如图5所示。
27.实施例3:马来酸盐晶型b的制备称取1 g由实施例1制得的游离碱化合物(api),加入8 ml四氢呋喃(thf),称取291.62 mg (1.2eq)的马来酸溶于12 ml的thf中,冰浴下将马来酸的thf溶液滴加到api溶液中,室温搅拌1 h后,于50℃反应16 h,后降温到室温,过滤,于50℃真空干燥20 h。得到1.1 g马来酸盐,收率为88.9%。xrpd显示,该马来酸盐和实施例2制备的马来酸盐晶型a不一致,命名为马来酸盐晶型b。
28.图6显示了马来酸盐晶型b的xrpd图谱。图7显示了马来酸盐晶型b的tga/dsc谱图。图8为马来酸盐晶型b的dvs曲线,dvs曲线显示在湿度80%下,增重0.1784%,几乎无引湿性。图9显示了马来酸盐晶型b 样品dvs测试前后的xrpd叠图,结果显示dvs测试前后晶型没有发生改变。
29.实施例4:马来酸盐晶型b的制备称取约100mg由实施例1制得的游离碱化合物(api),加入1.5 ml 2-甲基四氢呋喃(2-methf),然后称取1.2 eq(28.86 mg)马来酸溶于0.5 ml的2-methf中,-15℃下将马来酸的2-methf溶液滴加到api的2-methf混悬液中,滴加完毕,室温搅拌3 h,然后放在50℃搅拌16 h。降到室温过滤,冷的2-methf淋洗两遍,滤饼于50℃真空干燥20 h。得到约100 mg淡黄色固体。晶型与马来酸盐晶型b相同。
30.实施例5:富马酸盐晶型a的制备称取1 g 由实施例1制得的游离碱化合物(api),加入7 ml乙醇,称取288.9 mg (1.2eq)的富马酸溶于13 ml乙醇中,冰浴下将富马酸的乙醇溶液滴加到api的乙醇溶液中,室温搅拌1 h后,于50℃反应16 h,后降温到室温,过滤,于50℃真空干燥20 h,得到1.08 g富马酸晶型a,收率为87%。
31.富马酸晶型a的xrpd图谱如图10所示。图11显示了富马酸盐晶型a的tga/dsc谱图。dvs曲线如图12所示,显示在湿度80%下,增重0.4332%,略有引湿性。
32.实施例6:乙醇酸盐晶型a的制备称取1 g 由实施例1制得的游离碱化合物(api),溶于二氯甲烷(dcm)中,称取189.13 mg乙醇酸溶于dcm中,然后滴加到api的dcm溶液中,室温搅拌1 h后,于50℃反应16 h,后降温到室温,过滤,于50℃真空干燥20 h,得到710 mg乙醇酸盐晶型a。
33.乙醇酸盐晶型a的xrpd谱图如图13所示,图14显示了乙醇酸盐晶型a的tga/dsc谱图。dvs曲线如图15所示,结果显示在湿度为80%的条件下吸湿增重0.3754%,表明略有引湿性。
34.三、测试例1.不同盐晶型与游离碱化合物的溶解性比较分别称取约20毫克起始原料(游离碱晶型a)、实施例4制备的马来酸盐晶型b,实施例5制备的富马酸盐晶型a和实施例6制备的乙醇酸盐晶型a于样品瓶中,向各样品瓶中分别加入2毫升fassif、fessif、sgf、水和ph分别为1.2(氯化钾)、3.0(邻苯二甲酸氢钾)、4.5(三水乙酸钠)、6.8(磷酸二氢钾)、7.5(磷酸二氢钾)的缓冲液中,放入37℃水浴中搅拌。分别于2小时、24小时的时间点进行取样。所得滤液进行hplc的测试,剩余固体进行xrpd测试。测试结果如表1所示。
35.表1 不同盐晶型与游离碱化合物的溶解性比较
从表1的结果可以看出,所有样品除了在sgf和ph=1.2的缓冲液中具有一定的溶解度(溶解度最大的是马来酸盐晶型b,在sgf的溶解度达到0.4007mg/ml)之外,在其余缓冲液中的溶解度均很小。
36.为了初步筛选增溶剂对游离碱晶型a、马来酸盐晶型b,富马酸盐晶型a和乙醇酸盐晶型a的影响,通过观察法粗略测试样品的溶解度。取适量样品,然后分别加入不同种类的增溶剂——水、磺丁基倍他环糊精钠(20% sbecd)、羟丙基-β-环糊精(20% hpcd)、20% peg400、5% solutol hs15、1% 吐温-80、1%十二烷基硫酸钠和维生素e琥珀酸聚乙二醇酯(5%tpgs),最多加至8 ml,溶解度结果如表2所示。通过初步筛选发现,在1%十二烷基硫酸钠水溶液中,马来酸盐晶型b溶解度能达到1mg/ml以上。
37.表2
ꢀꢀ
样品在增溶剂中的粗略溶解度
2. 稳定性评估将一定量的样品(游离碱晶型a、马来酸盐晶型b、富马酸盐晶型a、乙醇酸盐晶型a)放入稳定性试验箱中,特定时间后取出检测hplc、xrpd。测试条件:25℃/60 %rh(敞口),取样时间:一周、两周、四周。结果如表3所示。
38.表3
ꢀꢀ
候选晶型的稳定性测试结果表3的稳定性结果显示,在25℃/60%rh下放置4周,游离碱晶型a降解0.02%,马来酸盐晶型b降解0.01%,富马酸盐晶型a降解0.15%,乙醇酸盐晶型a降解 0.02%。结果表明,马来酸盐晶型b最稳定。
39.混悬竞争实验
为了确定马来酸盐的最稳定晶型,将马来酸盐晶型a和晶型b进行混悬竞争实验。称取同等质量的马来酸盐晶型a和晶型b,分别加入到不同溶剂中,在50℃下进行混悬竞争打浆实验,于1天和3天检测晶型,结果如表4所示。
40.表4
ꢀꢀ
马来酸盐晶型a和晶型b的混悬竞争实验结果从表4的结果可以看出,晶型a在醋酸乙酯、二氯甲烷、2-甲基四氢呋喃、四氢呋喃中1天就转化为晶型b,在丙酮、正己烷和水中3天后晶型也完全转化为晶型b,故在上述溶剂中晶型b比晶型a更稳定。
41.以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1